图式 1.
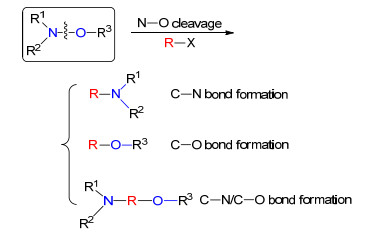
N—O键断裂方式
Scheme 1.
Type of N—O bond cleavage

N—O键化合物, 如肟、肟酯、肟醚、羟胺、羟肟酸、硝酮、吡啶或喹啉氮氧化合物、异噁唑以及异噁唑啉等, 不仅存在于很多天然产物中, 也是调节化合物生物活性的药效团, 由于其具有抗菌、消炎、抗癌等生物活性, 在医药领域中有着重要的应用价值[1~3].由于这类化合物中N—O键易于断裂, 也是有机合成中构建更复杂分子的重要砌块.因此, 发展选择性N—O键断裂的策略及其在有机合成的应用研究也是国际上研究的热点和难点之一.
综合文献报道, 目前N—O键化合物中N—O键断裂通常可以在无金属参与和金属催化下进行, 不论何种条件, N—O键断裂后通常存在三种方式(Scheme 1): (1)作为氮源, 在反应中形成新的C—N键; (2)作为氧源, 在反应中形成新的C—O键; (3)作为氮源和氧源, 同时形成新的C—N键和C—O键.通过三种断裂方式可以简便地构建胺类和含氮、氧等杂环化合物.
过渡金属催化C—C键、C—N键、C—O键的成键反应是当代有机合成的基石, 也是许多化学工作者研究的兴趣之一.尽管不同过渡金属, 如铜、铁、钯、铑、金、钌、铱、铂、钴、锰、镍等金属[4~13], 在N—O键断裂构建C—N键和C—O键方面也有很多的应用.但铜催化剂, 由于其含量丰富、易得, 在金属催化领域研究得较多[14~16], 而铜催化的N—O断裂策略, 在偶联反应、C—H键活化、环加成、烯烃/炔烃的双官能化以及利用N—O键断裂策略构建具有挑战性的复杂天然产物等领域, 近年来都取得了很大的进展.目前, 有关铜催化N—O键断裂及其应用研究方面的综述鲜有报道.[17]因此, 本文针对近些年来铜催化N—O键断裂策略的最新进展以及N—O键断裂在天然产物和药物分子合成中的应用进行了综述.
肟酯分别作为胺化试剂和内氧化剂, 除了安全、廉价外还有明显的优势:肟酯自身有着较强的氧化能力, 无需外加氧化剂即可实现金属的催化循环.在低价铜的作用下, 很容易实现N—O键的切断反应, 从而构建有用的氮杂环化合物.
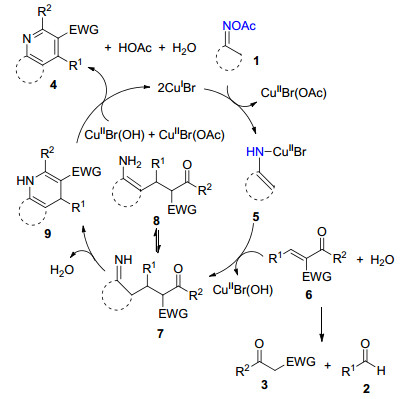
2015年, 江焕峰课题组[18]以CuBr作为催化剂, DMSO为溶剂, 碳酸锂作为碱, 发展了肟酯1、醛2和活性亚甲基化合物3的三组分串联反应, 生成多取代吡啶衍生物(表 1).该课题组利用金属铜盐作为催化剂, 成功地实现了N—O键断裂构建新的C—N键和C—C键, 生成多取代吡啶化合物4, 反应底物的官能团兼容性较好, 并以良好的产率得到各种骨架的吡啶化合物.
 下载:
导出CSV
下载:
导出CSV

|
作者进一步对CuBr催化N—O键断裂合成吡啶衍生物的反应提出了可能的机理(Scheme 2):一价铜首先切断化合物1的N—O键, 形成烯胺铜中间体5并产生二价铜物种, 中间体5对化合物2和3缩合生成的α, β-不饱和酮6进行1, 4-加成得到亚胺中间体7和互变异构体8, 中体间7发生分子内缩合脱水得到9, 中间体9在二价铜的氧化下芳构化生成吡啶化合物4.
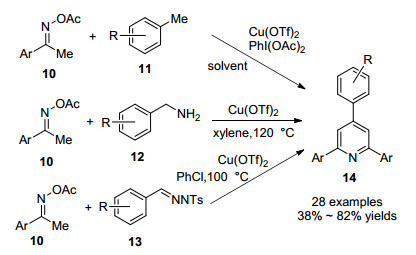
2016年, 陈保华课题组[19]发展了Cu(OTf)2催化肟乙酸酯10分别与甲苯衍生物11、苄胺12、对甲苯磺酰基腙13试剂反应生成2, 4, 6-三芳基吡啶衍生物的方法(Scheme 3).该方法使用廉价的铜盐催化肟乙酸酯sp3- C—H键发生氧化偶联反应, 以良好的产率得到目标化合物, 拓宽了肟酸酯通过N—O键断裂制备多取代吡啶的实用方法.
2017年, Yoshikai课题组[20]用六氟磷酸四乙腈铜催化肟酯15和α, β-不饱和酮亚胺16进行环化反应生成多取代吡啶衍生物17 (Eq. 1).该方法使用铜催化N—O键断裂策略形成烯胺铜为中间体, 再与α, β-不饱和酮亚胺16进行加成和缩合反应, 以较高的区域选择性和良好的产率得到多取代吡啶化合物.该方法底物范围广, 可以合成传统方法不能合成的吡啶化合物.
|
|
(1) |
2018年, 李兴伟课题组[21]报道了铜催化肟酯与β-CF3取代不饱和酮的[3+3]环加成反应.在不同的条件下, 可以一步构建含三氟甲基或二氟甲基的吡啶或吡啶酮等杂环化合物(Scheme 4).反应底物适用性广, 条件温和, 在合成含氟的氮杂环药物方面具有潜在的应用前景.
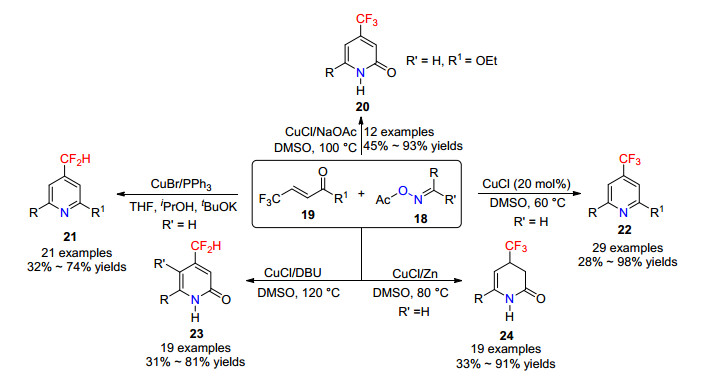
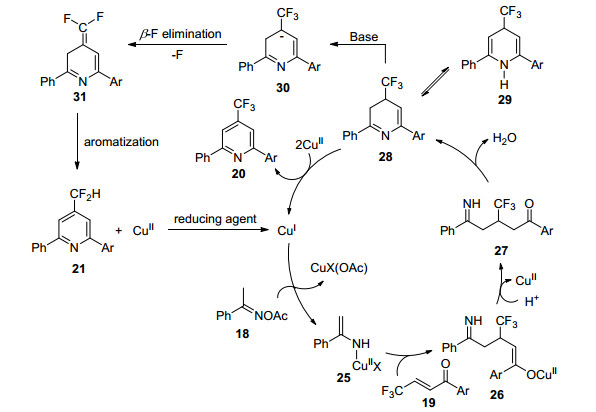
通过对机理的深入研究, 李兴伟课题组对一价铜催化肟酯与β-CF3取代α, β-不饱和酮化合物反应生成吡啶化合物20和21提出了可能的机理(Scheme 5):首先氯化亚铜与肟酯的N—O键发生反应形成烯胺铜物种25和醋酸铜盐, 烯胺铜25与酮19进行1, 4-加成反应生成烯醇物种26, 并异构化成酮27.酮27经分子内脱水形成二氢吡啶28及异构体29.二价铜继续氧化二氢吡啶 28得到含三氟甲基取代吡啶化合物, 二价铜被还原成一价铜, 进而完成催化循环.同时合适的碱和还原剂将二氢吡啶28脱氢, 再发生β-氟消除和芳构化生成4-二氟甲基吡啶21, 其中二价铜氧化或者HF消除反应是控制得到三氟甲基或二氟甲基吡啶化合物的关键因素.
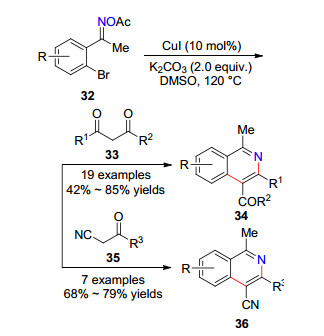
2016年, 江焕峰课题组[22]利用N—O键断裂策略和串联反应体系, 实现了邻溴芳基肟酯32分别与β-二酮化合物33和β-氰基酮35反应, 以中等到良好的产率生成官能团化的异喹啉衍生物34和36, 实现了两类异喹啉骨架化合物的合成(Scheme 6).
江焕峰课题组利用相同的策略, 进一步使用邻碘芳基肟酯37与吲哚38反应, 以良好的产率形成吲哚[1, 2-a]喹唑啉衍生物(Eq. 2).该合成方法简洁和实用, 为这类化合物在药物化学上的深入研究提供了实验基础.
|
|
(2) |
2018年, Bathula课题组[23]以碘化亚铜催化肟酯40和靛红41进行串联反应生成4-酰胺喹啉衍生物42 (Eq. 3), 该方法操作简单, 原子经济性高, 使用廉价的氧气做氧化剂参与反应.反应中靛红41发生了C—N键断裂和扩环反应形成喹啉衍生物.
|
|
(3) |
2016年, 邓国军课题组[24]报道了氯化铜催化2-取代吲哚43与肟酯44生成吡唑[1, 5-a]吲哚衍生物的方法(Eq. 4).该方法使用铜催化N—O键断裂引发自由基反应, 一步构建C—C和N—N键, 底物官能团兼容性较好, 反应条件温和, 原子利用率高.这一方法阐明了肟酯也可作为内氧化剂的新方式.
|
|
(4) |
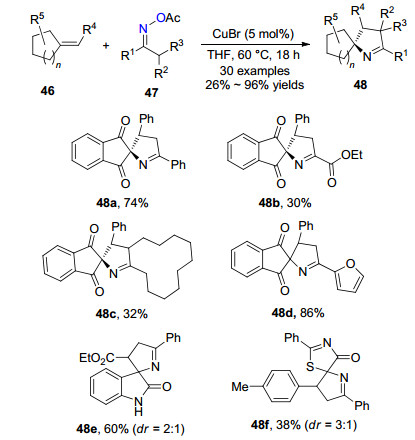
2017年, 魏晔课题组[25]报道了溴化铜催化肟酯47和活化烯烃46生成螺环吡咯啉化合物48 (Scheme 7).作者以5 mol%的溴化亚铜为催化剂, THF为溶剂, 60 ℃下反应, 以中等到优秀的产率得到目标化合物.反应的底物范围较广, 肟酸酯可以是链状或环状底物, 不同的烯烃底物都可以兼容, 但使用不对称的烯烃底物时, 螺环化合物的非对映选择性较差, 不同的底物可以分别产生含一至三个手性中心的产物.
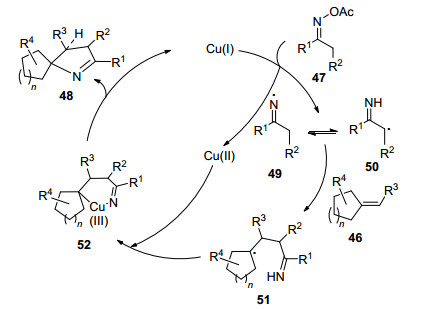
作者对反应机理进行了深入的研究, 对形成螺环吡咯啉化合物提出了可能的反应机理(Scheme 8).首先肟酸酯47在一价铜的作用下发生N—O键断裂形成亚胺自由基49或α-亚胺自由基50, 亚胺自由基对烯烃46进行自由基加成形成中间体51, 二价铜与中间体51形成三价铜中间体52, 52发生还原消除得到目标化合物48, 并再生一价铜, 实现催化循环.
2017年, 江焕峰课题组[26]以氯化亚铜催化肟酯53和三氟甲基酮54发生脱氢交叉偶联反应生成2-三氟甲基二氢吡咯-2-醇衍生物55 (Eq. 5).反应的底物范围较广, 都能以较高的非对映选择性(dr>15:1)和较好的产率得到各种含三氟甲基的吡咯啉化合物.作者认为该反应中肟酯是作为内氧化剂, 反应经过Cu(Ⅲ)/Cu(Ⅰ)的催化循环.
2018年, 魏晔课题组[27]报道了碘化亚铜催化马来酰亚胺57和肟酯56反应合成多取代苯环衍生物58的方法(Eq. 6).该策略是铜催化马来酰亚胺和肟酯发生[2+2+2]环加成反应, 一锅法构建三个C—C键, 底物范围较广, 产物的产率中等.
|
|
(5) |
|
|
(6) |
2018年, 谢应课题组[28]发展了铜和胺共催化α, β-不饱和醛60和α, β-不饱和肟酯59发生[3+2]环加成反应, 以较高的区域选择性生成四取代吡咯衍生物61的新方法(Eq. 7).这一新转化提供了一种合成多官能团吡咯的简易方法.
|
|
(7) |
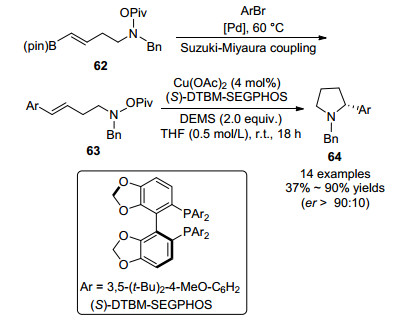
2018年, Buchwald课题组[29]报道了两步法合成α-芳基吡咯烷衍生物64的方法(Scheme 9).首先, 在钯催化下烯基硼酯62与溴代芳烃发生Suzuki-Miyaura偶联反应得到63.化合物63在醋酸铜催化下发生N—O键断裂, 进而发生分子内不对称胺氢化反应.反应在4 mol%醋酸铜为催化剂, S-DTBM-SEGPHOS为双膦配体, THF为溶剂, 室温下反应, 则以中等到优秀的产率得到产物.底物的适用性范围较广, 产物对映选择性都较高(er>90:10).
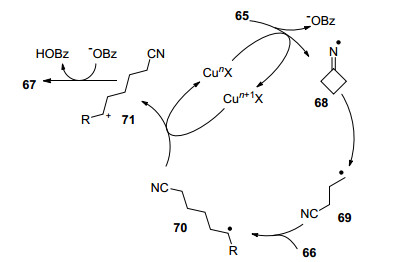
2017年, 史壮志课题组[30]报道了在1, 4-二氧六环和三氟甲苯(体积比1:1)的混合溶剂下, Cu(OTf)2催化环丁酮肟酯65和一般烯烃66反应生成腈类化合物67(表 2).该反应涉及N—O键和C—C键断裂, 高选择性地得到腈类化合物, 底物范围广, 不需要额外氧化剂和复杂配体, 产物的产率中等到优秀.该反应最大的优点是可以直接应用到天然产物及药物分子的后期修饰中, 为快速发现先导化合物提供了一条可靠的方法.
下载:
导出CSV

|
作者通过控制实验, 对反应提出了可能的机理(Scheme 10).首先环丁酮肟酯在铜的作用下发生N—O键断裂产生亚胺自由基68, 进而发生C—C键裂解产生烷基自由基69, 自由基69对烯烃进行加成生成更稳定自由基中间体70.在高价铜的氧化下, 自由基中间体70被氧化成碳正离子71并释放低价铜催化剂, 最后通过消除反应生成目标化合物67.
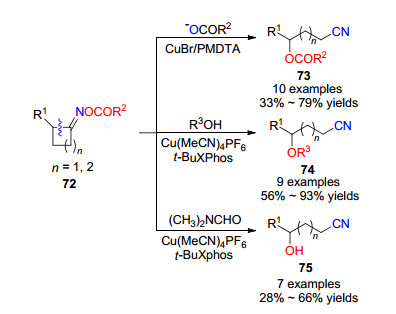
2018年, 卢忠林和刘强课题组[31]实现了在中性条件下铜盐催化环酮肟酯衍生物72开环反应生成腈类化合物73~75 (Scheme 11).反应条件温和, 化学选择性和区域选择性都很好.在五甲基二乙烯三胺(PMDTA)和Buchwald配体t-BuXphos可以分别实现不同类型亲核试剂的进攻产物.其中γ-和δ-羟基、烷氧基和酰氧基取代环酮肟酯兼容性很好, 可以合成各种官能团的腈类化合物.这一反应的另一个特点是:对于环张力较小的五元环酮肟酯, 在同样条件下也可以实现开环反应构建腈类化合物.
2018年, 江焕峰课题组[32]发展了碘化亚铜催化链状肟酯76发生N—O键和C—C键断裂形成腈类衍生物的方法, 以良好的产率高效地合成腈类衍生物.机理研究表明, 酮产物中的氧来源于空气中的氧原子(Eq. 8).
|
|
(8) |
2018年, 严汝龙课题组[33]通过Cu(OPiv)2催化环丁酮肟酯79发生开环反应, 并与邻氨基吡咯80环化生成4-丙氰基吡咯[1, 2-a]喹喔啉衍生物81 (Eq. 9).此过程中N—O键和C—C键断裂产生新的C—C和C—N键, 以良好的产率高效地合成新型含氰基的喹喔啉衍生物.
|
|
(9) |
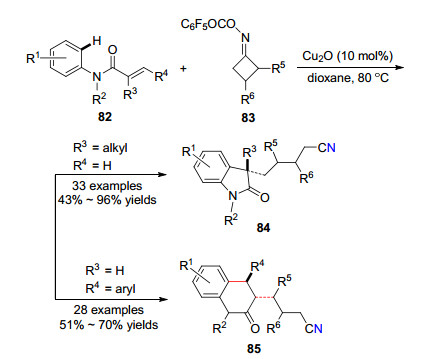
2018年, 郭丽娜课题组[34]报道了氧化亚铜催化α, β-不饱和芳基酰胺82与环丁酮肟酯83反应选择性地生成氰基吲哚-2-酮84或二氢喹啉衍生物85的方法(Scheme 12).反应通过开环、自由基加成和环化的串联过程.该策略具有反应条件温和, 底物范围宽等优点, 并能以中等到优秀的产率得到目标化合物.
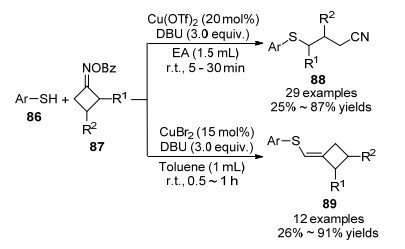
2018年, 严兆华和林森课题组[35]报道了Cu(OTf)2催化苯甲酰肟酯87与芳基硫酚86反应生成氰基硫醚衍生物88和环丁烯硫醚的方法(Scheme 13).该反应条件温和, 底物范围较宽, 都能以良好的产率得到产物.作者利用C—C键断裂和交叉偶联串联反应, 通过N—O键断裂产生亚胺自由基, 进而构建C—S/N—S键得到硫醚化合物.
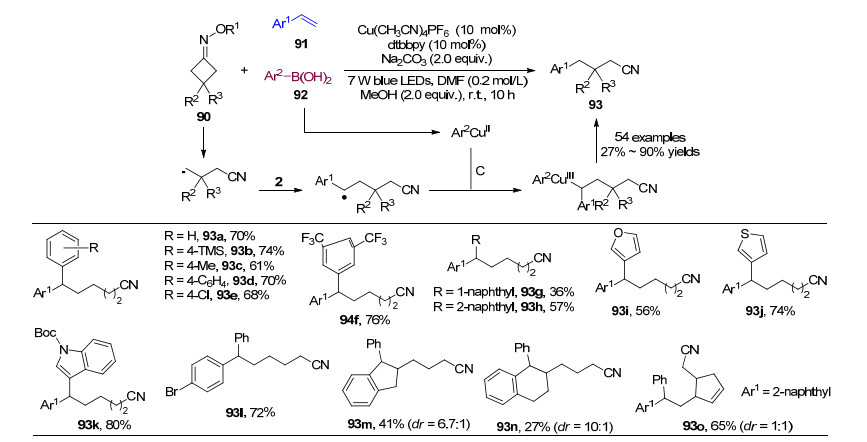
2018年, 陈加荣和肖文精课题组[36]通过光催化反应, 以Cu(CH3CN)4PF6和4, 4'-二叔丁基-2, 2'-联吡啶(dtb- bpy)配体为催化剂催化环丁酮肟酯90、苯乙烯9和芳基硼酸92三组分串联反应生成腈类化合物(Scheme 14), 该方法主要是通过光催化产生亚胺自由基, 进而发生C—C键断裂生成烷基自由基, 再对烯烃进行自由基加成, 与芳基铜物种形成三价铜, 还原消除得到目标化合物.
2019年, 袁宇和史壮志课题组[37]首次报道了环丁酮肟酯94进行分子内开环和重组形成3, 4-二氢萘-2-氰基衍生物95的方法(Eq. 10).反应只需要廉价的铜盐催化, 不需要配体、氧化剂、碱和有毒氰盐, 反应条件温和, 并以良好的产率得到萘基腈产物.
|
|
(10) |
同年, 余正坤课题组[38]报道了氯化亚酮催化烯胺硫酮97与环酮肟酯96反应生成氰基噻吩衍生物98的方法(Eq. 11).该方法反应条件温和, 操作简单, 有较好的区域选择性, 并以良好的产率得到产物.
|
|
(11) |
2019年, 刘峰课题组[39]报道了Cu(CH3CN)4PF6催化肟酯99与芳胺100生成含氰基胺类衍生物101的方法(Eq. 12).该反应以3, 3-二甲基-2, 2-联吡啶作配体, 碳酸钠作碱, 乙腈作溶剂, 室温反应, 底物适用性范围较广, 以良好到优秀的产率得到氰基芳胺类化合物.
|
|
(12) |
2016年, 江焕峰课题组[40]对肟酯在铜的催化下发生N—O键断裂产生烯胺铜中间体的策略进行了深入而系统的研究.作者利用碘化亚铜催化肟酯102和异硫氰酸酯103反应, 以良好到优秀的产率得到2-氨基噻唑类衍生物104 (Eq. 13).进一步研究发现[41], 在碘化亚铜和硫氰酸钾共同作用下, 肟酯102和酸酐105反应, 也可以形成2-氨基噻唑衍生物106 (Eq. 14), 旧的N—O键和C—S键断裂, 形成新的C—N和C—S键.
|
|
(13) |
|
|
(14) |
2018年, 江焕峰课题组[42]报道了溴化铜催化肟酯107与黄原酸盐108生成2-烷氧基噻唑衍生物109的合成方法(Eq. 15).该反应条件温和, 官能团兼容性很好, 不需添加碱和配体, 就能高效地合成噻唑2-烷氧基噻唑.
|
|
(15) |
2019年, 江焕峰课题组[43]发现以S8作为硫源, 可以实现CuOTf催化肟酯110、S8和芳基乙炔111生成苯并噻吩并噻唑衍生物112 (Eq. 16).反应中需要加入1, 5, 7-三叠氮双环[4.4.0]-癸-5-烯(TBD)作为添加剂.该方法的特点是底物范围较广, S8做为二硫源, 同时形成四根C—S键.
|
|
(16) |
2013年, 刘庆彬和席婵娟课题组[44]报道了氯化亚铜催化肟酯113与烯基锆试剂114发生偶联反应, 生成烯胺衍生物115 (Eq. 17).反应中用到的烯基锆试剂可以由炔和二茂锆现场生成.
|
|
(17) |
2017年, 江焕峰课题组[45]报道了碘化亚铜催化肟酯116与α-氧代羧酸117生成烯胺酮衍生物118 (Eq. 18).通过铜催化C—H键活化, N—O键和C—C键断裂构建新的C—C键, 从而生成烯胺酮衍生物.这一策略为合成烯胺酮提供了简洁的合成方法.
|
|
(18) |
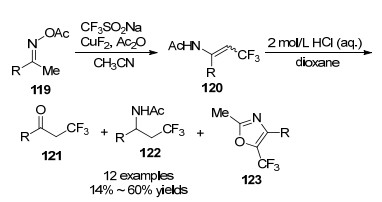
2017年, Selander课题组[46]以三氟甲磺酸钠盐为三氟甲基源, 氟化铜催化肟酯119与三氟甲磺酸钠盐可以生成三氟甲基烯胺酮衍生物120 (Scheme 15).得到的烯胺酮衍生物120在盐酸水解下, 可以分别生成含三氟甲基的酮121、胺122及噁唑123.这一策略为引入含三氟甲基提供了很好的合成方法.
2018年, 戴辉雄课题组[47]报道了铜催化C—H活化反应(Eq. 19).反应以噁唑啉作为导向基团, 肟酸酯125作为氮源, 实现了苯甲酰胺类底物124邻位的C—H键胺化反应, 产率中等到良好.该策略可以方便在芳环上引入氨基, 也可以在复杂天然产物及药物分子后期通过C—H活化进行修饰, 在分子中引入氨基.
|
|
(19) |
2016年, 关正辉课题组[48]报道了碘化亚铜催化肟酯127与吡啶或喹啉128通过脱氢氧化偶联反应生成咪唑[1, 2-a]吡啶衍生物的方法(Eq. 20).底物范围较广, 以良好到优秀的产率得到产物.
|
|
(20) |
2018年, 江焕峰课题组[49]发现肟酸酯130与芳基重氮盐131可以在氯化亚铜催化下发生偶联反应, 生成N-2-芳基-1, 2, 3-三唑衍生物132 (Eq. 21).该策略利用铜催化N—O键和C—N2键断裂构建了C—N键和N—N键, 该反应条件温和, 底物范围广, 以中等到优秀的产率得到目标化合物.
|
|
(21) |
2015年, 雷爱文课题组[50]报道了铜催化肟酸酯133与二苯基磷氧134进行偶联反应生成β-酮磷酸酯类衍生物135的方法(Eq. 22).反应中PCy3作为配体, 130 ℃下加热反应, 在铜的作用下分别产生亚胺自由基和膦氧自由基进行偶联反应, 在转化过程中涉及到N—O键断裂以及新C—P键的形成.
|
|
(22) |
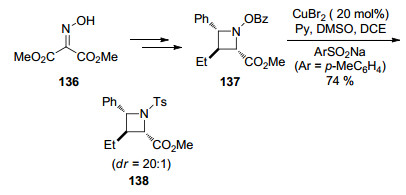
2017年, Anderson课题组[51]报道了醋酸铜催化肟136与烯基硼酸偶联反应生成氮杂环丁烷137, 在后续的转化中, 作者发现氮杂环丁烷137与芳基磺酸钠盐在铜盐作用下可以转化成N-磺酰基环丁烷138 (Scheme 16).反应中涉及到N—O键断裂以及C—S形成过程.该策略拓宽了合成氮杂环丁烷修饰药物分子的方法.
2016年, 严兆华和林森课题组[52]报道了铜催化肟酯139和香豆素140反应生成呋喃[3, 2-c]香豆素类衍生物141的方法(Eq. 23).在铜催化过程中, 涉及到N—O键、C—N键和O—H键断裂和C—C键和C—O键形成, 以良好的区域选择性得到2-或3-, 或2, 3-二取代基呋喃并[3, 2-c]香豆素化合物.
|
|
(23) |
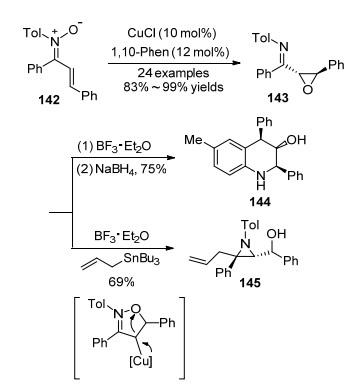
2013年, Anderson课题组[53]报道了氯化铜催化N-芳基硝酮142发生分子内氧转移反应生成α, β-环氧酮亚胺衍生物143的方法(Scheme 17).反应条件温和, 底物范围广, 以很好的非对映选择性和优秀的产率得到产物.所得α, β-环氧酮亚胺可以容易地转化成四氢喹啉144或三取代氮杂环丙烷145.
2015年, 万伯顺课题组[54]报道了铑和铜金属共同催化N-芳基硝酮146与炔烃147生成2, 3-二取代吲哚148的方法(Eq. 24).反应以1, 2-二氯乙烷(DCE)和1, 2-二乙氧基乙烷(EDE)为混合溶剂, 硝酮作为导向基团进行铑催化的C—H键活化, 取得了良好的区域选择性.
2016年, Furman课题组[55]报道了铜/N-PINAP络合物催化N-芳基硝酮149与末端炔150发生Kinugasa反应生成β-内酰胺衍生物151的方法(Eq. 25).但N-PINAP配体诱导能力只能得到良好的对映选择性.
2017年, Anderson课题组[56]报道了醋酸铜催化肟 152与烯基硼酸153发生Chan-Lam反应生成[4+2]环加成噁嗪衍生物154的新方法(Eq. 26).首先, 醋酸铜催化肟152与烯基硼酸153偶联得到噁嗪氮氧化物, 然后进一步在铜盐作用下发生N—O键断裂, 一步反应得到噁嗪化合物.
|
|
(24) |
|
|
(25) |
|
|
(26) |
2017年, 柳凌艳和李靖课题组[57]利用硝酮在反应中提供氧源, 发展了铜催化炔丙胺155与硝酮149发生环化生成四氢吡咯酮衍生物156的方法(Eq. 27).该反应经历铜催化N—O键断裂以及分子间胺氢化环化历程, 操作简单, 且以良好的产率得到四氢吡咯酮化合物.
|
|
(27) |
2017年, Ukaji课题组[58]报道了钯/铜共催化硝酮157邻位C—H键活化生成邻烯基化苯甲酰胺衍生物159的方法(Eq. 28).作者发现:其中醋酸抑制了1, 3-偶极子环加成反应, 有利于硝酮作为导向基从而发生C—H键活化.
|
|
(28) |
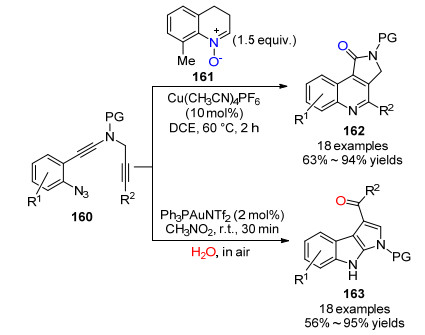
2017年, 叶龙武课题组[59]报道了叠氮二炔化合物160选择性环化反应生成喹啉及吲哚稠环衍生物的方法(Scheme 18).作者发现:以喹啉氮氧化物161提供氧源, 在Cu(CH3CN)4PF6 (10 mol%)催化下, 可以高效生成吡咯并[3, 4-c]喹啉酮化合物162.而以空气中的水作为氧源, 不需要额外的氧化剂, 在Ph3PAuNTf2 (2 mol%)催化下生成吡咯并[2, 3-b]吲哚化合物163.该策略是利用廉价的铜盐催化、喹啉氮氧化物为氧化剂氧化二炔类化合物, 环化反应表现出了很好的区域选择性.
同年, 该课题组[60]以Cu(CH3CN)4PF6 (10 mol%)作为催化剂, 喹啉氮氧化物161为氧化剂提供氧源, 可以实现对N-磺酰基炔酰胺化合物164进行双氧化反应生成α-酮酰亚胺衍生物165 (Eq. 29).该反应的官能团兼容性较好, 操作简便, 条件温和, 反应无需除水、除氧在空气中就可以进行.
|
|
(29) |
2018年, 王磊课题组[61]报道了铜催化喹啉氮氧化物166与苯甲酰羟胺167通过C—H活化生成2-氨基喹啉衍生物168的方法(Eq. 30).反应中1, 2-二氯乙烷不仅作为溶剂, 而且还作为还原试剂促进N—O键的断裂.该反应条件温和, 底物范围广, 以中等到良好的产率得到2-氨基喹啉.
|
|
(30) |
2018年, 许斌课题组[62]报道了醋酸铜催化芳基硝酮169与异腈酸酯170通过[3+1+1]环加成反应生成多取代吡咯衍生物171的方法(Eq. 31).作者使用红外光谱检测反应产生的中间体, 通过N—O键断裂构建新的C—C键.该方法的反应条件温和, 底物范围很广, 操作简单, 以良好的产率得到产物.
|
|
(31) |
2019年, 苏桂发和莫冬亮课题组[63]利用铜/铁共催化N-烯基-α, β-不饱和硝酮172和活化炔烃173反应合成吡咯稠环衍生物174(表 3).反应中铜盐需要先与1, 10-菲咯啉络合成铜催化剂.该反应经历了N-烯基-α, β-不饱和硝酮和炔烃发生[3+2]环加成/[3+3]重排反应得到含氮杂九元环化合物.氮杂九元环化合物在铜/铁的作用下发生N—O键断裂, 经过一系列转化得到吡咯稠环化合物.该反应条件温和, 底物范围广, 官能团兼容性良好.
下载:
导出CSV

|
同时, 作者还发现以醋酸酮为催化剂, PyBox为手性三氮配体, 首次实现了对氮杂九元环化合物169的动力学拆分, 拆分效率S值可以达到8, 氮杂九元环的ee值最高可以达到83%(表 4), 为进一步获得光学纯的氮杂九元环化合物提供了一条可选择的途径.
下载:
导出CSV

|
莫冬亮课题组[64]在此基础上, 进一步发展了在无金属条件下N-烯基α, β-不饱和硝基硝酮172和苯炔发生[7+2]环加成反应构建氮杂九元环化合物176.九元环化合物176在铜盐和铁盐的共催化下发生N—O键断裂, 经过系列转化, 以中等的产率得到吡咯连苯并吡喃177和吡咯连酚类化合物178(表 5).
下载:
导出CSV

|
苯并异噁唑类化合物不仅是很多复杂天然产物和药物分子的核心骨架, 同时也是有机合成中重要的中间体, 通过它可以构建多样性的分子骨架, 在反应中经过N—O键的断裂可以作为氮和氧源, 特别在合成氮杂环化合物领域有很广泛的应用[65, 66].
2016年, 李兴伟课题组[67]利用钴盐和铜盐共同催化芳基亚氨酸酯179的C—H活化, 以苯并异噁唑180为氮源, 经过N—O键的断裂生成新的C—N/N—N键, 以中等到优秀的产率合成1H-吲唑类衍生物181(表 6).
2017年, Samanta课题组[68]利用碘化亚铜催化喹啉氮氧化物182的2位C—H键活化, 以苯并噁唑183为氮源, 通过N—O键断裂高效地实现了芳胺化反应, 以很高的区域选择性获得喹啉氮氧胺化合物184 (Eq. 32).而得到的胺184通过简单的化学转化可以转化成多种多样的蒽醌类衍生物.
|
|
(32) |
2018年, Buchwald课题组[69]以醋酸铜为催化剂, (S)-DTBM-SEGPHOS为手性双膦配体, 1, 2-苯并噁唑186为氮源, 与烯烃或炔烃185发生氢胺化反应.通过N—O键断裂高效、高选择性地实现了氢胺化反应合成手性烷基胺化合物186 (Eq. 33).得到的亚胺能通过简单的化学转化可以转化成多种有用的手性芳基胺化合物.
|
|
(33) |
李兴伟课题组[70]利用同样的策略, 以醋酸亚铜为催化剂, (S, S)-Ph-BPE为手性双膦配体, 苯并噁唑189为氮源, 与烯烃188发生氢胺化反应(Eq. 34).通过N—O键断裂, 以优秀的区域选择性和优秀的对映选择性得到胺类化合物190.
|
|
(34) |
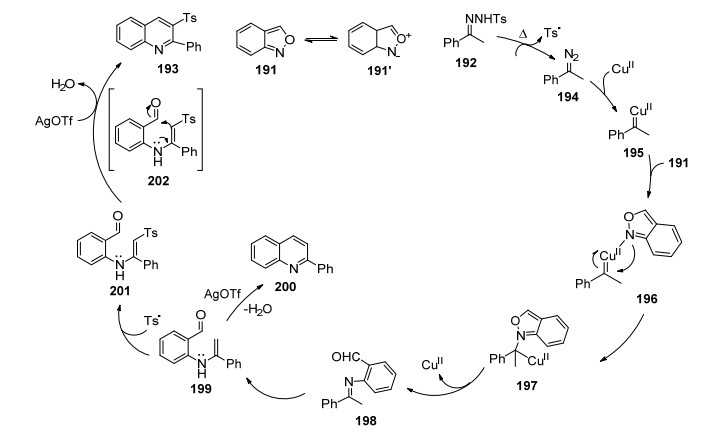
同年, 纪顺俊课题组[71]报道了二价铜催化苯并异噁唑191和对甲苯磺酰腙化合物192一步合成喹啉衍生物193的方法(表 7).该方法的反应条件温和, 二价铜催化苯并异唑啉中N—O键断裂, 同时新的C—C, C—N, C—S键形成, 是构建2, 3-二取代喹啉化合物的新策略.作者也对二价铜催化苯并异噁唑和腙一步合成喹啉衍生物的反应提出了可能的机理(Scheme 19).对甲苯磺酰肼192在加热条件下得到物种194和对甲苯磺酰自由基, 二价铜原子进攻物种194得到铜卡宾195并释放N2, 苯并异噁唑191与铜卡宾配位得196, 插入反应形成物种197, 随后N—O键断裂, 得到中间体物种198或异构体199, AgOTf促进环化反应就得到200.对甲苯磺酰自由基与199反应得到201. AgOTf促进化合物201进行分子内环化脱水得到目标分子193.
2018年, 陈超课题组[72]利用铜催化苯并氮杂异噁唑203与二芳基碘盐204反应生成吩嗪氮氧衍生物205的方法(Eq. 35).该方法是通过亲电芳基铜物种对苯并氮杂异噁唑203进行N—O键断裂, 高效、高区域选择性地合成吩嗪氮氧化合物.
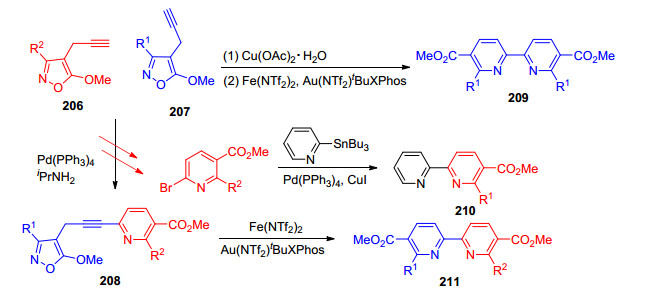
2019年, Khlebnikov课题组[73]利用过渡金属催化两分子异噁唑206和207通过N—O断裂后发生Eglinton反应、Sonogashira偶联反应、Stille偶联反应生成对称或不对称的2, 2-联吡啶配体(Scheme 20).该反应条件温和, 所得底物范围较广, 都能以良好的产率得到产物, 这一策略为对称或不对称的2, 2-联吡啶配体类配体的合成发展了一条简易的方法.
|
|
(35) |
2012年, Nakamura课题组[74]报道了以CuCl作为催化剂, 乙腈作溶剂, Cy2Nme作碱, 以O-炔丙基肟醚212为原料, 在120 ℃的条件下反应1~48 h, 以中等到优秀的产率生成环氧亚胺类化合物213(表 8).该反应的底物官能团兼容性较好, 取代基的电子效应对反应产率影响较小.
下载:
导出CSV

|
作者对反应提出了可能的机理(Scheme 21), 肟醚212在酮的作用下可以发生E/Z互变, 然后铜与炔键发生配位发生分子内5-endo-dig环化, 亚胺异构化成烯胺中间体以后, 发生N—O键的断裂形成铜卡宾中间体217, 氧原子进攻铜卡宾形成环氧铜中间体218, C—Cu键再发生质解就得到产物213.
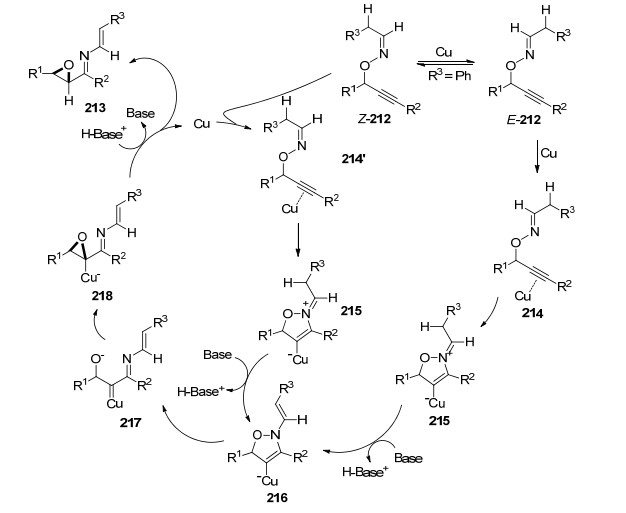
2013年, Nakamura课题组[75]进一步发展了铜催化炔丙基肟醚219和烯烃220反应生成多取代基噁唑嗪衍生物221的方法(Eq. 36).该方法经历了[2, 3]-重排、[3+2]环加成和1, 3氧迁移串联策略.通过N—O/C—O键断裂构建新C—N和C—O键合成七元氮杂环化合物.该反应条件温和, 底物范围广, 以优秀的区域选择性和非对映选择性得到目标产物.
2014年, Nakamura课题组[76]继续研究铜催化炔丙基肟醚222发生[2, 3]-重排反应生成二烯酰胺衍生物223 (Eq. 37).作者提出反应中铜催化炔丙基肟醚进行[2, 3]-重排反应, 然后发生N—O键发生断裂生成N-联烯基硝酮, 之后进行异构化得到二烯酰胺衍生物.
|
|
(36) |
|
|
(37) |
同年, 该课题组[77]发现了铜催化炔丙基肟醚224重排反应, 发现炔丙基肟醚与磺酰叠氮225反应可以生成α, β-不饱和脒类衍生物226 (Eq. 38).
|
|
(38) |
当磺酰叠氮换成异腈酸酯时, 该课题组[78]发展了以铜为催化剂, SPhos为配体, 催化炔丙基肟醚227与异氰酸酯228反应合成1, 6-二氢嘧啶衍生物的方法(Eq. 39).但反应的选择性较差, 同时得到不同异构体的1, 6-二氢嘧啶.
|
|
(39) |
2014年, Roy课题组[79]报道了钯盐和铜盐共同催化肟醚232发生分子内C—H键官能化、N—O键断裂和新的C—N键形成, 一步生成吲哚并异喹啉类衍生物233的新方法(Eq. 40).
|
|
(40) |
2015年, 王绍武课题组[80]发现碘化亚铜催化羟胺酸酯234与芳基铝试剂235可以发生偶联反应生成多种芳基胺衍生物236 (Eq. 41).以5 mol%碘化亚铜催化剂, THF作溶剂, 室温条件下进行反应, 底物范围广, 不需要额外的配体和碱, 就可以以中等到优秀的产率得到芳香胺化合物.
|
|
(41) |
2015年, Miura课题组[81]报道CuSCN或CuCl催化氧杂苯并降冰片烯双环烯烃237、二硼试剂和羟胺238发生氨基硼化反应(Eq. 42).这一方法可以同时向分子中高选择性地引入氨基和硼官能团, 所得化合物中硼官能团可以立体选择性地转化成更加有用的骨架分子.另外, 通过使用手性的双膦配体(R, R)-Ph-BPE还可以实现不对称胺硼化反应, 产物的er值可以高达94:6.
|
|
(42) |
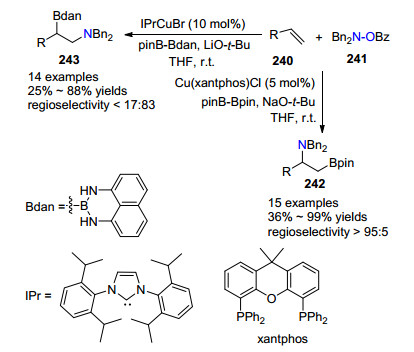
同年, Miura课题组[82]将胺硼化反应从桥环化合物拓展到一般的烯烃.利用配体和铜催化剂调控胺硼化反应的区域选择性(Scheme 22).作者发现:在氯化亚铜与xantphos双膦配体下, 未活化末端烯烃240和羟胺241发生胺硼化反应得到产物242; 而在溴化铜与氮杂环卜宾配体IPr为催化剂下, 可以相反区域选择性获得异构体胺硼化产物243.两个铜催化剂可以很好的控制区域选择性将多样的末端烯烃一步成为具有高价值的β-硼烷基胺化合物.
2016年, 王秋课题组[83]报道Cu(OTf)2催化不饱和羧酸244和苯甲酰羟胺245发生胺氧化反应生成氨基内酯衍生物246的方法(Eq. 43).同时作者也实现了分子间羧酸、烯烃、羟胺的三组分胺氧化反应.机理研究表明:苯甲酰羟胺245形成亲电胺化剂, 对双键进行分子内亲攻, 进而形成目标化合物.该反应条件温和, 底物范围广.这一策略为有效构建氨基内酯和1, 2-氨基醇衍生物提供一种新颖的合成方法.
|
|
(43) |
2017年, Nakamura课题组[84]报道了以NHC为配体, 铜盐催化N-烷氧基苯胺247发生[1, 3]-烷氧基重排反应生成2-烷氧基苯胺衍生物248的方法(Eq. 44).机理研究表明[1, 3]-烷氧基重排是分子内进行的重排.
|
|
(44) |
2018年, 游书力课题组[85]以醋酸铜为催化剂, 加入手性配体(R)-DTBM-SEGPHOS, 以苄基羟胺250为氮源, 通过对苯并呋喃去芳构化反应, 以中等的产率, 优秀的对映选择性得到手性胺化合物251(表 9), 产物的区域选择性优秀和对映选择性值较高(ee达94%).
2018年, Buchwald课题组[86]同样以醋酸铜为催化剂, 加入手性配体(R)-DTBM-SEGPHOS, 羟胺253为氮源, 对烯烃252进行不对称胺氢化反应, 以较好的区域选择性和对映选择性得到手性芳胺衍生物254(表 10).该反应还需要加入催化量的PPh3和当量的正丁醇为添加剂, 反应才能很好的进行.这一策略为手性胺化合物的合成提供了一种有效的方法.
2018年, Nakamura课题组[87]以IPrCuBr和AgBF4为催化剂催化芳基羟胺255发生分子内甲氧基的[1, 3]重排反应(Eq. 45).旧的N—O键和C—C断裂构建了新的C—C键和C—O键.该反应条件温和, 以良好的产率得到多取代的芳胺化合物256.
|
|
(45) |
2018年, 陈万芝和刘妙昌课题组[88]报道了一价铜催化羟胺257与氮杂环丙烯258反应, 高效、区域选择性地形成α-氨基肟衍生物259 (Eq. 46).该策略通过N—O和C—N键断裂构建新的C—N键和N—O键, 原子经济性高, 反应条件温和, 以良好到优秀的产率得到目标化合物.
|
|
(46) |
2019年, 陈应春和魏晔课题组[89]以醋酸亚铜为催化剂, dppe为配体, 催化邻异氰苯丙酸甲酯260和苯甲酰羟胺261反应生成二氢喹啉酮衍生物262 (Eq. 47).该反应涉及异氰插入苯甲酰羟胺中的N—O键、Mumm类型重排以及分子内亲核取代反应的串联策略.
|
|
(47) |
李兴伟课题组[90]利用铑盐和铜盐共同催化芳基亚胺酸酯263和亚硝基苯264反应, 在温和的中性条件下生成1H-吲唑类衍生物265的新方法(表 11).反应是铑和铜催化C—H活化键, 同时形成C—N、N—N键反应, 反应的官能团兼容性较好, 底物的范围较广.
可以看出, N—O键的断裂策略多种多样, 在有机合成中的应用非常的广. N—O键断裂后可以作为氮、氧源方便地引入到复杂的分子骨架中, 特别是在氮杂环化合物的合成中起到了很重要的作用.目前直接通过铜催化N—O键断裂应用到复杂天然产物及药物分子合成中的例子鲜有报道, 但近年来通过其它金属催化N—O断裂的策略在复杂天然产物及药物分子合成中的例子是有相关报道的.
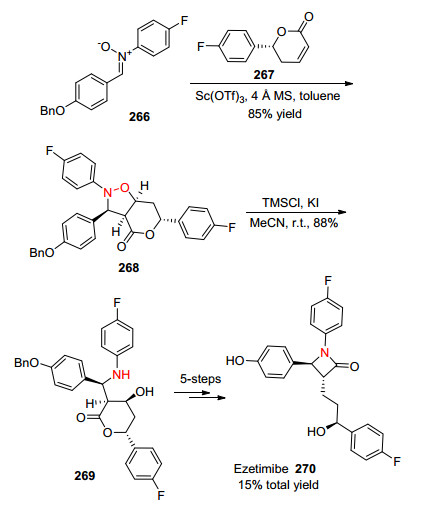
2013年, Chmielewski课题组[91]以硝酮266和内酯267为起始原料, 通过利用Kinugasa反应, 在Sc(OTf)3催化下以85%的产率得到化合物268, 再通过N—O键断裂得到269, 经过5步反应就可以合成药物分子Ezetimibe 270, 总产率为15% (Scheme 23).
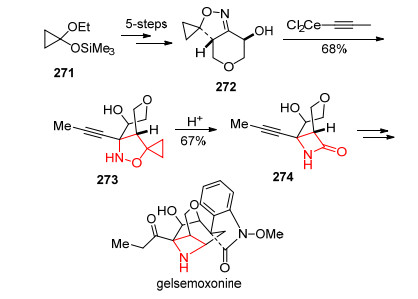
2015年, Carreira课题组[92]通过螺环丙烷异噁唑啉进行缩环反应, 成功地实现了gelsemoxonine的全合成(Scheme 24).反应以环丙烷271出发通过5步合成中间体272.化合物272进一步发生亲核加成反应, 以68%产率得到螺环丙烷异噁唑啉273.在酸性条件下273发生N—O键断裂的缩环反应, 以67%产率生成内酰胺274, 通过后续转化就可以实现gelsemoxonine的全合成.
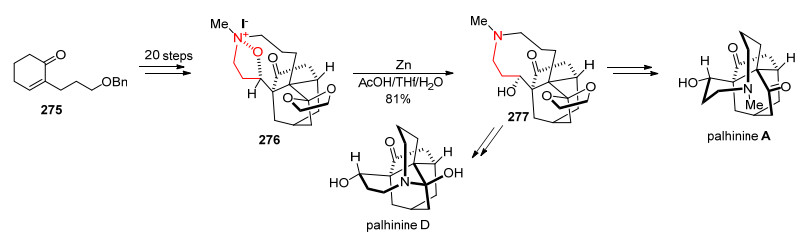
2017年, 樊春安课题组[93]以环己烯酮衍生物275出发, 通过20步反应得到关键的N—O化合物276.化合物276在Zn粉和醋酸还原条件下发生N—O键断裂得到化合物277.以化合物277开始进行后续转化, 可以分别实现对Palhinines A和D的不对称全合成(Scheme 25).
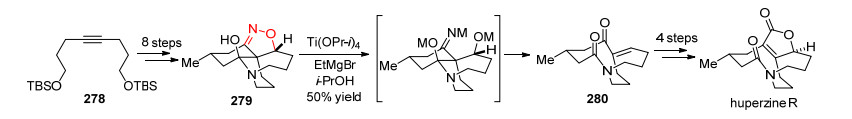
2018年, Fukuyama课题组[94]以炔278为原料, 通过8步反应得到N—O化合物279.通过N—O断裂不同条件下的尝试, 作者发现在Ti(OPr-i)4和EtMgBr作用下, 可以顺利地以50%的产率得到二酮化合物280, 再经过4步反应, 就可以实现对天然产物huperzine R的全合成(Scheme 26).
从上面的铜金属催化N—O键断裂进展来看, 经过化学家近几十年探索和研究, N—O键断裂的策略在C—H活化、环加成、重排反应、去芳构化、烯烃的双官能团化及多组分串联反应方面都取得了很大的进展, 通过N—O键断裂后快速构建新的C—N键、C—C键、C—O键和C−S键, 进而可以多样性地构建不同胺类、喹啉、吲哚、吡啶、噻唑等重要的氮杂环化合物, 成功地实现了通过N—O键断裂策略对天然产物及药物分子进行全合成.
尽管铜催化N—O键断裂策略方面取得了很大的进展, 也是一个充满活力的研究领域.但是在催化N—O键断裂的机理研究方面仍不够深入, 目前机理研究得较多的还是肟酯类化合物的N—O键断裂, 其它底物N—O键断裂的研究还较少.与此同时, 尽管其它过渡金属参与的N—O键断裂也取得了很多成绩, 但对映选择性的N—O断裂通常都是依靠手性底物进行控制, 而不对称催化的N—O键断裂仍然有待进一步探索, 需要发展新的催化体系或配体来解决这一问题. N—O键断裂策略的研究不仅具有重要的理论意义, 同时也有巨大的应用前景.我们相信, 经过有机化学家的不懈努力, N—O键断裂策略在有机合成中的应用将会取得更多成就.
Bolotin, D. S.; Bokach, N. A.; Demakova, M. Y.; Kukushkin, V. Y. Chem.Rev. 2017, 117, 13039. doi: 10.1021/acs.chemrev.7b00264
(a) Jiao, Y.-X.; Ma, X.-P.; Su, G.-F.; Mo, D.-L. Synthesis. 2017, 49, 933.
(b) Chen, N.; Xie, J. Org. Biomol. Chem. 2016, 14, 11028.
(a) Shi, W.-M.; Ma, X.-P.; Su, G.-F.; Mo, D.-L. Org. Chem.Front. 2016, 3, 116.
(b) Murahashi, S.-I.; Imada, Y. Chem. Rev. 2019, 119, 4684.
Lu, D.-F.; Zhu, C.-L.; Jia, Z.-X.; Xu, H. J. Am. Chem.Soc. 2014, 136, 13186. doi: 10.1021/ja508057u
Senadi, G. C.; Lu, T.-Y.; Dhandabani, G. K.; Wang, J.-J. Org.Lett. 2017, 19, 1172. doi: 10.1021/acs.orglett.7b00208
Neely, J. M.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 66. doi: 10.1021/ja3104389
Yeom, H.-S.; Shin, S. Acc. Chem. Res. 2014, 47, 966. doi: 10.1021/ar4001839
(a) Yao, C.-Z.; Xiao, Z.-F.; Liu, J.; Ning, X.-S.; Kang, Y.-B. Org.Lett. 2014, 16, 2498.
(b) Kumar, C. V. S.; Ramana, C. V. Org. Lett. 2015, 17, 2870.
(a) Zhou, J.; Shi, J.; Qi, Z.; Li, X.; Xu, H. E.; Yi, W. ACS Catal. 2015, 5, 6999.
(b) Xia, J.; Yang, X.; Li, Y.; Li, X. Org. Lett. 2017, 19, 3242.
Nakamura, I.; Sato, Y.; Terada, M. J. Am.Chem. Soc. 2009, 131, 4198. doi: 10.1021/ja900174t
Sivakumar, G.; Vijeta, A.; Jeganmohan, M. Chem.Eur. J. 2016, 22, 5899. doi: 10.1002/chem.201600471
Liu, X.-G.; Gao, H.; Zhang, S.-S.; Li, Q.; Wang, H. ACS Catal. 2017, 7, 5078. doi: 10.1021/acscatal.7b00677
(a) Yang, H.-B.; Pathipati, S. R.; Selander, N. ACS Catal. 2017, 7, 8441.
(b) Ding, D.; Wang, C. ACS Catal. 2018, 8, 111324.
Tang, X.; Wu, W.; Zeng, W.; Jiang, H. Acc.Chem. Res. 2018, 51, 1092. doi: 10.1021/acs.accounts.7b00611
(a) Ma, X.-P.; Liu, F.-P.; Mo, D.-L. Chin.J. Org. Chem. 2017, 37, 1069 (in Chinese).
(b) Lu, Q.; Yi, H.; Lei, A. Acta Chim. Sinica 2015, 73, 1245.
(c) Zhang, J.; Lu, Q.; Liu, C.; Lei, A. Chin. J. Org. Chem. 2015, 35, 743 (in Chinese).
(a) Ma, D.; Cai, Q. Acc. Chem. Res. 2008, 41, 1450.
(b) McCann, S. D.; Stahl, S. S. Acc. Chem. Res. 2015, 48, 1756.
Dong, X.; Liu, Q.; Dong, Y.; Liu, H. Chem.Eur. J. 2017, 23, 2481. doi: 10.1002/chem.201601607
Jiang, H.; Yang, J.; Tang, X.; Li, J.; Wu, W. J.Org. Chem. 2015, 80, 8763. doi: 10.1021/acs.joc.5b01621
Fu, Y.; Wang, P.; Guo, X.; Wu, P.; Meng, X.; Chen, B. J. Org. Chem. 2016, 81, 11671. doi: 10.1021/acs.joc.6b02081
Tan, W. W.; Ong, Y. J.; Yoshikai, N. Angew.Chem. Int. Ed. 2017, 56, 8240. doi: 10.1002/anie.201704378
Bai, D.; Wang, X.; Zheng, G.; Li, X. Angew.Chem. Int. Ed. 2018, 57, 6633. doi: 10.1002/anie.201802311
Jiang, H.; Yang, J.; Tang, X.; Wu, W. J. Org.Chem. 2016, 81, 2053. doi: 10.1021/acs.joc.5b02914
Ramaraju, A.; Chouhan, N. K.; Ravi, O.; Sridhar, B.; Bathula, S. R. Eur. J. Org. Chem. 2018, 2963.
Huang, H.; Cai, J.; Ji, X.; Xiao, F.; Chen, Y.; Deng, G.-J. Angew. Chem. Int. Ed. 2016, 55, 307. doi: 10.1002/anie.201508076
Zhao, B.; Liang, H.-W.; Yang, J.; Yang, Z.; Wei, Y. ACS Catal. 2017, 7, 5612. doi: 10.1021/acscatal.7b01876
Zhu, C.; Zhu, R.; Zeng, H.; Chen, F.; Liu, C.; Wu, W.; Jiang, H. Angew. Chem. Int. Ed. 2017, 56, 13324. doi: 10.1002/anie.201707719
Yang, J.; Zhao, B.; Xi, Y.; Sun, S.; Yang, Z.; Ye, Y.; Jiang, K.; Wei, Y. Org. Lett. 2018, 20, 1216. doi: 10.1021/acs.orglett.8b00141
Xie, Y.; Li, Y.; Chen, X.; Liu, Y.; Zhang, W. Org.Chem. Front. 2018, 5, 1698. doi: 10.1039/C8QO00204E
Dai, X.-J.; Engl, O. D.; León, T.; Buchwald, S. L. Angew. Chem. Int. Ed. 2019, 58, 3407. doi: 10.1002/anie.201814331
Zhao, B.; Shi, Z. Angew. Chem. Int.Ed. 2017, 56, 12727. doi: 10.1002/anie.201707181
Ai, W.; Liu, Y.; Wang, Q.; Lu, Z.; Liu, Q. Org.Lett. 2018, 20, 409. doi: 10.1021/acs.orglett.7b03707
Zhu, C.; Chen, F.; Liu, C.; Zeng, H.; Yang, Z.; Wu, W.; Jiang, H. J. Org. Chem. 2018, 83, 14713. doi: 10.1021/acs.joc.8b02103
An, Z.; Jiang, Y.; Guan, X.; Yan, R. Chem.Commun. 2018, 54, 10738. doi: 10.1039/C8CC06256K
Wu, J.; Zhang, J.-Y.; Gao, P.; Xu, S.-L.; Guo, L.-N. J. Org. Chem. 2018, 83, 1046. doi: 10.1021/acs.joc.7b02714
He, M.; Yan, Z..; Zhu, F.; Lin, S. J. Org.Chem. 2018, 83, 15438. doi: 10.1021/acs.joc.8b02707
Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem. Int. Ed. 2018, 57, 15505. doi: 10.1002/anie.201809820
Wang, P.; Zhao, B.; Yuan, Y.; Shi, Z. Chem.Commun. 2019, 55, 1971. doi: 10.1039/C8CC10109D
He, Y.; Lou, J.; Wu, K.; Wang, H.; Yu, Z. J.Org. Chem. 2019, 84, 2178. doi: 10.1021/acs.joc.8b03175
Min, Q.-Q.; Li, N.; Chen, G.-L.; Liu, F. Org.Chem. Front. 2019, 6, 1200. doi: 10.1039/C9QO00235A
Tang, X.; Zhu, Z.; Qi, C.; Wu, W.; Jiang, H. Org.Lett. 2016, 18, 180. doi: 10.1021/acs.orglett.5b03188
Tang, X.; Yan, J.; Zhu, Z.; Zheng, M.; Wu, W.; Jiang, H. J. Org. Chem. 2016, 81, 11461. doi: 10.1021/acs.joc.6b02124
Zhu, Z.; Tang, X.; Cen, J.; Li, J.; Wu, W.; Jiang, H. Chem. Commun. 2018, 54, 3767. doi: 10.1039/C8CC00445E
Zhou, P.; Huang, Y.; Wu, W.; Yu, W.; Li, J.; Zhu, Z.; Jiang, H. Org. Biomol. Chem. 2019, 17, 3424. doi: 10.1039/C9OB00377K
Liu, H.; Yan, X.; Chen, C.; Liu, Q.; Xi, C. Chem.Commun. 2013, 49, 5513. doi: 10.1039/c3cc41574k
Zhu, Z.; Tang, X.; Li, J.; Li, X.; Wu, W.; Deng, G.; Jiang, H. Chem. Commun. 2017, 53, 3228 doi: 10.1039/C7CC00260B
Yang, H.-B.; Selander, N. Org. Biomol.Chem. 2017, 15, 1771. doi: 10.1039/C7OB00203C
Xu, L.-L.; Wang, X.; Ma, B.; Yin, M.-X.; Lin, H.-X.; Dai, H.-X.; Yu, J.-Q. Chem. Sci., 2018, 9, 5160. doi: 10.1039/C8SC01256C
Ren, Z.-H.; Zhao, M.-N.; Yi, Y.; Wang, Y.-Y.; Guan, Z.-H. Synthesis 2016, 48, 1920. doi: 10.1055/s-0035-1561950
Zhu, C.; Zeng, H.; Chen, F.; Liu, C.; Zhu, R.; Wu, W.; Jiang, H. Org. Chem. Front. 2018, 5, 571. doi: 10.1039/C7QO00874K
Ke, J.; Tang, Y.; Yi, H.; Li, Y.; Cheng, Y.; Liu, C.; Lei, A. Angew. Chem. Int. Ed. 2015, 54, 6604. doi: 10.1002/anie.201501287
Reidl, T. W.; Son, J.; Wink, D. J.; Anderson, L. L. Angew. Chem. Int. Ed. 2017, 56, 11579. doi: 10.1002/anie.201705681
He, M.; Yan, Z.; Wang, W.; Zhu, F.; Lin, S. Tetrahedron Lett. 2018, 59, 3706. doi: 10.1016/j.tetlet.2018.09.007
Mo, D.-L.; Anderson, L. L. Angew. Chem. 2013, 52, 6722. doi: 10.1002/anie.201301963
Yan, H.; Wang, H.; Li, X..; Xin, X.; Wang, C.; Wan, B. Angew. Chem. Int. Ed. 2015, 54, 10613. doi: 10.1002/anie.201503997
Wolosewicz, K.; Michalak, M.; Adamek, J.; Furman, B. Eur. J. Org. Chem. 2016, 2212. doi: 10.1002/chin.201636098/full
Son, J.; Kim, K. H.; Mo, D.-L.; Wink, D. J.; Anderson, L. L. Angew. Chem. Int. Ed. 2017, 56, 3059. doi: 10.1002/anie.201611791
Kong, Y.; Liu, Y.; Wang, B.; Li, S.; Liu, L.; Chang, X.; Li, J. Adv. Synth. Catal. 2018, 360, 1240. doi: 10.1002/adsc.201701476
Hasegawa, M.; Nomoto, A.; Soeta, T.; Ukaji, Y. Chem.Lett. 2017, 46, 45. doi: 10.1246/cl.160821
Shen, W.-B.; Sun, Q.; Li, L.; Liu, X.; Zhou, B.; Yan, J.-Z.; Lu, X.; Ye, L.-W. Nat. Commun. 2017, 8, 1748. doi: 10.1038/s41467-017-01853-1
Liu, X.; Zhang, Z.-X.; Zhou, B.; Wang, Z.-S.; Zheng, R.-H.; Ye, L.-W. Org. Biomol. Chem. 2017, 15, 10156. doi: 10.1039/C7OB02728A
Wang, Z.; Han, M.-Y.; Li, P.; Wang, L. Eur.J. Org. Chem. 2018, 5954.
Tian, Z.; Xu, J.; Liu, B.; Tan, Q.; Xu, B. Org.Lett. 2018, 20, 2603. doi: 10.1021/acs.orglett.8b00798
Zou, N.; Jiao, J.-W.; Feng, Y.; Pan, C.-X.; Liang, C.; Su, G.-F.; Mo, D.-L. Org. Lett. 2019, 21, 481. doi: 10.1021/acs.orglett.8b03767
Ma, X.-P.; Li, L.-G.; Zhao, H.-P.; Du, M.; Liang, C.; Mo, D.-L. Org. Lett. 2018, 20, 4571. doi: 10.1021/acs.orglett.8b01761
Hu, F.; Szostak, M. Adv. Synth. Catal. 2015, 357, 2583. doi: 10.1002/adsc.201500319
Li, L.; Tan, T.-D.; Zhang, Y.-Q.; Liu, X.; Ye, L.-W. Org. Biomol.Chem. 2017, 15, 8483. doi: 10.1039/C7OB01895A
Li, L.; Wang, H.; Yu, S.; Wang, X.; Li, X. Org.Lett. 2016, 18, 3662. doi: 10.1021/acs.orglett.6b01716
Biswas, A.; Karmakar, U.; Nandi, S.; Samanta, R.J. Org. Chem. 2017, 82, 8933. doi: 10.1021/acs.joc.7b01343
Guo, S. Yang, C. J.; Buchwald, S. L. J. Am.Chem. Soc. 2018, 140, 15976. doi: 10.1021/jacs.8b10564
Xie, F.; Shen, B.; Li, X. Org. Lett. 2018, 20, 7154. doi: 10.1021/acs.orglett.8b03093
Wang, F.; Xu, P.; Wang, S.-Y.; Ji, S.-J. Org. Lett. 2018, 20, 2204. doi: 10.1021/acs.orglett.8b00525
Sheng, J.; He, R.; Xue, J.; Wu, C.; Qiao, J.; Chen, C. Org. Lett. 2018, 20, 4458. doi: 10.1021/acs.orglett.8b01748
Galenko, E. E.; Novikov, M. S.; Shakirova, F. M.; Shakirova, J. R.; Kornyakov, I. V.; Bodunov, V. A.; Khlebnikov, A. F. J.Org. Chem. 2019, 84, 3524. doi: 10.1021/acs.joc.9b00115
Nakamura, I.; Iwata, T.; Zhang, D.; Terada, M. Org.Lett. 2012, 14, 206. doi: 10.1021/ol203001w
Nakamura, I.; Kudo, Y.; Terada, M. Angew. Chem. Int. Ed. 2013, 52, 7536. doi: 10.1002/anie.201302751
Nakamera, I.; Ishida, Y.; Terada, M. Org. Lett. 2014, 16, 2562. http://www.ncbi.nlm.nih.gov/pubmed/24749932
Zhang, D.; Nakamura, I.; Terada, M. Org. Lett. 2014, 16, 5184. doi: 10.1021/ol502541w
Nakamura, I.; Onuma, T.; Zhang, D.; Terada, M. Tetrahedron Lett. 2014, 55, 1178. doi: 10.1016/j.tetlet.2013.12.105
Hazra, S.; Mondal, B.; Rahaman, H.; Roy, B. Eur.J. Org. Chem. 2014, 2806. https://www.researchgate.net/publication/260531055_Copper-_and_Palladium-Cocatalyzed_Intramolecular_CH_FunctionalizationCN_Bond_Formation_A_Route_to_the_Synthesis_of_Indoloisoquinoline_Derivatives
Zhou, S.; Yang, Z.; Chen, X.; Li, Y.; Zhang, L.; Fang, H.; Wang, W.; Zhu, X.; Wang, S. J. Org. Chem. 2015, 80, 6323. doi: 10.1021/acs.joc.5b00767
Sakae, R.; Hirano, k.; Satoh, T.; Miura, M. Angew.Chem. Int. Ed. 2015, 54, 613.
Sakae, R.; Hirano, K.; Miura, M. J. Am.Chem. Soc. 2015, 137, 6460. doi: 10.1021/jacs.5b02775
Hemric, B. N.; Shen, K.; Wang, Q. J. Am.Chem. Soc. 2016, 138, 5813. doi: 10.1021/jacs.6b02840
Nakamura, I.; Jo, T.; Ishida, Y.; Tashiro, H.; Terada, M. Org. Lett. 2017, 19, 3059. doi: 10.1021/acs.orglett.7b01110
Xu, Q.-F.; Liu, Q.-Q.; Zhang, X.; You, S.-L. Angew.Chem. Int. Ed. 2018, 57, 15204. doi: 10.1002/anie.201809003
Ichikawa, S.; Zhu, S.; Buchwald, S. L. Angew. Chem. Int. Ed. 2018, 57, 8714. doi: 10.1002/anie.201803026
Ishida, Y.; Nakamura, I.; Terada, M. J. Am.Chem. Soc. 2018, 140, 8629. doi: 10.1021/jacs.8b03669
Wu, F.; Zhang, M.; Zhou, W.; Chen, W.; Liu, M.; Wu, H. J. Org. Chem. 2018, 83, 5999. doi: 10.1021/acs.joc.8b00605
Yang, Z.; Jiang, K.; Chen, Y.-C.; Wei, Y. J.Org. Chem. 2019, 84, 3725. doi: 10.1021/acs.joc.9b00262
Wang, Q.; Li, X. Org. Lett. 2016, 18, 2102. doi: 10.1021/acs.orglett.6b00727
Śnieżek, M.; Stecko, S.; Panfil, I.; Furman, B.; Chmielewski, M. J. Org. Chem. 2013, 78, 7048. doi: 10.1021/jo400807c
Diethelm, S.; Carreira, E. M. J. Am.Chem. Soc. 2015, 137, 6084. doi: 10.1021/jacs.5b02574
Wang, F.-X.; Du, J.-Y.; Wang, H.-B.; Zhang, P.-L.; Zhang, G.-B.; Yu, K.-Y.; Zhang, X.-Z.; An, X.-T.; Cao, Y.-X.; Fan, C.-A.J. Am. Chem. Soc. 2017, 139, 4282. doi: 10.1021/jacs.6b13401
Nomura, T.; Yojoshima, T.; Fukuyama, T. Org.Lett. 2018, 20, 119. doi: 10.1021/acs.orglett.7b03555

图式 2 铜催化三组分串联反应合成吡啶机理
Scheme 2 Proposed mechanism of copper-catalyzed three component cascade reaction

图式 3 铜催化肟乙酸酯氧化偶联反应
Scheme 3 Coupling reaction of copper-catalyzed oxime acetates oxidative

图式 4 铜催化[3+3]反应合成含氟吡啶或吡啶酮
Scheme 4 Copper-catalyzed [3+3] reaction to synthesize pyridines or pyridones containing fluoro-atom

图式 5 铜催化[3+3]反应合成含氟吡啶或吡啶酮反应机理
Scheme 5 Proposed mechanism for copper(Ⅰ)-catalyzed [3+3] reaction to synthesize pyridines or pyridones containing fluoro-atom

图式 6 铜催化N—O断裂和环化串联反应
Scheme 6 Copper-catalyzed N—O bond cleavage and annulation cascade reaction

图式 9 铜催化N—O键断裂不对称合成吡咯啉
Scheme 9 Copper-catalyzed N—O bond cleavage to synthesize chiral pyrrolines

图式 10 铜催化N—O/C—C键断裂合成烷基腈反应机理
Scheme 10 Proposed mechanism of copper-catalyzed N—O/ C—C bond cleavage to synthesize alkylnitriles

图式 11 铜催化N—O/C—C键断裂合成烷基腈
Scheme 11 Copper-catalyzed N—O/C—C bond cleavage to synthesize alkylnitriles

图式 12 铜催化活性烯烃的氰基烷基化反应
Scheme 12 Copper-catalyzed cyanoalkylarylation of activated alkenes

图式 13 铜催化N—O键/C—C键断裂合成硫醚化合物
Scheme 13 Copper-catalyzed N—O/C—C bond cleavage to synthesize thioether compounds

图式 14 铜/光催化N—O/C—C键断裂合成烷基腈
Scheme 14 Copper/visible light-catalyzed N—O/C—C bond cleavage to synthesize alkylnitriles

图式 15 铜催化肟酯和三氟甲磺酸盐的反应
Scheme 15 Copper-catalyzed oxime ester with trifluoromethyl- sulfonate reaction

图式 16 铜催化N—O键断裂/C—S键形成合成多取代氮杂环丁烷
Scheme 16 Copper-catalyzed N—O bond cleavage/C—S bond formation to synthesize substituted azetidines

图式 17 铜催化N-芳基硝酮重排反应合成α, β-环氧亚胺化合物
Scheme 17 Copper-catalyzed N-aryl nitrones to synthesize α, β-epoxyketimines

图式 18 铜或金催化叠氮二炔可控合成N杂环
Scheme 18 Copper or gold catalyzed azido-diynes to synthesize controllable N-heterocycles

图式 19 Cu(Ⅱ)催化N—O键断裂合成喹啉反应机理
Scheme 19 Proposed mechanism of copper (Ⅱ)-catalyzed N—O bond cleavage to synthesize quinolines

图式 20 N—O键断裂合成2, 2-联吡啶化合物
Scheme 20 synthesis of 2, 2-bipyridines via N—O bond cleavage

图式 21 铜催化N—O键断裂合成N-烯基环氧亚胺机理
Scheme 21 Proposed mechanism of copper-catalyzed N—O bond cleavage to prepare N-alkenyl oxiranylketimines

图式 22 铜催化区域选项性未活化末端烯烃胺硼化
Scheme 22 Copper-catalyzed regiodivergent aminoboration of unactivated terminal alkenes

图式 24 N—O键断裂全合成gelsemoxonine
Scheme 24 N—O bond cleavage to total synthesis of gelsemoxonine

图式 25 N—O键断裂全合成Palhinines A和D
Scheme 25 N—O bond cleavage to total synthesis of palhinines A and D

图式 26 N—O键断裂全合成huperzine R
Scheme 26 N—O bond cleavage to total synthesis of huperzine R
表 1 铜催化三组分串联反应合成多取代吡啶化合物
Table 1. Copper-catalyzed three component cascade reaction to prepare polysubstituted pyridines
|
|
 下载: 导出CSV
下载: 导出CSV
表 2 铜催化N—O/C—C键断裂合成烷基腈
Table 2. Copper-catalyzed N—O/C—C bond cleavage to synthesize alkylnitriles
|
|
下载: 导出CSV
表 3 铜/铁共催化N—O键断裂合成吡咯稠环衍生物
Table 3. Cu/Fe-cocatalyzed N—O bond cleavage to synthesize fused pyrroles
|
|
下载: 导出CSV
表 4 铜催化氮杂九元环的动力学拆分
Table 4. Copper-catalyzed kinetic resolution of nine-membered N-heterocycles
|
|
下载: 导出CSV
表 5 铜/铁催化N—O键断裂生成吡咯化合物
Table 5. Cu/Fe-cocatalzyed N—O bond cleavage to synthesize pyrroles
|
|
下载: 导出CSV
表 8 铜催化N—O键断裂合成N-烯基环氧亚胺
Table 8. Copper-catalyzed N—O cleavage to prepare N-alkenyl oxiranylketimines
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们