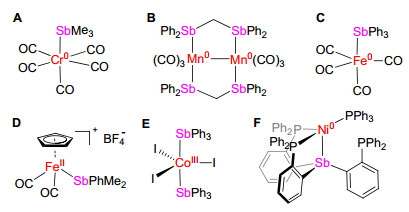
图 1.
含有机锑配体的3d金属配合物示例
Figure 1.
Examples of 3d metal complexes bearing stibine ligation

在第十五族元素P, As, Sb, Bi配体中, 有机锑(SbR3)是被研究相对较少的一类配体.有机锑配体是σ-给体型配体, 其给电子能力较有机膦和有机砷配体弱[1, 2].已报导的含有机锑配体的配合物以4d和5d后过渡金属配合物为主, 而3d金属配合物相对较少[1~4].这些3d金属配合物以低价金属羰基配合物居多, 配位数多为5和6. 图 1中列举了一些示例(A[5], B[6], C[7], D[8], E[9]和F[10]).有机锑的弱场(weak-field ligand)性质可能使得这类配体在开壳型(open-shell)过渡金属配合物, 特别是高自旋型(high-spin)过渡金属配合物的合成方面具有价值[11~13].然而, 可能与高自旋型过渡金属配合物易发生配体解离反应这一问题相关, 文献中关于开壳型含有机锑配体的金属配合物的报道极少.据我们所知, 顺磁性的三价钴配合物[Co(SbMe3)2I3]可能是唯一结构得到单晶X射线衍射表征的例子(图 1, 化合物E)[9].该化合物的固态样品在室温下的磁矩为4.4 μB.作者推测其存在高自旋Co(III)与中间自旋态或低自旋态Co(III)的平衡.
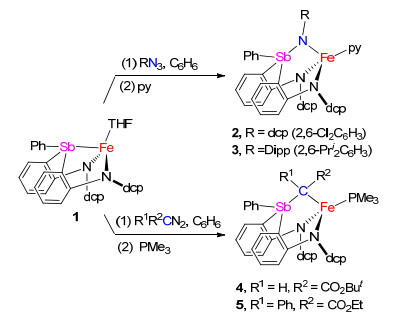
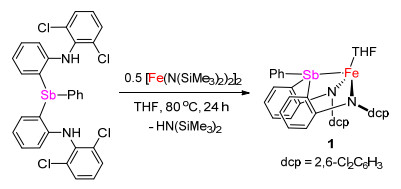
这里将报道一个新型含锑配体二(2-(N-(2', 6'-二氯苯基)胺基)苯基)苯基锑及其亚铁配合物的合成和结构表征.我们发现这种二胺基-锑配体可与二价铁配位形成四配位的高自旋亚铁配合物, 其Fe—Sb距离长达0.2792(1) nm.该亚铁配合物与有机叠氮化合物和重氮化合物反应, 可生成相应的二胺基-锑亚胺和二胺基-锑叶立德配位的亚铁配合物.
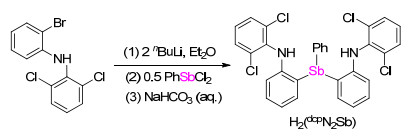
二(2-(N-(2', 6'-二氯苯基)胺基)苯基)苯基锑的合成采用我们之前报道的合成二胺基-膦和胂配体类似的方法[14, 15].如Scheme 1所示, 化合物(2, 6-Cl2C6H3)(2- BrC6H4)NH与2 equiv. nBuLi反应原位生成二锂化合物(2, 6-Cl2C6H3)(2-LiC6H4)NLi.该二锂化合物进一步与PhSbCl2(0.5 equiv.)反应, 所生成的混合物经水解, 柱层析分离后以53%的收率得到二(2-(N-(2', 6'-二氯苯基)胺基)苯基)苯基锑[H2(dcpN2Sb)].
H2(dcpN2Sb)为白色固体, 在空气中稳定, 易溶于二氯甲烷、四氢呋喃, 难溶于正己烷, 无色针状晶体由二氯甲烷溶液挥发溶剂得到. H2(dcpN2Sb)通过核磁共振、元素分析和单晶X射线衍射进行了表征(CCDC号码为1909387). H2(dcpN2Sb)的分子结构显示, Sb中心呈三角锥构型(图 2).三个Sb—C(Ar)键键长相近, 平均值为0.2154(5) nm.三个C—Sb—C键角也相近, 平均值为95.97(16)°.这些数值与三芳基锑化合物Sb(Mes)2Ph[16]中的Sb—C(Ar)键长(0.217 nm)和C—Sb—C键角(102°)相当.三芳基锑中较小的C—Sb—C键角与Sb的5s和5p轨道能级相差较大, 杂化度低相关[1].
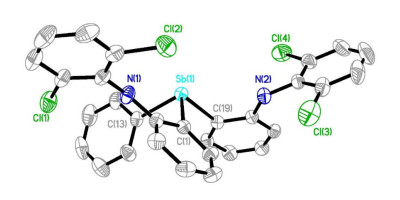
Selected distances (nm) and angles (°): Sb(1)—C(1) 0.2153(4), Sb(1)—C(13) 0.2161(5), Sb(1)—C(19) 0.2149(4), C(1)—Sb(1)—C(13) 94.67(16), C(1)—Sb(1)—C(19) 97.15(15), C(13)—Sb(1)—C(19) 96.09(16)
(二胺基-锑)亚铁配合物的合成可通过H2(dcpN2Sb)与[Fe(N(SiMe3)2)2]2的胺消除反应实现.在80 ℃下二胺基-锑配体H2(dcpN2Sb)与[Fe(N(SiMe3)2)2]2 (0.5 equiv.)在四氢呋喃中反应24 h, 可以定量生成二胺基-锑配位的亚铁配合物[(κ3-N, N, Sb-dcpN2Sb)Fe(THF)] (1) (Scheme 2) (CCDC号码为1909388).配合物1在四氢呋喃中溶解度较好, 在甲苯、苯和乙醚中溶解度较差.将其溶于四氢呋喃中可得到红色溶液, 通过正戊烷扩散重结晶, 得到其红色晶状固体, 产率为83%.
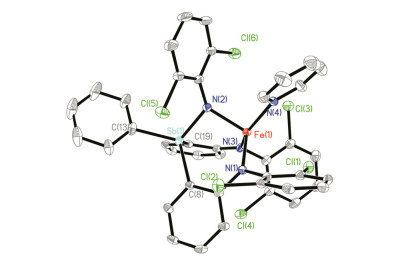
配合物1的分子结构通过单晶X-射线衍射得到确认.如图 3所示, 配合物1中Fe中心除与三齿配体[dcpN2Sb]2-中的两个胺基氮原子[N(1)、N(2)]和桥联锑原子配位外, 还同时与一个THF 分子以及[dcpN2Sb]2-中一个Cl原子作用(Cl1), 表现出扭曲的三角双锥构型.配合物1中Fe—Sb距离为0.2792(1) nm, 明显长于低自旋零价铁锑烷配合物[Fe(CO)4(SbPh3)] (0.2472(1) nm)[7]、二价铁锑烷配合物[CpFe(CO)2(SbPhMe2)][BF4] (0.2485(1) nm)[8]和[(CpFe(CO)2)2(O((CH2)2SbMe2)2)][BF4]2 (0.2486 (2) nm)[8]中的Fe—Sb键键长, 但比两原子的共价半径之和略小(0.285 nm)[17], 表明Fe和Sb之间可能存在一定的轨道相互作用.与配合物1中较长的Fe—Sb作用相关, 其Fe—Sb—C(Ph)键角达到172°, 远大于[Fe(CO)4-(SbPh3)] (117°)[7]和[CpFe(CO)2(SbPhMe2)][BF4] (113°)[8]中相应键角.锑配体部分的三个C—Sb—C键角为97.65(7)°, 100.29(7)°和100.80(7)°, 比配体H2(dcpN2Sb)中C—Sb—C键角稍大[平均值为95.97(16)°]. Fe与Cl(1)间距离为0.2718(0) nm, 比两原子的共价半径之和略长(0.240 nm)[17], 但比两原子的范德华半径之和(0.426 nm)短[18], 说明Fe—Cl之间仅存在弱的相互作用.配合物1中两个Fe—N键的平均键长(0.1967(2) nm)也与亚铁配合物[(κ3-N, N, P-dfpN2P)Fe(THF)] (0.2030(2) nm)[19]和[(κ3-N, N, As-dfpN2As)Fe(THF)2] (0.2038(7) nm)[15]中相应键长相当.这些结构特点显示, 配合物1应为高自旋型亚铁配合物(S=2).与此推测一致, 配合物1的溶液相磁矩μeff为4.7(2) μB (C6D6溶液), 与高自旋亚铁中心的自旋磁矩相当.
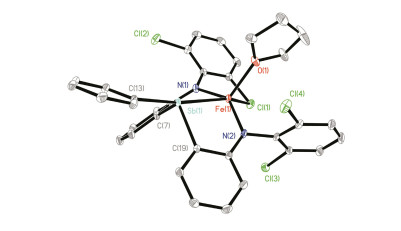
Selected distances (nm) and angles (°): Sb(1)—Fe(1) 0.2792(1), Fe(1)—N(1) 0.1973(2), Fe(1)—N(2) 0.1961(1), Fe(1)—O(1) 0.2047(1), Fe(1)—Cl(1) 0.2718(0), Sb(1)—C(7) 0.2139(2), Sb(1)—C(13) 0.2146(2), Sb(1)—C(19) 0.2136(1), C(13)—Sb(1)—Fe(1) 171.88(5), N(1)—Fe(1)—O(1) 111.41(7), N(2)—Fe(1)—O(1) 107.80(7), N(2)—Fe(1)—N(1) 140.49(7), C(7)—Sb(1)—C(13) 97.65(7), C(7)—Sb(1)—C(19) 100.29(7), C(13)—Sb(1)—C(19) 100.80(7)
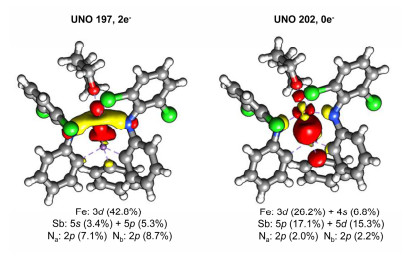
配合物1中Fe—Sb之间存在轨道相互作用, 这一猜测得到了理论计算的支持.密度泛函理论计算显示, 配合物1在S=2态下金属中心为(3dz2)2(3dxz, 3dyz)2(3dx2-y2, 3dxy)2形式的高自旋二价铁构型.除这些以3d轨道为主的UNO (unrestricted natural orbital)轨道外, 双占据的分子轨道UNO197和空轨道UNO202既包含Fe 3d轨道成分, 也有相当的配位原子Sb和N的p轨道成分(图 4), 可看作是铁中心与配体场轨道的σ-成键和σ*-反键形式的相互作用.需要指出的是, 高自旋配合物中金属与配位原子间较远的距离使得轨道间的最大有效重叠难以实现.因此, Fe—Sb, Fe—N和Fe—O的Mayer键级仅为0.39, 0.71和0.21.
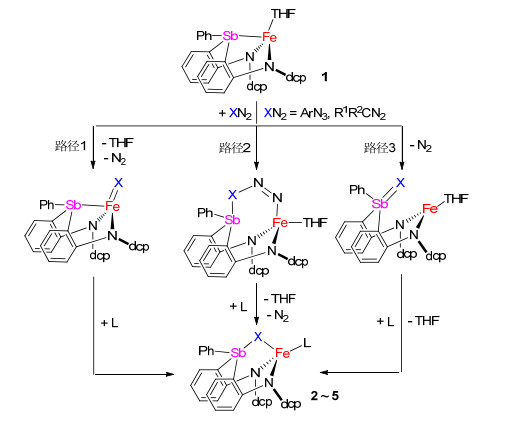
最近的一些研究显示, 配合物中锑配体的锑中心可与外加试剂反应形成高配位锑-过渡金属配合物[4].例如, Gabbaï等[20]发现含锑桥联膦配体的Pt(II)配合物[(κ4-Sb, P, P, P-(Sb(o-C6H4PPh2)3))Pt(CNcy)][SbF6]2与KF反应生成锑原子上含氟负离子配位的配合物[(κ4-Sb, P, P, P-(FSb(o-C6H4PPh2)3))Pt(CNCy)][SbF6], Au(I)配合物[(κ3-Sb, P, P-(Sb(o-C6H4PPh2)3))Au(Cl)]与PhICl2反应生成锑被氧化的Sb(V)-Au(I)配合物[(κ3-Sb, P, P-(Cl2Sb(o-C6H4PPh2)3))Au(Cl)][21].与此相关, 我们发现二胺基-膦配体和二胺基-胂配体支撑的亚铁配合物, 可与有机叠氮化合物和有机重氮化合物反应生成相应的磷亚胺、砷亚胺和磷叶立德配位的亚铁配合物[14, 15, 19].受这些工作的启发, 我们进一步研究了二胺基-锑配体支撑的亚铁配合物1与有机叠氮化合物和有机重氮化合物反应, 发现它们可以生成二胺基-锑亚胺和二胺基-锑叶立德配位的亚铁配合物.
配合物1与2, 6-二氯苯基叠氮(dcpN3)的苯溶液在室温下反应释放大量气泡并生成深绿色溶液.反应混合物进一步与吡啶(1 equiv.)作用后, 经重结晶等后处理操作可分离得到二胺基-锑亚胺配位的亚铁配合物[(κ3-N, N, N-dcpN2SbNdcp)Fe(py)] (2) (CCDC号码为1909389), 产率为70%.配合物1也可以与2, 6-二异丙基苯基叠氮(DippN3)反应生成二胺基-锑亚胺配位的亚铁配合物[(κ3-N, N, N-dcpN2SbNDipp)Fe(py)] (3) (CCDC号码为1909390), 产率为61%.配合物2和3的溶液相磁矩(C6D6溶液中)分别为4.9(2)和4.8 (1) μB, 表明其为高自旋态(S=2)亚铁配合物.这两个(二胺基-锑亚胺)亚铁配合物的结构均通过单晶X射线衍射得到确认, 图 5展示的是化合物2的分子结构.配合物2分子中的Fe中心与一个三齿二胺基-锑亚胺配体[dcpN2SbNdcp]2-和一个吡啶配位, 表现为三角锥构型, 新形成的二胺基-锑亚胺配体中的Sb中心为四面体构型. Sb—N键键长为0.1942(1) nm, 与已报道的锑亚胺化合物Mes3SbNCOCl3 (0.199 nm)[22]和(2-MeC6H4)3SbNSO2CF3 (0.196 nm)[23]中Sb—N键长较为接近, 小于典型的Sb—N单键的平均键长0.201~0.208 nm[24~27].
Selected distances (nm) and angles (°): Sb(1)—N(2) 0.1942(1), Fe(1)—N(1) 0.2044(2), Fe(1)—N(2) 0.2023(2), Fe(1)—N(3) 0.2031(2), Fe(1)—N(4) 0.2154(2), Sb(1)—C(12) 0.2094(2), Sb(1)—C(13) 0.2106(3), Sb(1)—C(19) 0.2088(2), N(1)—Fe(1)—N(4) 95.45(8), N(2)—Fe(1)—N(1) 102.94(8), N(2)—Fe(1)—N(3) 101.93(8), N(2)—Fe(1)—N(4) 91.47(8), N(3)—Fe(1)—N(1) 108.40(8), N(3)—Fe(1)—N(4) 148.91(8), Sb(1)—N(2)—Fe(1) 96.73(8), N(2)—Sb(1)—C(13) 114.00(10)
配合物1也可与重氮化合物, 如HC(N2)CO2But、PhC(N2)CO2Et, 反应.通过加入PMe3到反应体系中辅助重结晶, 可以70%的产率分离得到锑叶立德配位的亚铁配合物[(κ3-N, N, C-dcpN2SbCR1, R2)Fe(PMe3)] (R1=H, R2=CO2But, 4; R1=Ph, R2=CO2Et, 5) (Scheme 3).配合物4和5的溶液相磁矩[5.1(1)和4.7(3) μB]表明其为高自旋态(S=2)亚铁配合物.配合物4 (CCDC号码为1909391)和5 (CCDC号码为1909392)的分子结构还进一步通过单晶X射线衍射得到确定.这两个配合物分子中围绕铁中心的键长键角参数非常接近.以配合物4为例, 图 6展示了其分子结构.配合物4中的Fe和Sb中心均为四面体构型. Fe与叶立德C[Fe(1)—C(1)]的键长为0.2191(2) nm, 与我们报道的二胺基-磷叶立德亚铁配合物[(κ3-N, N, C-dfpN2PCTol2)Fe(THF)]中Fe—C键键长(0.2138(4) nm)相当[19].锑叶立德部分的Sb—C键键长为0.2064(2) nm, 与已报道的一些锑叶立德化合物中的Sb—C键平均键长0.204 nm相近[28, 29].值得指出的是配合物2~5中较短的Sb—N(亚胺)和Sb—C(叶立德)键长并不能说明其为Sb=N和Sb=C双键. Koketsu等[30]的理论计算研究表明, 在H3M=NH (M=P, As, Sb, Bi)中离子形式(M+—N-)是主导的共振结构. Lammertsma等[31]对H3P=NH的理论研究也指出, H3P=NH中较短的P—N键主要是由两基团间的库伦相互作用导致, 而其中的π-键贡献较小.基于这些认识, 同时考虑到亚铁中心Fe2+可以稳定极化的共振结构N(R)-—SbAr3+和C(R1R2)-—SbAr3+, 我们认为2~5中Sb—N和Sb—C键主要是单键性质.
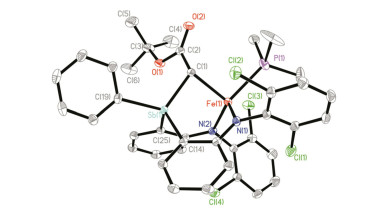
Selected distances (nm) and angles (°): Sb(1)—C(1) 0.2064(2), Sb(1)—C(14) 0.2106(2), Sb(1)—C(19) 0.2117(2), Sb(1)—C(25) 0.2095(2), Fe(1)—P(1) 0.24311(7), Fe(1)—N(1) 0.2005(1), Fe(1)—N(2) 0.1996(1), Fe(1)—C(1) 0.2191(2), C(14)—Sb(1)—C19 109.90(9), C(25)—Sb(1)—C(14) 110.21(8), C(25)—Sb(1)—C(19) 105.58(9), Sb(1)—C(1)—Fe(1) 89.12(8), N(2)—Fe(1)—Sb(1) 77.62(6), N(2)—Fe(1)—N(1) 111.32(8), N(2)—Fe(1)—C(1) 109.83(8)
我们推测配合物2~5的形成可能涉及到铁亚胺和铁卡宾中间体.如Scheme 4中路径1所示, (二胺基-锑)亚铁配合物先与有机叠氮和重氮反应脱除氮气生成二胺基-锑配位的铁亚胺和铁卡宾中间体.该活性中间体进一步发生分子内从铁到锑的基团转移反应, 从而形成锑亚胺-和锑叶立德-亚铁配合物.另外, 配合物2~5的形成也可能由锑与铁协同与有机叠氮和重氮作用, 然后脱除氮气而实现(Scheme 4, 路径2).因为配体H2(dcpN2Sb)不能与dcpN3、DippN3以及PhCN2CO2Et在温和条件下反应, 我们推测2~5的形成应该不是经由锑烷直接与有机叠氮和重氮反应生成锑亚胺和锑叶立德, 然后再与铁中心配位的途径(Scheme 4, 路径3).这些反应机理的揭示还需要进一步研究.
通过H2(dcpN2Sb)与[Fe(N(SiMe3)2)2]2的胺消除反应实现了有机锑配位的高自旋亚铁配合物[(κ3-N, N, Sb-dcpN2Sb)Fe(THF)] (1)的合成, 并对其分子结构和基团转移反应进行了研究.配合物1中Fe—Sb键长长达0.2792(1) nm.理论计算显示Fe和Sb之间存在较弱的σ-相互作用.化合物1可与有机叠氮化合物以及重氮化合物反应, 生成二胺基-锑亚胺和二胺基-锑叶立德配位的高自旋亚铁配合物[(κ3-N, N, N-dcpN2SbNR)Fe(py)] (R=dcp, 2; R=Dipp, 3)和[(κ3-N, N, C-dcpN2SbCR1, R2)Fe(PMe3)] (R1=H, R2=CO2But, 4; R1=Ph, R2=CO2Et, 5).这些基团转移反应的实现表明, 有机锑配体在具有金属-配体协同反应性质的催化剂设计方面拥有前景.
1H NMR和13C NMR在VARIAN Mercury 400 MHz, Agilent 400 MHz或Bruker 400 MHz核磁共振仪上测定. 1H NMR和13C NMR以氘代试剂的残余峰定标, 顺磁性化合物氢谱峰的半峰宽用ν1/2表示.元素分析由中国科学院上海有机化学研究所元素分析组测定.低温单晶X射线衍射在Bruker APEX CCD或Bruker D8 Venture衍射仪上测定, 具体方法:于手套箱中挑选合适的单晶用Paratone-N型油脂包裹, 将单晶放置在玻璃纤维顶端并置于低温氮气气流中, 在石墨单色Mo-Kα射线照射下, 用Bruker APEX-II CCD或Bruker D8 Venture衍射仪收集晶体的衍射数据.红外光谱在Bruker TENSOR 27 ART-FTIR上测定.溶液相磁矩在室温下通过Evans方法测定[32, 33].所有与配合物相关实验均在氮气氛围下, 采用标准Schlenk操作或在有氮气氛围的Vigor手套箱中进行.所用溶剂在使用前都通过有机溶剂纯化系统(Innovative Technology)除水, 而后在真空线上氮气鼓泡除去空气, 再经活化后的分子筛进一步干燥, 存储在手套箱中使用. C6D6经Na-K合金干燥后真空转移, 储存在手套箱里使用. (2, 6-Cl2C6H3)(2-BrC6H4)- NH[14]、PhSbCl2[34]、[Fe(N(SiMe3)2)2]2[35]均根据文献方法制备.配合物1的理论计算利用ORCA 3.0软件[36], 采取B3LYP方法进行[37, 38].用于计算的配合物1的原子坐标直接根据其晶体结构未进行进一步的几何优化. C、H、N原子采取def2-SVP基组, Fe和Sb原子采取def2-TZVP基组[39, 40].
将化合物(2, 6-Cl2C6H3)(2-BrC6H4)NH (3.8 g, 12 mmol)加入到50 mL Schlenk反应瓶中, 氮气保护下用乙醚(10 mL)溶解.将该溶液冷却到-78 ℃后, 缓慢加入正丁基锂的正己烷溶液(2.5 mol/L, 9.6 mL, 24 mmol).将反应液缓慢恢复至室温, 搅拌过夜.再于-78 ℃下将PhSbCl2 (1.62 g, 6.0 mmol)的四氢呋喃(10 mL)溶液缓慢加入到反应体系中.待其缓慢恢复至室温反应18 h后, 加入饱和碳酸氢钠溶液(5 mL)和水(100 mL)淬灭反应.分出有机层, 水层用二氯甲烷萃取(200 mL×3).合并的有机相用水和饱和食盐水各洗一次, 无水硫酸钠干燥, 然后过滤.滤液经减压除去溶剂可得到红棕色油状液体.该液体经硅胶柱层析分离[淋洗剂V(正己烷):V(二氯甲烷)=6:1]可得淡黄色粉末状固体.将固体溶于二氯甲烷, 经挥发溶剂重结晶, 可得无色晶状固体2.12 g, 即为产物H2(dcpN2Sb), 产率为53%. m.p. 181~182 ℃; 1H NMR (400 MHz, CDCl3, 293 K) δ: 7.73 (d, J=2.9 Hz, 2H), 7.45 (dd, J=8.5, 5.4 Hz, 5H), 7.29 (dd, J=11.5, 8.0 Hz, 6H), 7.03 (t, J=7.3 Hz, 2H), 6.97 (t, J=8.0 Hz, 2H), 6.64 (d, J=8.0 Hz, 2H), 5.99 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 148.2, 137.7, 137.2, 137.2, 135.9, 129.9, 129.5, 129.3, 129.1, 128.8, 128.6, 124.3, 123.6, 118.1; IR (KBr) ν: 3348, 3321, 3061, 3001, 1568, 1487, 1471, 1452, 1437, 1407, 1287, 1246, 1217, 1188, 1149, 1114, 1090, 1064, 1019, 996, 879, 868, 846, 767, 752, 734, 725, 717, 697, 659, 607, 564, 519, 484, 457, 444 cm-1. Anal. calcd for C30H21Cl4N2Sb: C 53.53, H 3.14, N 4.16; found C 53.32, H 3.37, N 4.19.
向25 mL Schlenk反应瓶中加入配体H2(dcpN2Sb) (673 mg, 1.0 mmol), [Fe(N(SiMe3)2)2]2 (377 mg, 0.50 mmol)和四氢呋喃(20 mL).搅拌均匀得黄绿色澄清溶液.溶液在80 ℃油浴下加热反应24 h后变为深红色溶液.反应液冷却至室温后, 减压除去溶剂.残余的红色固体用正己烷和乙醚洗涤, 减压干燥后得黄色粉末状固体.通过正戊烷扩散至其四氢呋喃溶液得到配合物1的红色晶状固体0.66 g, 产率83%. m.p.>201 ℃ (dec.); 1H NMR (400 MHz, d8-THF, 293 K) δ: 49.22 (ν1/2=77.64 Hz), 36.09 (ν1/2=102.28 Hz), 34.45 (ν1/2=50.83 Hz), 20.72 (ν1/2=32.08 Hz), 19.97 (ν1/2=74.13 Hz), 9.20 (ν1/2=91.16 Hz), 6.56 (ν1/2=Hz), -2.03 (ν1/2=30.59 Hz), -29.31 (ν1/2=75.38 Hz), -50.30 (ν1/2=56.83 Hz); IR (KBr) ν: 3038, 2977, 1568, 1550, 1477, 1453, 1427, 1292, 1192, 1154, 1116, 1082, 1065, 1042, 1018, 997, 915, 860, 776, 748, 730, 695, 551, 523, 458, 449 cm-1. Magnetic susceptibility (d8-THF, 293 K): μeff=4.7(2) μB. Anal. calcd for C34H27Cl4FeN2OSb: C 51.11, H 3.41, N 3.51; found C 50.83, H 3.44, N 3.47.
室温下, 向配合物1 (150 mg, 0.19 mmol)的苯(4 mL)悬浊液中缓慢滴加2, 6-二氯苯基叠氮(dcpN3) (35 mg, 0.19 mmol)的苯溶液(1 mL).反应体系有气泡产生, 颜色由红棕色逐渐变为深绿色.滴加完毕后, 反应液在室温下继续搅拌2 h, 再向其中加入吡啶(22 mg, 0.28 mmol).反应液颜色变为深棕绿色.反应4 h后经减压除去溶剂得棕色残渣.残渣用少量正己烷和乙醚洗涤, 过滤抽干后得绿色固体.将固体溶于适量四氢呋喃, 用正戊烷扩散重结晶, 可得配合物2的黄绿色晶状固体127 mg, 产率为70%. m.p.>208 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 44.56 (ν1/2=72.42 Hz), 32.58 (ν1/2=38.62 Hz), 30.42 (ν1/2=41.63 Hz), 28.71 (ν1/2=96.16 Hz), 5.84 (ν1/2=107.17Hz), 4.28 (ν1/2=49.62 Hz), 3.17 (ν1/2=49.62 Hz), -4.17 (ν1/2=124.96 Hz), -5.51 (ν1/2=51.34 Hz), -12.28 (ν1/2=194.22 Hz), -30.75 (ν1/2=67.40 Hz), -37.56 (ν1/2=77.03 Hz), -38.90 (ν1/2=39.77 Hz); IR (KBr) ν: 3054, 2954, 2923, 2856, 2360, 1600, 1569, 1548, 1463, 1421, 1303, 1275, 1215, 1193, 1161, 1083, 1067, 1038, 1017, 998, 859, 765, 751, 733, 701, 625, 522, 445, 430 cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=4.9(2) μB. Anal. calcd for C41H27Cl6- FeN4Sb: C 50.98, H 2.82, N 5.80; found C 50.99, H 3.18, N 5.36.
室温下, 向配合物1 (150 mg, 0.19 mmol)的苯(4 mL)悬浊液中缓慢滴加2, 6-二异丙基苯基叠氮(DippN3) (38 mg, 0.188 mmol)的苯溶液(1 mL).反应体系有气泡产生, 颜色由红棕色逐渐变为深棕色.滴加完毕后, 反应液在室温下继续搅拌2 h, 再向其中加入吡啶(22 mg, 0.28 mmol).反应液颜色变为深红棕色.反应4 h后经减压除去溶剂得棕色残渣.残渣用少量正己烷和乙醚洗涤, 过滤抽干后得红棕色固体.将固体溶于适量四氢呋喃, 用正戊烷扩散重结晶, 可得配合物3的红色晶状固体109 mg, 产率为59%. m.p.>185 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 127.04 (very broad), 46.48 (ν1/2=183.18 Hz), 33.69 (ν1/2=67.36 Hz), 32.32 (ν1/2=42.62 Hz), 31.58 (ν1/2=74.23 Hz), 28.92 (ν1/2=171.84 Hz), 8.91 (ν1/2=125.86 Hz), 4.68 (ν1/2=169.69 Hz), 3.78 (ν1/2=23.85 Hz), 2.02 (ν1/2=31.60 Hz), -0.81 (ν1/2=39.45 Hz), -2.53 (ν1/2=123.92 Hz), -13.82 (ν1/2=170.67 Hz), -27.01 (ν1/2=107.97 Hz), -30.71 (ν1/2=64.62 Hz), -38.03 (ν1/2=80.80 Hz); IR (KBr) ν: 3049, 2958, 2924, 2865, 1604, 1574, 1547, 1486, 1456, 1424, 1358, 1313, 1299, 1279, 1263, 1231, 1191, 1162, 1100, 1067, 1039, 1012, 997, 856, 770, 751, 730, 696, 511, 442 cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=4.8(1) μB. Anal. calcd for C47H41Cl4FeN4Sb: C 57.53, H 4.21, N 5.71; found C 57.05, H 4.32, N 5.37.
室温下, 向配合物1 (100 mg, 0.125 mmol)的苯(4 mL)悬浊液中缓慢滴加HCN2CO2But (18 mg, 0.125 mmol)的苯溶液(1 mL), 反应体系有气泡产生, 颜色由橙红色逐渐变为绿色.滴加完毕, 室温下继续反应4 h后, 向其中加入PMe3 (19 mg, 0.25 mmol), 反应液变为红棕色.反应4 h后经减压除去溶剂得棕色残渣.残渣用少量正己烷和乙醚洗涤, 过滤抽干后得棕黄色固体.将固体溶于适量四氢呋喃, 用正戊烷扩散重结晶, 可得配合物4的红棕色晶状固体79 mg, 产率为69%. m.p.>198 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 66.57 (ν1/2=259.40 Hz), 43.22 (ν1/2=74.28 Hz), 40.52 (ν1/2=64.56 Hz), 34.43 (overlapped), 34.25 (overlapped), 32.05 (ν1/2=37.68 Hz), 28.68 (ν1/2=49.83 Hz), 24.01 (ν1/2=57.84 Hz), 21.08 (ν1/2=52.87 Hz), 8.28 (ν1/2=20.29 Hz), 5.48 (ν1/2=24.13 Hz), 3.61 (ν1/2=39.98 Hz), -0.17 (ν1/2=49.98 Hz), -6.74 (ν1/2=191.19 Hz), -22.61 (ν1/2=117.14 Hz), -24.79 (ν1/2=122.13 Hz), -44.24 (ν1/2=32.79 Hz), -46.81 (ν1/2=32.07 Hz), -48.57 (ν1/2=39.12 Hz), -56.87 (ν1/2=40.82 Hz); IR (KBr) ν: 3040, 2962, 2908, 1649, 1581, 1569, 1546, 1479, 1461, 1427, 1364, 1307, 1279, 1261, 1239, 1210, 1149, 1116, 1066, 1021, 998, 980, 967, 864, 783, 766, 755, 733, 694, 599, 447 cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=5.1(1) μB. Anal. calcd for C39H38Cl4FeN2O2PSb: C 51.07, H 4.18, N 3.05; found C 51.19, H 4.15, N 2.76.
将配合物1 (170 mg, 0.21 mmol)与PhCN2CO2Et(40 mg, 0.21 mmol)在苯(5 mL)中的悬浊液, 于60 ℃油浴中加热反应2 h.反应体系颜色由橙红色逐渐变为深棕色.反应液冷至室温后向其中加入PMe3(32 mg, 0.42 mmol), 搅拌4 h后, 减压除去溶剂, 用少量正己烷和乙醚洗涤残渣, 过滤抽干后得红棕色粉末状固体.将固体溶于适量四氢呋喃, 用正戊烷扩散重结晶, 可得配合物5的橙红色晶状固体142 mg, 产率为70%. m.p.>210 ℃(dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 66.23 (ν1/2=341.55 Hz), 33.22 (ν1/2=450.51 Hz), 25.66 (ν1/2=161.81 Hz), 11.52 (peak overlapped), 9.87 (peak overlapped), 9.17 (ν1/2=101.17 Hz), 5.31 (ν1/2=11.27 Hz), 3.69 (ν1/2=317.26 Hz), -12.81 (ν1/2=183.36 Hz); IR (KBr) ν: 2990, 2971, 2946, 2906, 1668, 1504, 1476, 1463, 1443, 1417, 1389, 1368, 1326, 1304, 1292, 1206, 1169, 1099, 1057, 1028, 1006, 997, 970, 949, 895, 870, 854, 815, 782, 771, 746, 714, 698, 620, 597, 574, 476, 455, 418 cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=4.7(3) μB. Anal. calcd for C43H38Cl4FeN2O2PSb: C 53.51, H 3.97, N 2.90; found C 53.84, H 3.93, N 2.91.
辅助材料(Supporting Information) 新化合物的核磁谱图、IR谱图、配合物1的前线分子轨道图和Mayer键级图(PDF), 配体H2(dcpN2Sb)以及配合物1~5的晶体结构数据已上传至英国剑桥晶体数据库(Cambridge Crystallographic Data Centre), CCDC号码为1909387-1909392.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Levason, W.; Mcauliffe, C. A. Acc. Chem. Res. 1978, 11, 363. doi: 10.1021/ar50129a007
Champness, N. R.; Levason, W. Coord. Chem. Rev. 1994, 133, 115. doi: 10.1016/0010-8545(94)80058-8
Levason, W.; Reid, G. Coord. Chem. Rev. 2006, 250, 2565. doi: 10.1016/j.ccr.2006.03.024
Jones, J. S.; Gabbai, F. P. Acc. Chem. Res. 2016, 49, 857. doi: 10.1021/acs.accounts.5b00543
Breunig, H. J.; Borrmann, T.; Lork, E.; Moldovan, O.; Raţ, C. I.; Wagner, R. P. J. Organomet. Chem. 2009, 694, 427. doi: 10.1016/j.jorganchem.2008.11.022
Genge, A. R. J.; Holmes, N. J.; Levason, W.; Webster, M. Polyhedron 1999, 18, 2673. doi: 10.1016/S0277-5387(99)00168-0
Bryan, R. F.; Schmidt, W. C. J. Chem. Soc., Dalton Trans. 1974, 2337.
Benjamin, S. L.; Karagiannidis, L.; Levason, W.; Reid, G.; Rogers, M. C. Organometallics 2011, 30, 895. doi: 10.1021/om1010148
Godfrey, S. M.; McAuliffe, C. A.; Pritchard, R. G. J. Chem. Soc., Chem. Commun. 1994, 45.
Jones, J. S.; Wade, C. R.; Gabbai, F. P. Angew. Chem., Int. Ed. 2014, 53, 8876. doi: 10.1002/anie.201404156
Mo, Z.; Deng, L. Coord. Chem. Rev. 2017, 350, 285. doi: 10.1016/j.ccr.2017.07.007
Poli, R. Chem. Rev. 1996, 96, 2135. doi: 10.1021/cr9500343
Power, P. P. Chem. Rev. 2012, 112, 3482. doi: 10.1021/cr2004647
Xiao, J.; Deng, L. Dalton Trans. 2013, 42, 5607. doi: 10.1039/c3dt50518a
Zhao, M.; Mao, G.; Liu, Y.; Xiao, J.; Deng, L. Chin. J. Org. Chem. 2018, 38, 1656. doi: 10.6023/cjoc201803015
Bojan, V. R.; Fernandez, E. J.; Laguna, A.; Lopez-de-Luzuriaga, J. M.; Monge, M.; Olmos, M. E.; Puelles, R. C.; Silvestru, C. Inorg. Chem. 2010, 49, 5530. doi: 10.1021/ic1003484
Slater, J. C. J. Chem. Phys. 1964, 41, 3199. doi: 10.1063/1.1725697
Alvarez, S. Dalton Trans. 2013, 42, 8617. doi: 10.1039/c3dt50599e
Liu, J.; Hu, L.; Wang, L.; Chen, H.; Deng, L. J. Am. Chem. Soc. 2017, 139, 3876. doi: 10.1021/jacs.7b00484
You, D.; Yang, H. F.; Sen, S.; Gabba , F. P. J. Am. Chem. Soc. 2018, 140, 9644. doi: 10.1021/jacs.8b05520
Wade, C. R.; Gabbai, F. P. Angew. Chem., Int. Ed. 2011, 50, 7369. doi: 10.1002/anie.201103109
Matano, Y.; Nomura, H.; Suzuki, H.; Shiro, M.; Nakano, H. J. Am. Chem. Soc. 2001, 123, 10954. doi: 10.1021/ja003623l
Matano, Y.; Nomura, H.; Suzuki, H. Inorg. Chem. 2000, 39, 1340. doi: 10.1021/ic991120e
Edwards, A. J.; Paver, M. A.; Raithby, P. R.; Russell, C. A.; Wright, D. S. J. Chem. Soc., Dalton Trans. 1993, 2257.
Edwards, A. J.; Paver, M. A.; Rennie, M.-A.; Raithby, P. R.; Russell, C. A.; Wright, D. S. J. Chem. Soc., Dalton Trans. 1994, 2963.
Burford, N.; Macdonald, C. L. B.; Robertson, K. N.; Cameron, T. S. Inorg. Chem. 1996, 35, 4013. doi: 10.1021/ic951532x
Burford, N.; Cameron, T. S.; Lam, K. C.; LeBlanc, D. J.; Macdonald, C. L. B.; Phillips, A. D.; Rheingold, A. L.; Stark, L.; Walsh, D. Can. J. Chem. 2001, 79, 342. doi: 10.1139/v01-057
Glidewell, C.; Lloyd, D.; Metcalfe, S. Tetrahedron 1986, 42, 3887. doi: 10.1016/S0040-4020(01)87542-6
Ferguson, G.; Glidewell, C.; Gosney, I.; Lloyd, D.; Metcalfe, S.; Lumbroso, H. J. Chem. Soc., Perkin Trans. 2 1988, 1829.
Koketsu, J.; Ninomiya, Y.; Suzuki, Y.; Koga, N. Inorg. Chem. 1997, 36, 694. doi: 10.1021/ic951220u
Sudhakar, P. V.; Lammertsma, K. J. Am. Chem. Soc. 1991, 113, 1899. doi: 10.1021/ja00006a005
Evans, D. F. J. Chem. Soc. 1959, 2003.
Sur, S. K. J. Magn. Reson. 1989, 82, 169.
Chalmers, B. A.; Buhl, M.; Arachchige, K. S. A.; Slawin, A. M. Z.; Kilian, P. Chem. Eur. J. 2015, 21, 7520. doi: 10.1002/chem.201500281
Olmstead, M. M.; Power, P. P.; Shoner, S. C. Inorg. Chem. 1991, 30, 2547. doi: 10.1021/ic00011a017
Neese, F. ORCA-an ab initio, Density Functional and Semi-empirical Program Package (v.3.0.3), Max-Planck Institute for Bioinorganic Chemistry, Mülheim an der Ruhr, Germany, 2015.
Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B:Condens. Matter Mater. Phys. 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
Schafer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829. doi: 10.1063/1.467146
Schafer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571. doi: 10.1063/1.463096

Scheme 1 二胺基-锑配体H2(dcpN2Sb)的合成
Scheme 1 Synthesis of the diamido-stibine ligand H2(dcpN2Sb)

图 2 配体H2(dcpN2Sb)的分子结构
Figure 2 Molecular structure of H2(dcpN2Sb)
Selected distances (nm) and angles (°): Sb(1)—C(1) 0.2153(4), Sb(1)—C(13) 0.2161(5), Sb(1)—C(19) 0.2149(4), C(1)—Sb(1)—C(13) 94.67(16), C(1)—Sb(1)—C(19) 97.15(15), C(13)—Sb(1)—C(19) 96.09(16)

图 3 配合物1的分子结构
Figure 3 Molecular structure of 1
Selected distances (nm) and angles (°): Sb(1)—Fe(1) 0.2792(1), Fe(1)—N(1) 0.1973(2), Fe(1)—N(2) 0.1961(1), Fe(1)—O(1) 0.2047(1), Fe(1)—Cl(1) 0.2718(0), Sb(1)—C(7) 0.2139(2), Sb(1)—C(13) 0.2146(2), Sb(1)—C(19) 0.2136(1), C(13)—Sb(1)—Fe(1) 171.88(5), N(1)—Fe(1)—O(1) 111.41(7), N(2)—Fe(1)—O(1) 107.80(7), N(2)—Fe(1)—N(1) 140.49(7), C(7)—Sb(1)—C(13) 97.65(7), C(7)—Sb(1)—C(19) 100.29(7), C(13)—Sb(1)—C(19) 100.80(7)

图 4 配合物1中涉及金属-配体成键作用的分子轨道
Figure 4 Selected molecular orbitals of 1 involving metal-ligand bonding and anti-bonding interactions

图 5 配合物2的分子结构
Figure 5 Molecular structure of 2
Selected distances (nm) and angles (°): Sb(1)—N(2) 0.1942(1), Fe(1)—N(1) 0.2044(2), Fe(1)—N(2) 0.2023(2), Fe(1)—N(3) 0.2031(2), Fe(1)—N(4) 0.2154(2), Sb(1)—C(12) 0.2094(2), Sb(1)—C(13) 0.2106(3), Sb(1)—C(19) 0.2088(2), N(1)—Fe(1)—N(4) 95.45(8), N(2)—Fe(1)—N(1) 102.94(8), N(2)—Fe(1)—N(3) 101.93(8), N(2)—Fe(1)—N(4) 91.47(8), N(3)—Fe(1)—N(1) 108.40(8), N(3)—Fe(1)—N(4) 148.91(8), Sb(1)—N(2)—Fe(1) 96.73(8), N(2)—Sb(1)—C(13) 114.00(10)

Scheme 3 二胺基-锑配位的亚铁配合物1与有机叠氮和重氮化合物的反应
Scheme 3 Reactions of 1 with organic azides and diazo compounds

图 6 配合物4的分子结构
Figure 6 Molecular structure of 4
Selected distances (nm) and angles (°): Sb(1)—C(1) 0.2064(2), Sb(1)—C(14) 0.2106(2), Sb(1)—C(19) 0.2117(2), Sb(1)—C(25) 0.2095(2), Fe(1)—P(1) 0.24311(7), Fe(1)—N(1) 0.2005(1), Fe(1)—N(2) 0.1996(1), Fe(1)—C(1) 0.2191(2), C(14)—Sb(1)—C19 109.90(9), C(25)—Sb(1)—C(14) 110.21(8), C(25)—Sb(1)—C(19) 105.58(9), Sb(1)—C(1)—Fe(1) 89.12(8), N(2)—Fe(1)—Sb(1) 77.62(6), N(2)—Fe(1)—N(1) 111.32(8), N(2)—Fe(1)—C(1) 109.83(8)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

