Scheme 1.
Different C—C bond cleavage methods of ketones
Triethylamine Promoted the C-C Bond Cleavage of α-Halo Ketones: α-Acetoxyaryl Ketone Synthesis
Maorui Wang , Yuzheng Wu , Jian Yao , Li Deng , Yingming Pan , Kebin Huang , Haitao Tang

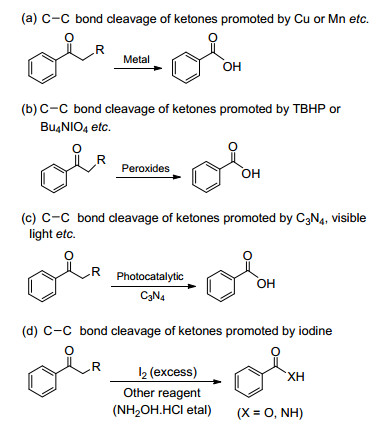
The C—C bond cleavage plays an important role in organic chemistry because this process generates many compounds.[1] However, breaking C—C bonds under mild conditions remains a challenge because of the thermodynamically stability and kinetically inert characteristics. Traditional methods of inducing C—C bond cleavage rely on harsh reaction conditions, such methods include the use of salts and peroxides or vapor phase cracking at high temperatures.[2~4] Wang and Ishii et al.[5] studied Cu- or Mn-catalyzed C—C bond cleavage reactions from ketones to benzoic acids (Scheme 1a). Shen and Lee et al.[6] reported equivalent t-butyl hydroperoxide (TBHP) or Bu4NIO4 oxidative C—C bond cleavage reactions from ketones to benzoic acids (Scheme 1b). Wang et al.[7] reported visible light-catalyzed C—C bond cleavage reactions from ketones to benzoic acids, but this method requires heterogeneous C3N4 material as a catalyst (Scheme 1c). Bathula and Wu et al.[8] presented the use of excess iodine to promote the oxidative C—C bond cleavage reactions from alkyl aryl ketones to benzoic acids or benzamides (Scheme 1d). Therefore, the development of novel metal-free, simple, inexpensive methods for C—C bond cleavage of ketone compounds is important for organic chemists.
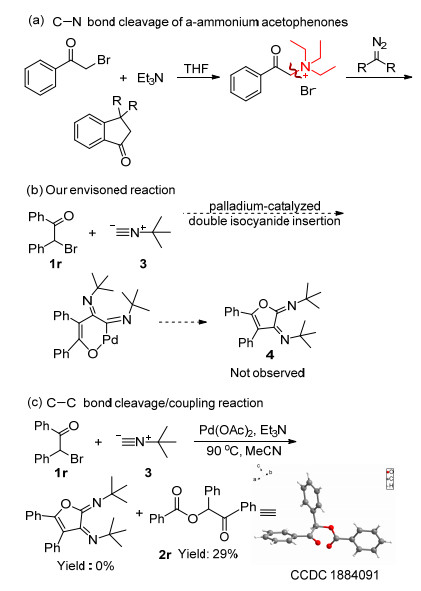
α-Halo ketones are a very important organic synthon that can undergo a series of transformations.[9] In 2015, Li's group[10] studied the synthesis of benzocyclopentanones by C—N bond cleavage from benzoylammonium salt which was synthesized from triethylamine and α-halo ketones (Scheme 2a). A variety of substrates were previously reported to react with isonitrile to synthesize many compounds with biologically active backbones.[11] The diphenylfuran derivative 4 from 2-bromo-1, 2-dipheny-lethan-1-one (1r) and isonitrile 3 were also synthesized. First, the substrates 1r and 3 were treated with 0.4 mmol, 1.5 mL of MeCN, and 10 mol% Pd(OAc)2. Product 4 was not formed, and 2-oxo-1, 2-diphenylethyl benzoate (2r) was obtained with 29% yield (Schemes 2b, 2c). The structure of 2-oxo-1, 2-diphenylethyl benzoate was confirmed by single-crystal X-ray analysis.
A literature survey of the reaction was conducted, which indicated that product 4 required the addition of an oxidant or a rare iron and copper complex metal when undergoing similar reactions.[12] The reaction of cleavage of C—C bond of α-halo ketones by triethylamine followed by dimerization to the α-acetoxyaryl ketone was not reported. Thus, we decided to investigate the dimerization of 2-bromo-1-phenylethan-1-one by C—C bond cleavage of 2-bromo-1-phenylethan-1-one in the presence of Et3N.
The reaction parameters of 1a dimerization were studied using 2-bromo-1-phenylethan-1-one (1a) as a model compound (Table 1). When the reaction was treated with Pd(OAc)2 (10 mol%) and Et3N (0.1 mmol) in MeCN at 90 ℃, product 2a was obtained with 44% yield. When the reaction was carried out in the absence of Pd(OAc)2, product 2a was still obtained with 46% yield. The result concluded that Pd(OAc)2 had no effect on the reaction (Table 1, Entries 1~2). Et3N is increased to 2 equiv., and the yield of 2a is increased to 66%. (Table 1, Entries 3~5). The organic base (NHEt2, (CH3)3CNH2, DBU) was then screened and the yield of 2a did not increase (Table 1, Entries 6~8). When the inorganic bases (NaOH, Cs2CO3) were screened, the reaction was not promoted (Table 1, Entries 9~10). After screening for the best base, the best solvent was found to be MeCN (Table 1, Entries 11~14). The temperature was further screened and the yield of 2a increased to 89% at 120 ℃ (Table 1, Entry 15). Therefore, the optimal conditions for preparing 2a were as follows: 1a as substrate, MeCN as solvent at 120 ℃ in Et3N for 8 h (Table 1, Entry 10).
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entry | Catalyst | Base (equiv.) | Solvent | Yieldb/% |
| 1 | Pd(OAc)2 | Et3N (0.5) | MeCN | 44c |
| 2 | — | Et3N (0.5) | MeCN | 46 |
| 3 | — | Et3N (1) | MeCN | 55 |
| 4 | — | Et3N (2) | MeCN | 66 |
| 5 | — | Et3N (3) | MeCN | 60 |
| 6 | — | Et2NH (2) | MeCN | 45 |
| 7 | — | (CH3)3CNH2 (2) | MeCN | Trace |
| 8 | — | DBU (2) | MeCN | NR |
| 9 | — | NaOH (2) | MeCN | NR |
| 10 | — | Cs2CO3 (2) | MeCN | Trace |
| 11 | — | Et3N (2) | MeCN/H2Od | 43 |
| 12 | — | Et3N (2) | 1, 4-Dioxane | NR |
| 13 | — | Et3N (2) | DMF | Trace |
| 14 | — | — | N(Et)3 | 23 |
| 15 | — | Et3N (2) | MeCN | 89e |
| a Reaction conditions: 1a (0.2 mmol), Et3N (0.4 mmol), solvent (1.5 mL), 90 ℃, 8 h. b Isolated yield. c With 3 equiv. of 2-isocyano-2-methylpropane and Pd(OAc)2 (10 mol %). d MeCN/H2O (V:V=2:1). e 120 ℃. | ||||
Under the optimal reaction conditions, the substrate range in which different α-haloketones were dimerized to construct a plurality of α-acetoxyaryl ketones was explored (Table 2). When X=Br, the 2a with 89% yield was separated, and when X=Cl, 2a was obtained with a slightly reduced yield (81%) (Table 2, 2a). When R1 is H and R2 is substituted Ar, a series of phenyl groups with electron donating groups (R=2-OCH3, 3-OCH3, 4-OCH3, 4-CH3, 4-t-Bu) led to high yields of 2b~2f. The steric hindrance exerted a certain influence on the yield. When R=2-OC-H3, the yield decreased. When R1 is H and R2 is substituted Ar, a series of phenyl groups with electron withdrawing groups (R=2-F, 4-Br, 4-OCF3, 4-CN, 3-NO2, 4-F) produced 2g~2l. When R1 is H and R2 is substituted Ar, the yield when R is an electron-donating group is higher than that electron-withdrawing group. When R1 is H and R2 is disubstituted Ar (R=3, 4-(OCH3)2, 3, 4-F2), the reaction proceeded smoothly and 2m~2n were obtained in moderate yield. When alkyl ketones were used as substrates, the reactions failed (2t and 2u).
Fused or heterocyclic substrates, including 2-bromo-1-(thiophen-2-yl)ethan-1-one, 2-bromo-1-(naphthalen-2-yl)-ethan-1-one, and 1-(benzofuran-2-yl)-2-bromoethan-1-one, were also tolerated (2o~2q). Furthermore, when R is H of the benzene ring, R1 is a methyl group or an Ar substituent, the substrates 1r and 1s can give the desired products 2r and 2s in moderate yield (2r~2s).
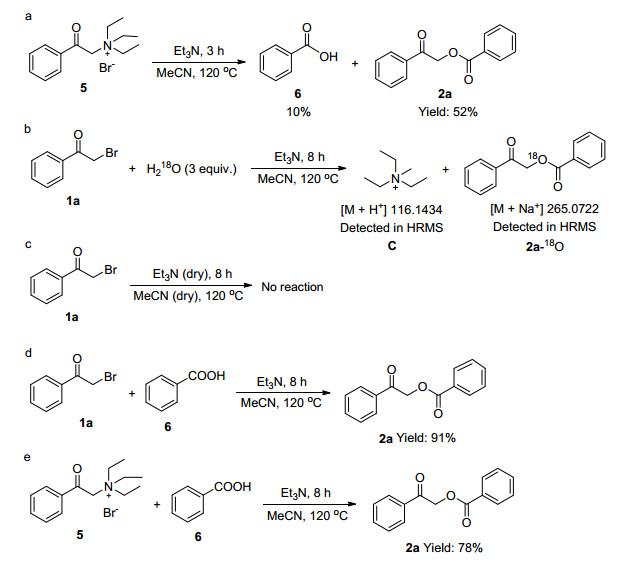
Control experiments were conducted to study the reaction mechanism (Scheme 3). First, N, N, N-triethyl-2-oxo-2-phenylethan-1-ammonium 5 was used as the reaction substrate to obtain 6 in 10% yield. The result indicated that when 5 reacted with water, 2a was obtained in 52% yield (Scheme 3a). 18O-labeled water was added to 2-bromo-1-phenylethan-1-one (1a) under the optimal conditions, and compound C was observed by HRMS. Triethylamine promoted the C—C bond cleavage of 1a. At the same time, 18O-labeled compound 2a-18O was detected by HRMS, indicating water participation (Scheme 3b). If this reaction was conducted in dry Et3N and CH3CN, the reaction failed (Scheme 3c), which further demonstrated that water played a key role in this reaction. When the benzoic acid 6 and 2-bromo-1-phenylethan-1-one (1a) or α-ammonium acetophenone (5) were used as the reaction substrates, 2-oxo-2-phenylethyl benzoate (2a) was obtained with 91% and 78% yields. These results indicated that benzoic acid 6 and α-ammonium acetophenone 5 are the key intermediates for the formation of 2-oxo-2-phenylethyl benzoate (2a) (Scheme 3d). To further study the mechanism, we conducted a crossover experiment, which obtained the target products A and B (A:B=100:46, determined by LC-HRMS). This result indicated that the reaction was an intermolecular process and the substrate with electron donating group was more convenient for the reaction.
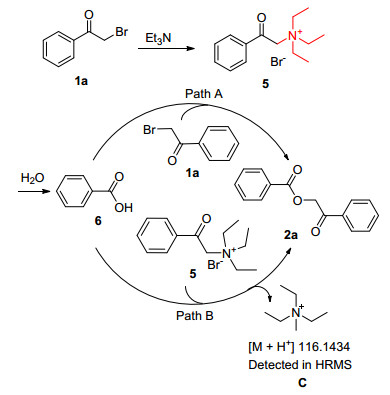
Analysis of the reaction mixture by HRMS showed the presence of compound C and intermediate 6. On the basis of the controlled experiments, two rational mechanisms of the reaction can be proposed (Scheme 4). Intermediate 5, initially formed by adding Et3N to 2-bromo-1-phenylethan-1-one 1a, could react with water to form intermediate 6, which then reacts with 1a to obtain 2a (Path A) and with 5 to obtain 2a (Path B).
This study proposes a simple method that does not require metals, oxidants, and water for the cleavage of C—C bond of α-halo ketones by trimethylamine, then followed by dimerization to obtain valuable α-acetoxyaryl ketones. The method not only provides a new approach to synthesize α-acetoxyaryl ketone, but also expands the application of C—C bond cleavage of α-halo ketones.
Unless otherwise noted, all reagents and solvents were obtained commercially and used without further purification. Column chromatography on silica gel (300-400 mesh) was carried out using technical grade 60~90 ℃ petroleum ether (distillated prior to use) and analytical grade EtOAc (without further purification). 1H NMR and 13C NMR spectra were recorded on a 400 MHz spectrometer. 1H NMR spectra were referenced to CDCl3 (δ: 7.26), and 13C NMR spectra were referenced to CDCl3 (δ: 77.0). HRMS spectra were recorded with a Micromass QTOF2 Quadrupole/Time-of-Flight Tandem mass spectrometer using electron spray ionization.
A solution of α-halo ketones 2-bromo-1-phenylethan-1-one (0.2 mmol), NEt3 (0.45 mmol), and solvent (1.5 mL) was placed in a 15 mL test tube and stirred at 120 ℃ for 8 h. When the reaction was completed (monitored by TLC, about 8 h), the mixture was concentrated and the residue was chromatographed through silica gel eluted with ethyl acetate/petroleum ether to obtain 2-oxo-2-phenylethyl benzoate.
2-Oxo-2-phenylethyl benzoate (2a): Yield 89%, 42.7 mg. Yellow solid, m.p. 114.6~115.8 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.15 (d, J=7.0 Hz, 2H), 7.98 (d, J=7.5 Hz, 2H), 7.64~7.58 (m, 2H), 7.53~7.46 (m, 4H), 5.58 (s, 2H); 13C NMR (125 MHz, CDCl3) δ: 192.1, 166.0, 134.3, 133.9, 133.3, 130.0, 129.4, 128.9, 128.4, 127.8, 66.4. HRMS (ESI) calcd for C15H12O3Na [M+Na]+ 263.0684, found 263.0679.
2-(2-Methoxyphenyl)-2-oxoethyl 2-methoxybenzoate (2b): Yield 71%, 42.6 mg. Yellow solid, m.p. 75.4~77.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.74 (d, J=7.6 Hz, 1H), 7.66~7.62 (m, 1H), 7.54~7.46 (m, 2H), 7.41~7.34 (m, 2H), 7.18~7.10 (m, 2H), 5.54 (s, 2H), 3.83 (d, J=2.4 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 191.9, 165.8, 159.9, 159.5, 135.5, 130.6, 129.8, 129.4, 122.3, 120.3, 120.2, 120.0, 114.2, 112.1, 66.5, 55.4, 55.4. LC-MS (ESI) calcd for C17H17O5 [M+H]+ 301.1071, found 301.1074.
2-(3-Methoxyphenyl)-2-oxoethyl 3-methoxybenzoate (2c): Yield 83%, 49.8 mg. Yellow solid, m.p. 66.1~68.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.75 (d, J=7.6 Hz, 1H), 7.65 (s, 1H), 7.57~7.48 (m, 2H), 7.43~7.34 (m, 2H), 7.19~7.09 (m, 2H), 5.55 (s, 2H), 3.85 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 191.9, 165.9, 160.0, 159.6, 135.6, 130.7, 129.9, 129.5, 122.4, 120.4, 120.2, 120.1, 114.2, 112.1, 66.6, 55.5, 55.4. LC-MS (ESI) calcd for C17H17O5 [M+H]+ 301.1071, found 301.1074.
2-(4-Methoxyphenyl)-2-oxoethyl 4-methoxybenzoate (2d): Yield 90%, 54.0 mg. Brown solid, m.p. 207.8~209.0 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=8.8 Hz, 2H), 7.93 (d, J=8.8 Hz, 2H), 6.93 (t, J=8.6 Hz, 4H), 5.48 (s, 2H), 3.86 (d, J=3.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 190.9, 165.7, 164.0, 163.6, 132.0, 130.1, 127.4, 121.9, 114.0, 113.6, 66.0, 55.4, 55.4. LC-MS (ESI) calcd for C17H17O5 [M+ H]+ 301.1071, found 301.1072.
2-Oxo-2-(p-tolyl)ethyl 4-methylbenzoate (2e): Yield 86%, 46.1 mg. Brown solid, m.p. 139.0~142.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J=8.0 Hz, 2H), 7.83 (d, J=8.0 Hz, 2H) 7.25~7.21 (m, 4H), 5.49 (s, 2H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 191.9, 166.1, 144.8, 144.0, 131.9, 130.0, 129.5, 129.1, 127.9, 126.7, 66.3, 21.7, 21.7. LC-MS (ESI) calcd for C17H17O3 [M+ H]+ 269.1172, found 269.1170.
2-(4-(tert-Butyl)phenyl)-2-oxoethyl 4-(tert-butyl)-benzoate (2f): Yield 80%, 56.4 mg. Brown solid, m.p. 196.2~198.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=8.4 Hz, 2H), 7.92 (d, J=8.4 Hz, 2H), 7.54~7.47 (m, 4H), 5.55 (s, 2H), 1.35 (s, 18H); 13C NMR (100 MHz, CDCl3) δ: 191.8, 166.1, 157.7, 157.0, 131.9, 129.9, 127.8, 126.7, 125.8, 125.4, 66.3, 35.2, 35.1, 31.1, 31.0. LC-MS (ESI) calcd for C23H29O3 [M+H]+ 353.2117, found 353.2111.
2-(3-fluorophenyl)-2-oxoethyl 3-fluorobenzoate (2g): Yield 84%, 46.4 mg. Yellow solid, m.p. 127.6~129.7 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.94~7.90 (m, 1H), 7.83~7.78 (m, 1H), 7.74 (d, J=7.8 Hz, 1H), 7.67~7.63 (m, 1H), 7.52~7.43 (m, 2H), 7.35~7.28 (m, 2H), 5.55 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 190.6 (d, J=3 Hz), 164.8 (d, J=3 Hz), 162.8 (d, J=247.5 Hz), 162.5 (d, J=246 Hz), 136.0 (d, J=7.5 Hz), 131.3 (d, J=7.5 Hz), 130.7 (d, J=7.5 Hz), 130.1 (d, J=7.5 Hz), 125.7 (d, J=3 Hz), 123.5 (d, J=3 Hz), 121.1 (d, J=21 Hz), 120.5 (d, J=21 Hz), 116.8 (d, J=22.5 Hz), 114.6 (d, J=22.5 Hz), 66.6. HRES (ESI) calcd for C15H10F2O3Cl [M+ Cl]- 311.287 found 311.0292.
2-(4-Bromophenyl)-2-oxoethyl 4-bromobenzoate (2h): Yield 88%, 69.7 mg. Yellow solid, m.p. 128.2~130.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.00~7.96 (m, 2H), 7.84~7.79 (m, 2H), 7.66~7.59 (m, 4H), 5.51 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 191.0, 165.3, 132.9, 132.3, 131.9, 131.4, 129.3, 128.7, 128.1, 66.4. HRES (ESI) calcd for C15H10Br2O3Na [M+Na]+ 418.8894, found 418.8889.
2-Oxo-2-(4-(trifluoromethoxy)phenyl)ethyl 4-(trifluoro-methoxy) benzoate (2i): Yield 81%, 66.1 mg. Yellow solid, m.p. 133.8~135.7 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.219 (d, J=8.5 Hz, 2H), 8.02 (d, J=8.5 Hz, 2H), 7.32 (dd, J=18.5, 8.5 Hz, 4H), 5.56 (s, 2H); 13C NMR (125 MHz, CDCl3) δ: 190.4, 164.8, 153.2 (d, J=1.6 Hz), 153.0 (d, J=1.6 Hz), 132.3, 131.0 (d, J=262.3 Hz), 127.6, 123.4 (d, J=4.6 Hz), 121.3 (d, J=5.0 Hz), 120.5 (d, J=42.2 Hz), 119.2 (d, J=5.5 Hz), 117.2 (d, J=5.9 Hz), 66.4. HRES (ESI) calcd for C17H10F6O5Na [M+Na]+ 431.0330, found 431.0333.
2-(4-Cyanophenyl)-2-oxoethyl 4-cyanobenzoate (2j): Yield 63%, 36.5 mg. Yellow solid, m.p. 107.8~109.6 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.25~8.21 (m, 2H), 8.06 (d, J=8.5 Hz, 2H), 7.86~7.78 (m, 4H), 5.60 (s, 2H); 13C NMR (125 MHz, CDCl3) δ: 190.4, 164.3, 136.9, 132.8, 132.4, 130.5, 128.3, 117.8, 117.5, 117.5, 117.0, 66.8. HRES (ESI) calcd for C17H10N2O3Cl [M+Cl]- 325.0380, found 325.0385.
2-(3-Nitrophenyl)-2-oxoethyl 3-nitrobenzoate (2k): Yield 64%, 42.2 mg. Yellow solid, m.p. 123.2~125.3 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.96 (s, 1H), 8.79 (s, 1H), 8.52~8.44 (m, 3H), 8.31 (d, J=8.0 Hz, 1H), 7.79~7.70 (m, 2H), 5.68 (s, 2H); 13C NMR (125 MHz, CDCl3) δ: 189.6, 163.9, 148.5, 148.3, 135.6, 135.1, 133.4, 130.8, 130.4, 129.9, 128.3, 128.0, 125.0, 122.7, 66.9. LC-MS (ESI) calcd for C15H10N2O7Na [M+Na]+ 353.0386, found 353.0397.
2-(4-Fluorophenyl)-2-oxoethyl 4-fluorobenzoate (2l): Yield 53%, 37.5 mg. Yellow solid, m.p. 165.8~167.3 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.18~8.13 (m, 2H), 8.02~7.98 (m, 2H), 7.21~7.12 (m, 4H), 5.53 (s, 2H); 13C NMR (125 MHz, CDCl3) δ: 190.5, 166.2 (d, J=254.7 Hz), 166.1(d, J=253.2 Hz), 165.1, 132.6 (d, J=9.4 Hz), 130.7 (d, J=3.2 Hz), 130.5 (d, J=9.5 Hz), 125.6 (d, J=3.0 Hz), 116.2 (d, J=22.1 Hz), 115.7 (d, J=22.1 Hz), 66.3. HRES (ESI) calcd for C15H10F2O3Na [M+Na]+ 299.0496, found 299.0488.
2-(3, 4-Dimethoxyphenyl)-2-oxoethyl 3, 4-dimethoxy-benzoate (2m): Yield 61%, 43.9 mg. Yellow solid, m.p. 155.6~157.3 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.79 (dd, J=8.5, 1.5 Hz, 1H), 7.63~7.52 (m, 3H), 6.91 (d, J=8.5 Hz, 2H), 5.51 (s, 2H), 3.95~3.92 (m, 12H); 13C NMR (125 MHz, CDCl3) δ: 190.9, 165.8, 153.9, 153.3, 149.3, 148.6, 127.5, 124.1, 122.3, 121.9, 112.2, 110.3, 110.2, 110.0, 66.1, 56.1, 56.0, 56.0. LC-MS (ESI) calcd for C19H21O7 [M+H]+ 361.1282, found 361.1284.
2-(3, 4-Difluorophenyl)-2-oxoethyl 3, 4-difluorobenzoate (2n): Yield 73%, 45.6 mg. Yellow solid, m.p. 74.8~76.7 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.98~7.92 (m, 2H), 7.84~7.80 (m, 1H), 7.77~7.74 (m, 1H), 7.35~7.27 (m, 2H), 5.53 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 189.3, 164.0 (d, J=1.7 Hz), 154.9 (dd, J=29.9, 12.7 Hz), 153.2 (dd, J=28.2, 12.8 Hz), 151.2 (dd, J=78.4, 13.1 Hz), 149.5 (dd, J=76.4, 13.0 Hz), 131.0 (t, J=4.1 Hz), 127.0 (dd, J=7.5, 3.6 Hz), 126.1 (dd, J=5.7, 3.6 Hz), 124.8 (dd, J=7.6, 3.6 Hz), 119.3 (dd, J=18.8, 1.6 Hz), 118.0 (d, J=18.0 Hz), 117.6(d, J=18.0 Hz), 117.3 (dd, J=18.2, 1.7 Hz), 66.4. HRES (ESI) calcd for C15H8F4O3C1 [M+ Cl]- 347.0098, found 347.0104.
2-(Naphthalen-2-yl)-2-oxoethyl 2-naphthoate (2o): Yield 56%, 38.1 mg. Brown solid, m.p. 222.9~224.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.76 (s, 1H), 8.52 (s, 1H), 8.18 (dd, J=8.8, 1.6 Hz, 1H), 8.06~7.88 (m, 7H), 7.66~7.54 (m, 4H), 5.78 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 192.1, 166.3, 135.9, 135.8, 132.5, 132.4, 131.7, 129.6, 129.5, 128.9, 128.4, 128.3, 127.9, 127.8, 127.1, 126.7, 125.4, 123.4, 66.6. LC-MS (ESI) calcd for C23H17O3 [M+H]+ 341.1172, found 341.1170.
2-(Benzofuran-2-yl)-2-oxoethyl benzofuran-2-carboxylate (2p): Yield 76%, 48.7 mg. Black solid, m.p. 201.8~203.6 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.75~7.69 (m, 3H), 7.67 (d, J=0.6 Hz, 1H), 7.63~7.59 (m, 2H), 7.54~7.47 (m, 2H), 7.36~7.32 (m, 2H), 5.61 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 183.0, 158.7, 155.9, 155.6, 150.2, 144.5, 128.8, 128.0, 126.9, 126.7, 124.2, 123.9, 123.5, 123.0, 115.3, 113.6, 112.5, 112.4, 66.4. LC-MS (ESI) calcd for C19H13O5 [M+H]+ 321.0757, found 321.0759.
2-Oxo-2-(thiophen-2-yl)ethyl thiophene-2-carboxylate (2q): Yield 81%, 40.8 mg. Yellow solid, m.p. 139.5~141.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.93~7.89 (m, 1H), 7.81 (d, J=3.6 Hz, 1H), 7.72 (d, J=4.8 Hz, 1H), 7.63~7.60 (m, 1H), 7.19~7.12 (m, 2H), 5.41 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 185.2, 161.4, 140.4, 134.4, 134.4, 133.2, 132.5, 132.1, 128.3, 127.9, 66.2. LC-MS (ESI) calcd for C11H9O3S2 [M+H]+ 252.9988, found 252.9989.
2-Oxo-1, 2-diphenylethyl benzoate (2r): Yield 77%, 48.7 mg. Yellow solid, m.p. 118.6~120.7 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.16~8.13 (m, 2H), 8.03~8.01 (m, 2H), 7.61~7.51 (m, 4H), 7.46~7.36 (m, 7H), 7.12 (s, 1H); 13C NMR (150 MHz, CDCl3) δ: 193.6, 166.0, 134.6, 133.7, 133.5, 133.3, 129.9, 129.3, 129.3, 129.1, 128.8, 128.6, 128.4, 77.9. LC-MS (ESI) calcd for C21H17O3 [M+H]+317.1172, found 317.1170.
1-Oxo-1-phenylpropan-2-yl benzoate (2s): Yield 65%, 33.0 mg. White solid, m.p. 97.6~99.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12~8.06 (m, 2H), 8.03~7.98 (m, 2H), 7.63~7.55 (m, 2H), 7.52~7.42 (m, 4H), 6.24~6.18 (m, 1H), 1.67 (d, J=6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 196.7, 166.0, 134.5, 133.6, 133.3, 129.9, 129.5, 128.8, 128.5, 128.4, 71.9, 17.2. LC-MS (ESI) calcd for C16H15O3 [M+H]+ 255.1016, found 255.1016.
Supporting Information Copies of 13C NMR and 1H NMR spectra for products, and MP and MS data of all products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410.
(b) Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613.
(c) Wang, T.; Jiao, N. Acc. Chem. Res. 2014, 47, 1137.
(d) Murakami, M.; Ishida, N. J. Am. Chem. Soc. 2016, 138, 13759.
(e) Tiwari, B.; Zhang, J.; Chi, Y. R. Angew. Chem. Int. Ed. 2012, 51, 1911.
(f) Sai, M.; Yorimitsu, H.; Oshima, K. Angew. Chem. Int. Ed. 2011, 50, 3294.
(g) Chen, W.-L.; Wu, S.-Y.; Mo, X.˗L.; Wei, L.˗X.; Liang, C.; Mo, D.˗L. Org. Lett. 2018, 20, 3527.
(a) Tobisu, M.; Chatani, N. Chem. Soc. Rev. 2008, 37, 300.
(b) Qin, C.; Shen, T.; Tang, C.; Jiao, N. Angew. Chem. Int. Ed. 2012, 51, 6971.
(c) Qin, C.; Zhou, W.; Chen, F.; Ou, Y.; Jiao, N. Angew. Chem. Int. Ed. 2011, 50, 12595.
(d) Klein, J. E. M. N.; Plietker, B. Org. Biomol. Chem. 2013, 11, 1271.
(e) Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597.
(f) Bart, S. C.; Chirik, P. J. J. Am. Chem. Soc. 2003, 125, 886.
(g) Seiser, T.; Cramer, N. J. Am. Chem. Soc. 2010, 132, 5340.
(h) Seiser, T.; Saget, T.; Tran, D. N.; Cramer, N. Angew. Chem. Int. Ed. 2011, 50, 7740.
(i) Jun, C-H. Chem. Soc. Rev., 2004, 33, 610.
(a) Kumar, K. A. A.; Venkateswarlu, V.; Vishwakarma, R. A.; Sawant, S. D. Synthesis 2015, 47, 3161.
(b) Zhang, J.; Jiang, J.; Li, Y.; Zhao, Y.; Wan, X. Org. Lett. 2013, 15, 3222.
Liu, L.; Ishida, N.; Murakami, M. Angew. Chem. Int. Ed. 2012, 51, 2485. doi: 10.1002/anie.201108446
(a) Wang, M.; Lu, J.; Zhang, X.; Li, L.; Li, H.; Luo, N.; Wang, F. ACS Catal. 2016, 6, 6086.
(b) Liu, H.; Wang, M. Li, H.; Luo, N.; Xu, S.; Wang, F. J. Catal. 2017, 346, 170.
(c) Wang, M.; Lu, J.; Li, L.; Li, H.; Liu, H.; Wang, F. J. Catal. 2017, 348, 160.
(d) Anjum, A.; Srinivas, P. Chem. Lett. 2001, 9, 900.
(e) Baruah, S.; Borthakur, S.; Gogoi, S. Chem. Commun. 2017, 53, 9133.
(f) Nakamura, R.; Obora, Y.; Ishii, Y. Adv. Synth. Catal. 2009, 351, 1677.
(g) Minisci, F.; Recupero, F.; Pedulli, G. F.; Lucarini, M. J. Mol. Catal. A 2003, 204~205, 63.
(h) Minisci, F.; Recupero, F.; Fontana, F.; Bjϕrsvik, H˗R.; Liguori, L. Synlett. 2002, 610.
(i) Sathyanarayana, P.; Ravi, O.; Muktapuram, P. R.; Bathula, S. R. Org. Biomol. Chem. 2015. 13, 9681.
(a) Xu, L.; Wang, S.; Chen, B.; Li, M.; Hu, X.; Hu, B.; Jin, L.; Sun, N.; Shen, Z. Synlett. 2018, 1505.
(b) Shaikh, T. M. A.; Sudalai, A. Eur. J. Org. Chem. 2008, 4877.
(c) Santaniello, E.; Manzocchi, A.; Farachi, C. Synthesis 1980, 563.
(d) Lee, J. C.; Lee, J. M. Synth. Commun. 2006, 36, 1071.
(e) Lee, J. C.; Choi, J. H.; Lee, Y. C. Synlett. 2001, 1563.
(a) Liu, H.; Li, H.; Lu, J.; Zeng, S.; Wang, M.; Luo, N.; Xu, S.; Wang, F. ACS Catal. 2018, 8, 4761.
(b) Hirashima, S˗i.; Nobuta, T.; Tada, N.; Itoh, A. Synlett 2009, 2017.
(c) Yamaguchi, T.; Matsusaki, Y.; Tada, N.; Miura, T.; Itoh, A. Photochem. Photobiol. Sci. 2013, 12, 417.
(a) Sathyanarayana, P.; Upare, A.; Ravi, O.; Muktapuram, P. R.; Bathula, S. R. RSC Adv. 2016, 6, 22749.
(b) Angeles, N. A.; Villavicencio, F.; Guadarrama, C.; Corona, D.; Cuevas˗Yañez, E. J. Braz. Chem. Soc. 2010, 21, 905.
(c) Cao, L.; Ding, J.; Gao, M.; Wang, Z.; Li, J.; Wu, A. Org. Lett. 2009, 11, 3810.
(d) Rajendar, K.; Kant, R.; Narender, T. Adv. Synth. Catal. 2013, 355, 3591.
(a) Caputo, D. F. J.; Arroniz, C.; Dürr, A. B.; Mousseau, J. J.; Stepan, A. F.; Mansfield, S. J.; Anderson, E. A. Chem. Sci. 2018, 9, 5295.
(b) Ram, R. N.; Soni, V. K. J. Org. Chem. 2015, 80, 8922.
(c) Luo, J.; Zhang, X.; Zhang, J. ACS Catal. 2015, 5, 2250.
(d) Xu, G˗C.; Yu, H˗L.; Zhang, X˗Y.; Xu, J˗H. ACS Catal. 2012, 2, 2566.
(e) Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem. Int. Ed. 2011, 50, 951.
(f) Jiang, H.; Bak, J. R.; López˗Delgado, F. J.; Jørgensen, K. A. Green Chem. 2013, 15, 3355.
(g) Zhou, F.; Hu, X.; Gao, M.; Cheng, T.; Liu, G. Green Chem. 2016, 18, 5651.
(h) Toma, T.; Shimokawa, J.; Fukuyama, T. Org. Lett. 2007, 9, 3195.
Yu, S.; Liu, S.; Lan, Y.; Wan, B.; Li, X. J. Am. Chem. Soc. 2015, 137, 1623. doi: 10.1021/ja511796h
(a) Tong, W.; Li, W.˗H.; He, Y.; Mo, Z.˗Y.; Tang, H.˗T.; Wang, H.˗S.; Pan, Y.˗M. Org. Lett. 2018, 20, 2494.
(b) He, Y.; Wang, Y.; Liang, X.; Huang, B.; Wang, H.; Pan, Y.˗M.; Org. Lett. 2018, 20, 7117.
(c) Huang, X.˗Y.; Ding, R.; Mo, Z.˗Y.; Xu, Y.˗L.; Tang, H.˗T.; Wang, H.˗S.; Chen, Y.˗Y.; Pan, Y.˗M. Org. Lett. 2018, 20, 4819.
(d) Zhang, B.; Studer, A. Chem. Soc. Rev. 2015, 44, 3505.
(e) Wang, X.; Xiong, W.; Huang, Y.; Zhu, J.; Hu, Q.; Wu, W.; Jiang, H. Org. Lett. 2017, 19, 5818.
(f) Hu, W.; Li, J.; Xu, Y.; Li, J.; Wu, W.; Liu, H.; Jiang, H. Org. Lett. 2017, 19, 678.
(g) Peng, J.; Gao, Y.; Hu, W.; Gao, Y.; Hu, M.; Wu, W.; Ren, Y.; Jiang, H. Org. Lett. 2016, 18, 5924.
(a) Tan, L.; Chen, C.; Liu, W. Beilstein J. Org. Chem. 2017, 13, 1079.
(b) Jia, W˗G.; Zhang, H.; Li, D˗D.; Yan, L˗Q. RSC Adv. 2016, 6, 27590.
Table 1. Optimization of the reaction conditionsa
| | ||||
| Entry | Catalyst | Base (equiv.) | Solvent | Yieldb/% |
| 1 | Pd(OAc)2 | Et3N (0.5) | MeCN | 44c |
| 2 | — | Et3N (0.5) | MeCN | 46 |
| 3 | — | Et3N (1) | MeCN | 55 |
| 4 | — | Et3N (2) | MeCN | 66 |
| 5 | — | Et3N (3) | MeCN | 60 |
| 6 | — | Et2NH (2) | MeCN | 45 |
| 7 | — | (CH3)3CNH2 (2) | MeCN | Trace |
| 8 | — | DBU (2) | MeCN | NR |
| 9 | — | NaOH (2) | MeCN | NR |
| 10 | — | Cs2CO3 (2) | MeCN | Trace |
| 11 | — | Et3N (2) | MeCN/H2Od | 43 |
| 12 | — | Et3N (2) | 1, 4-Dioxane | NR |
| 13 | — | Et3N (2) | DMF | Trace |
| 14 | — | — | N(Et)3 | 23 |
| 15 | — | Et3N (2) | MeCN | 89e |
| a Reaction conditions: 1a (0.2 mmol), Et3N (0.4 mmol), solvent (1.5 mL), 90 ℃, 8 h. b Isolated yield. c With 3 equiv. of 2-isocyano-2-methylpropane and Pd(OAc)2 (10 mol %). d MeCN/H2O (V:V=2:1). e 120 ℃. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们