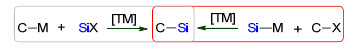
图式 1.
交叉偶联构建碳硅键
Scheme 1.
Cross-coupling of Si—C bond

有机硅化合物因其特殊的性质而广泛应用于合成化学、材料、药物和农药化学等领域, 特别在国防尖端科技、航空航天特种材料等尖端领域[1].有机硅通常是由活泼金属试剂与硅亲电试剂发生取代反应来制备, 但由于其效率低、底物范围窄和普适性差等原因, 难以满足当前有机硅需求.烯烃氢硅化反应是有机硅合成的重要途径之一, 其区域选择性和异构化问题也取得新进展[2].碳氢键直接硅化合成有机硅的新方法[3], 但底物范围仍待发展.交叉偶联反应是高效构建碳碳键的重要方法, 近年来, 利用其构建与碳同族的碳硅键也取得了显著进展[4].通常可以由碳金属试剂和硅亲电试剂或者碳亲电试剂与硅金属试剂间直接偶联来制备(Scheme 1).本论文关注有机硅金属试剂参与碳硅偶联合成有机硅烷研究进展, 将从有机硅硼试剂、有机硅镁试剂、有机硅锌试剂、联硅试剂、有机硅铝试剂和有机硅锂试剂参与偶联构建碳硅键合成有机硅烷方面最新研究进展.
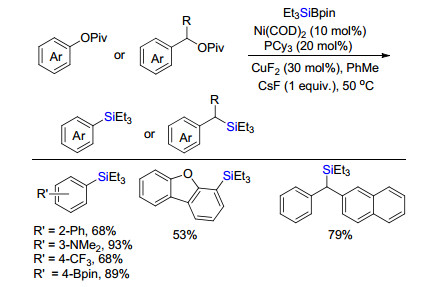
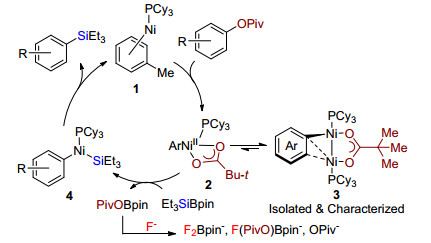
2014年, Martin课题组[5a]报道了Ni/Cu催化碳氧键活化硅化反应(Scheme 2).在Ni(COD)2和配体PCy3以及CuF2协同催化下, 特戊酰芳香酯或苄酯的碳氧键被活化, 与有机硅硼试剂Et3SiBpin交叉偶联生成芳基或苄基硅烷.该反应底物范围广, 官能团兼容性好.直到2018年, 该课题组[5b]进一步通过关键中间体分离和反应性验证、动力学研究和理论计算, 详细阐述了合理的催化机理(Scheme 3).零价镍与三环己基膦和溶剂甲苯配位生成中间体1, 随后特戊酸酯对其氧化加成生成活性中间体2, 有趣的是通过单晶衍射和验证实验发现双镍中间体3, 而且3与2存在平衡, 更趋向稳定的3.随后硅硼试剂与2发生转金属形成含硅金属中间体4, 最后, 还原消除释放硅化产物和活性中间体1完成催化循环.
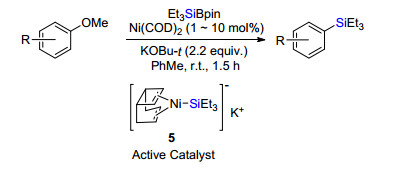
2017年, 该课题组[6]进一步实现了无配体参与下, 镍催化芳基甲醚惰性碳氧键活化与硅硼试剂偶联(Scheme 4).该反应底物普适性好, 取代萘甲醚、芳甲醚、烯基甲醚、甚至苄甲醚碳氧键都能被活化转化为相应硅烷.初步机理研究推测, 该反应重要活性催化中间体是含辛二烯配体的镍硅金属阴离子团5, 但是并没有确凿的晶体结构或其他实验数据证实.
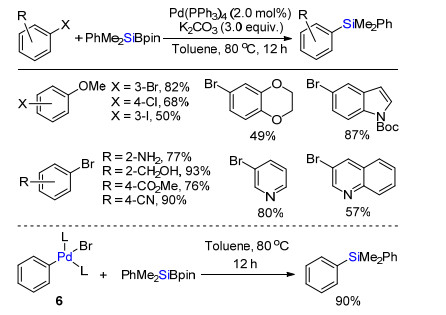
2015年, 何伟课题组[7]报道首次实现钯催化卤代芳烃与有机硅硼试剂类Suzuki偶联(Scheme 5).底物拓展发现官能团普适性好, 系列卤代烃(Cl, Br或I)都能被活化转化为芳基硅烷, 芳烃的取代基为给电子或吸电子基团都能够兼容该体系.苯并1, 4-二氧六环、吲哚环、吡啶环和喹啉等杂环在该体系耐受.机理研究发现, 零价钯与溴代苯氧化加成生成的苯基钯溴代物中间体6与PhMe2SiBpin反应高效转化为目标产物.证实该反应经历传统Suzuki偶联类似的转金属过程.
碳氟键键能高, 难活化的惰性化学键, 实现其活化和官能团化是非常具有挑战的一项工作.近两年, 氟代烃与有机硅硼试剂脱氟硅化反应也取得重大突破.
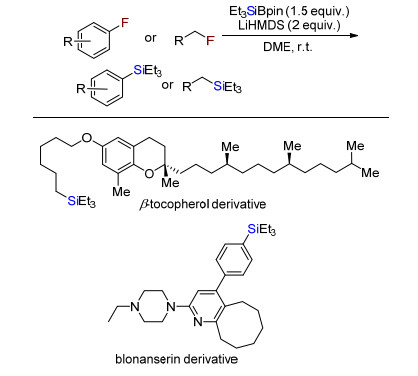
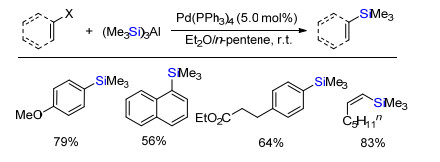
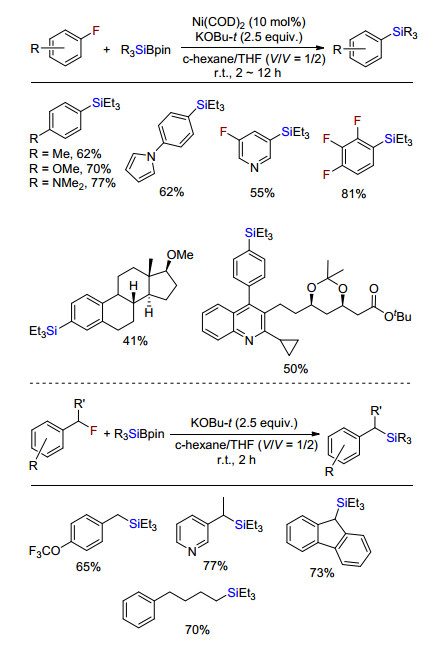
2018年, Shibata课题组[8]首次实现了惰性的氟代烃的脱氟硅化反应(Scheme 6).无需任何配体参与下, 零价镍催化剂Ni(COD)2催化活化氟代芳烃碳氟键与有机硅硼试剂脱氟偶联得到有机硅烷.多氟化合物也能够选择性的实现单个脱氟硅化反应.更有趣的是, 在没有过渡金属镍存在下, 氟代烷烃与有机硅硼试剂在只有叔丁醇钾强碱存在下, 也能发生脱氟硅化反应合成烷基硅.该方法成功应用于药物及其衍生物的脱氟硅化反应, 可潜在应用于药物或者复杂分子后期官能团修饰.
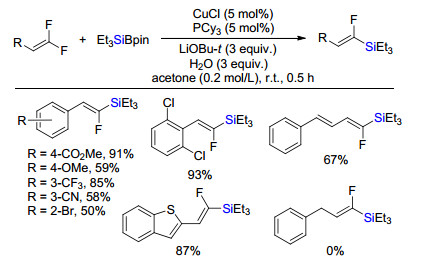
2018年, 王宏根课题组[9]报道了铜催化偕二氟取代烯烃与有机硅硼烷实现单脱氟硅化反应(Scheme 7).得到高选择性的Z型脱氟硅化产物.该反应中底物范围普适性好, 系列取代芳烃、杂环取代的偕二氟烯烃或者取代二烯烃都能够兼容.但是苄基取代偕二氟烯烃无反应活性, 推测π共轭体系在该反应中很重要.实验和理论计算证实该反应是经历铜硅金属试剂对烯烃的插入和后续顺式共平面β氟消除反应机理.
2019年, Martin课题组[10]报道无过渡金属催化的氟代芳烃或烷烃与硅硼试剂脱氟硅化反应(Scheme 8).碱六甲基二硅氨基锂(LHMDS)促进氟代芳烃或者烷烃与有机硅硼试剂脱氟硅化反应.该反应条件简单、温和、无需过渡金属和配体且底物范围广.该反应成功应用于具有生物活性等复杂结构分子后期结构的脱氟硅化.
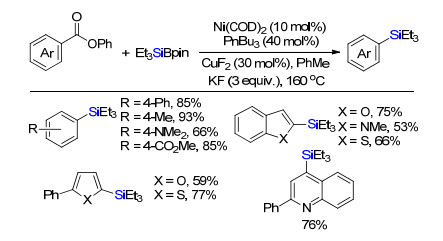
除了卤代烃作为硅化反应的碳亲电试剂, 其他碳亲电试剂也被逐渐开发作为合成子. 2016年, Rueping等[11]报道Ni/Cu催化芳基甲酸苯酯与Suginome硅硼试剂脱羧偶联构建碳硅键(Scheme 9).该反应底物范围广, 各类拉电子或给电子基团都能很好兼容.此外, 各类杂环都能够很好兼容, 例如苯并呋喃、苯并噻吩、吲哚、噻吩、呋喃和喹啉等杂环, 转化得到相应的硅烷.同年, 史壮志课题组同样报道了Ni/Cu协同催化酯活化和脱羰硅化反应[12], 不同在于该体系用双齿膦配体双二环己基膦乙烷.这两例反应中铜催化剂的作用是活化有机硅硼试剂, 原位生成的铜硅中间体与金属镍物种发生转金属化, 进而实现还原消除构建碳硅键.
有机硅硼试剂参与C(sp3)—Si偶联合成有机硅烷是近几年取得突破性进展, 特别是与惰性卤代烷烃或者类卤化合物.这是因为卤代烃碳卤键键能高难活化且易于发生β消除反应, 此外, 硅原子取代基的自身位阻效应导致其金属试剂转金属过程困难.
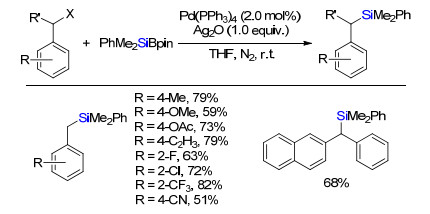
2015年, 罗德平和徐允河课题组报道了苄基卤代物与硅硼试剂在催化剂Pd(PPh3)4和Ag2O条件下偶联生成苄基硅烷(Scheme 10)[13].芳环上取代基团官能团兼容性好, 系列吸电子或者供电子取代基都能很好兼容, 且反应效率受取代基在芳基位置影响较小.二芳基苄氯同样能够在该体系反应, 以中等收率得到二芳基取代苄硅烷.该反应中Ag2O的作用是与苄基钯卤中间体的卤离子配体解离生成含苄基钯氧中间体, 随后与有机硅硼试剂发生σ复分解得到含硅钯中间体, 进一步还原消除得到苄基硅烷.
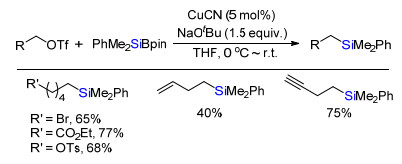
2016年, Oestreich课题组[14]报道铜催化一级烷烃的三氟甲磺酸酯与有机硅硼试剂交叉偶联生成C(sp3)—Si键合成烷基硅(Scheme 11).该反应底物普适性好, 不论在钯催化体系易于被活化的酯基、磺酸酯基和溴等取代基, 还是高活性的烯烃和炔烃等官能团都能在该体系能兼容.
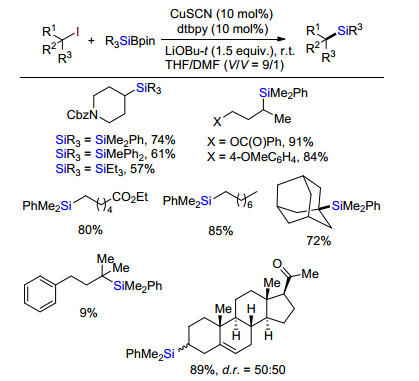
同年, Oestreich课题组[15]报道首例铜催化碘代烷烃与有机硼硅试剂交叉偶联合成烷基硅化合物(Scheme 12).底物考察发现, 无论链状或环状的一级或二级碘烷烃都具有非常好的反应活性, 官能团普适性好.然而, 位阻大的三级烷基碘代物, 除了金刚烷碘代物外, 反应性活性低.天然产物甾体类卤代物同样能够发生硅化反应得到烷基硅化合物.通过理论计算和实验验证, 该反应是经历自由基反应历程.
随后, Oestreich课题组[16]发展脂肪酸与N-羟基邻苯二甲酰亚胺(NHPI)的酯化物与有机硼硅试剂脱羧交叉偶联合成烷基硅化合物(Scheme 13).在Cu/dtbpy或PCy3条件下, 各类羧酸衍生物脱羧硅化得到有机硅化合物.底物拓展发现系列氨基酸, 烯烃、吲哚等官能团都能够很好兼容, 转化为二级烷烃或一级烷烃取代的烷基硅.同样, 由于羧酸α碳为季碳时, 其位阻效应导致反应活性显著降低, 只有金刚烷取代羧酸衍生物以中等分离收率得到有机硅.通过机理研究, 该反应经历自由基历程.
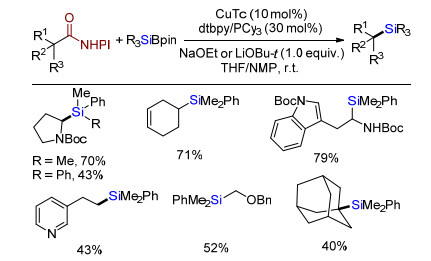
2017年, Oestreich课题组[17]报道手性α-三氟甲磺酸酯取代的烷基氰化物或者羧酸酯与有机硅硼试剂交叉偶联合成构型完全翻转的手性烷基硅烷(Scheme 14).在CuCN催化下, 该手性底物与有机硅硼试剂发生碳硅偶联, 得到立体专一的构型完全翻转手性硅烷.该方法底物普适性好, 提供了一种通过交叉偶联合成手性烷基硅的新方法.
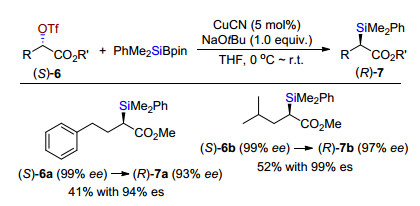
1983年Oshima课题组[18]报道首例钯催化有机硅镁试剂与烯基或烯丙基磷酸酯交叉偶联合成有机硅烷(Scheme 15).该反应具有非常好的立体选择性, Z或E构型的烯基磷酸酯在PdCl2(PPh3)2催化下与有机硅镁试剂PhMe2SiMgMe偶联得到相应构型的目标产物.构型保持.
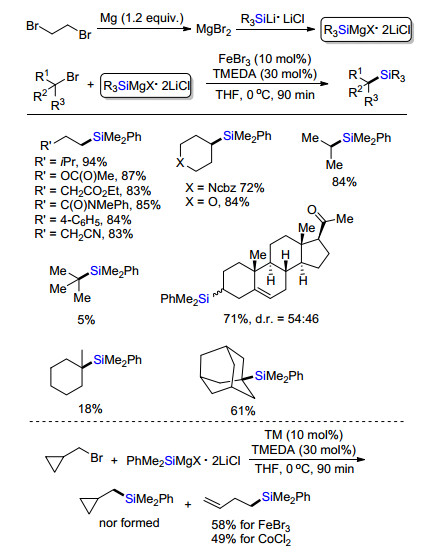
有机硅格氏试剂相对于碳格氏试剂, 其研究和应用非常少.其主要原因是由于有机硅格氏试剂制备相对复杂, 通过常规格氏试剂制备方法难以得到有机硅格氏试剂, 而是往往发生武滋反应得到联硅试剂.其次是因为有机格氏试剂溶解性非常低, 且在醚类溶剂中的稳定性差, 易于使得四氢呋喃环开环.直到2018年, Oestreich课题组[19]报道了铁或钴催化有机硅镁试剂与烷基溴代物交叉偶联合成烷基硅(Scheme 16).新制备的有机硅锂试剂与原位制备的MgBr2的四氢呋喃溶液制备有机硅格氏试剂.各类有机硅格式试剂被合成并研究了它们的稳定性.底物筛选发现一级和二级烷烃都能够很好地在该反应体系展现非常好的反应活性, 得到相应的烷基硅试剂.然而, 叔碳亲电试剂, 因其位阻效应, 在该体系中兼容性不好, 只有位阻相对较小的金刚烷溴代物有较好的反应活性.该方法成功应用于甾体类等天然产物的修饰.进一步反应机理研究, 环丙烷甲基溴代物在标准反应条件下反应, 得到环丙烷开环的硅化产物, 推测该反应经历自由基反应历程.
2005年, Oestreich课题组[20]首次实现铜催化烯丙基醋酸酯或者氨基甲酸酯与有机硅锌试剂交叉偶联合成烯丙基硅烷(Scheme 17).该反应避免当量铜盐的使用, 同时立体选择性好.
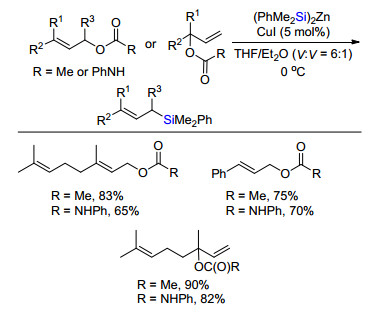
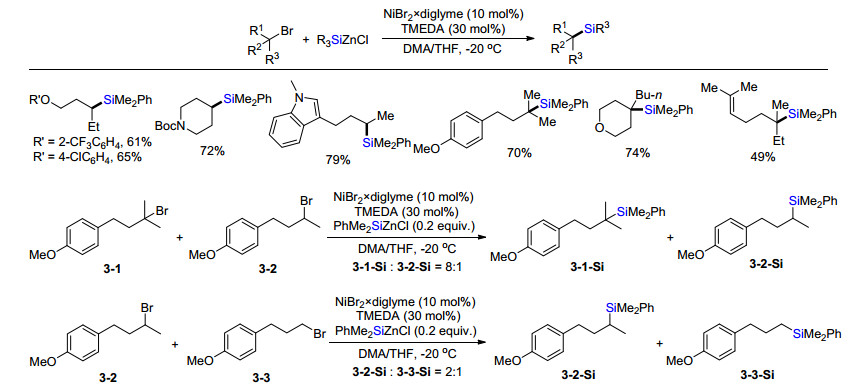
2016年, Fu课题组[21]首次实现了镍催化有机硅锌金属试剂与二级或三级烷基溴代物偶联形成C(sp3)—Si键制备烷基硅化合物(Scheme 18).条件优化发现最佳反应温度为-20 ℃, 控制性实验发现该反应对水和空气并不非常敏感, 对反应效率影响不大.底物拓展发现链状或者环状的溴代物, 一级或者二级溴代物都能高效转化为相应硅烷.特别指出的是, 在该体系中, 三级烷基溴代物转化为相应的有机硅化合物的效率均高于Oestreich报道有机硅硼作为亲核硅试剂的体系[15, 16], 且杂环吲哚类化合物也能够很好兼容.通过取代甲基不同的三个底物3-1、3-2和3-3竞争实验发现, 碳溴键相连的取代甲基越多, 反应效率越高, 这符合自由基稳定性的规律.当四甲基哌啶氧化物(TEMPO)加入到标准反应中, 反应淬灭.基于这些实验, 文章推断该反应是经历自由基反应历程.
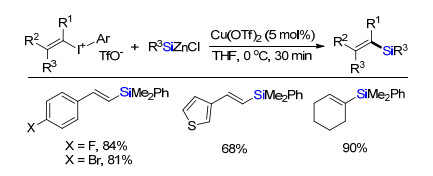
2018年, Oestreich课题组[22]报道了铜催化有机硅锌试剂与烯基高价碘化合物偶联生成C(sp2)—Si键合成烯基硅化合物(Scheme 19).取代苯乙烯、杂环取代烯烃或环状多取代烯烃都能很好地兼容, 转化为烯基硅化合物.该方法发展了一类系统合成烯基取代硅烷新方法.
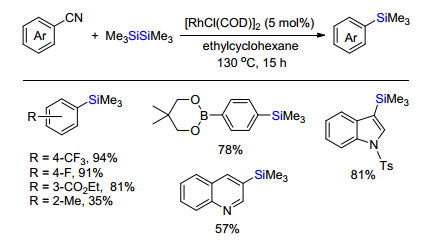
惰性联硅试剂例如六甲基联硅烷广泛应用于与碳亲电试剂偶联合成有机硅烷. Bokermann等[23a]首次报道过渡金属催化卤代芳烃或卤代烷烃偶联与联硅烷直接偶联.后来Nagai等[23b]报道了Pd(PPh3)4催化卤代芳烃与六甲基联硅烷偶联生成芳基取代三甲基硅.随后, 大量卤代烃与联硅烷偶联合成有机硅烷被报道. 2008年, Chatani课题组[23c]报道铑催化芳基氰化物与联硅烷偶联制备硅烷(Scheme 20).各类含拉电子或者给电子基团的芳烃都能够兼容, 除了邻位甲基取代影响收率外, 其他取代基取代位置对反应效率影响不大.需要提及的是含硼酸新戊二酯的取代基、吲哚或喹啉等杂环都能够在该体系中耐受.
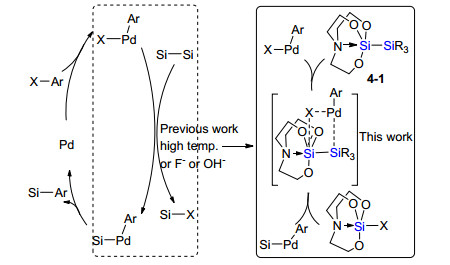
通常对称非活化的联硅试剂参与偶联需要加入过量的活化试剂氟盐或者高温来促进转金属过程. 2015年, Osuka课题组[23d]设计合成了不对称活化的四取代硅联硅试剂4-1, 在没有任何活化试剂且温和反应条件下, 芳基氯代物与该不对称联硅试剂4-1能够发生偶联生成芳基硅烷(Scheme 21).该体系官能团兼容性好, 甚至一些常用于交叉偶联的硼试剂或者硅氧基取代基团都能很好兼容.实验和理论计算发现, 该反应中具有路易斯酸性硅原子和卤代烃中的氯原子之间相互作用极大促进了不对称联硅试剂向芳基钯中间体转金属, 有利于硅化反应的发生.
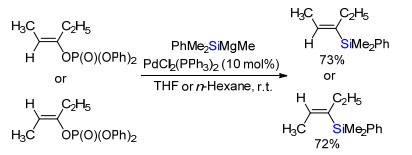
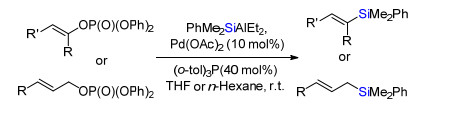
1983年, Oshima及合作者[18]报道了钯催化烯基或烯丙基磷酸酯与有机硅铝试剂PhMe2SiAlEt2交叉偶联合成烯基或烯丙基硅烷(Scheme 22).
1983年, Trost课题组[24]报道了钯催化三甲基硅基铝与芳基溴代物或者烯基碘代物交叉偶联合成芳基/烯基硅烷(Scheme 23).在Pd(PPh3)4催化下, 乙醚和正己烷溶剂中, 室温条件下就能实现各类溴代芳烃或碘代烯烃与有机硅铝试剂交叉偶联.
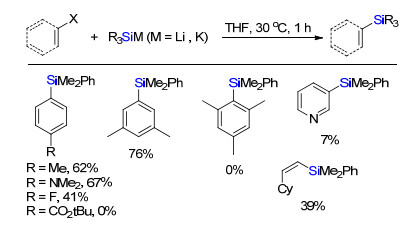
2017年, Ito课题组[25]报道了有机硅锂试剂在无过渡金属参与下与溴代芳烃或者碘代烯烃直接交叉偶联生成芳基硅或者烯基硅烷(Scheme 24).底物筛选发现, 给电子基团如甲基, 二甲氨基等都能兼容, 当为氟原子时, 反应收率显著下降.如果为甲酸叔丁酯取代基或者杂环吡啶时候, 反应不发生, 进一步与碘代烯烃反应得到构型保持的硅化产物.
本文综述了有机硅金属试剂参与交叉偶联构建碳硅键合成有机硅最新研究进展.从有机硅金属试剂分类, 分别介绍6大类有机硅金属试剂参与的偶联反应.同时从碳亲电试剂中碳杂键活化难易程度进行归纳它们硅化反应方面进展.目前, 有机硅金属试剂通过交叉偶联合成有机硅烷方面的研究报道相对较少: (1)试剂种类而言, 有机硅金属试剂相对碳金属试剂种类少, 商品化有机硅金属试剂缺乏, 制备这些试剂比碳金属试剂困难; (2)试剂稳定性而言, 有机硅金属试剂比碳金属试剂稳定性差, 难以储存备用; (3)反应机理研究少, 有机硅金属试剂参与交叉偶联合成有机硅烷反应机理仍待研究, 导致反应体系仍相对少; (4)含硅金属有机络合物相关研究仍相对较少, 从而导致其应用研究匮乏.这些原因也是未来丰富有机硅金属有机化学和有机硅合成的发展方向.近几年碳硅键交叉偶联的突破性进展将促进该领域蓬勃发展和带动有机硅合成和应用研究.此外, 碳硅交叉偶联反应将促进基于碳硅键成键各类新模式的研究和开发.
Brook, M. A. Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000, Chapter 5.
(a) Tondreau, A. M.; Atienza, C. C. H.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Delis, J. G. P.; Chirik, P. J. Science 2012, 335, 567.
(b) Peng, D.; Zhang, Y.; Du, X.; Zhang, L.; Leng, X.; Walter, M. D.; Huang, Z. J. Am. Chem. Soc. 2013, 135, 19154.
(c) Buslov, I.; Becouse, J.; Mazza, S.; Montandon-Clerc, M.; Hu, X. Angew. Chem. Int. Ed. 2015, 54, 14523.
(d) Chen, J.; Cheng, B.; Cao, M.; Lu, Z. Angew. Chem. Int. Ed. 2015, 54, 4661.
(e) Sun, J.; Deng, L. ACS Catal. 2016, 6, 290.
(f) Cheng, B.; Lu, P.; Zhang, H.; Cheng, X.; Lu, Z. J. Am. Chem. Soc. 2017, 139, 9439.
(g) Yang, Y.; Song, R.-J.; Li, Y.; Ouyang, X.-H.; Li, J.-H.; He, D.-L. Chem. Commun. 2018, 54, 1441.
(h) Liu, J.; Chen, W.; Li, J.; Cui, C. ACS Catal. 2018, 8, 2230.
(i) Huai, G. Z.; Teng, H. -L.; Luo, Y.; Lou, S.-J.; Nishiura, M.; Hou, Z. Angew. Chem. Int. Ed. 2018, 57, 12342.
(a) Cheng, C.; Hartwig, J. F. Chem. Rev. 2015, 115, 8946.
(b) Toutov, A. A.; Liu, W.-B.; Betz, K. N.; Fedorov, A.; Stoltz, B. M.; Grubbs, R. H. Nature 2015, 518, 80.
(c) Tobisu, M.; Onoe, M.; Yusuke Kita, Y.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 7506.
(d) Ihara, H.; Suginome, M. J. Am. Chem. Soc. 2009, 131, 7502.
(e) Onoe, M.; Baba, K.; Kim, Y.; Kita, Y.; Mamoru Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2012, 134, 19477.
(f) Liang, Y.; Geng, W.; Wei, J.; Xi, Z. Angew. Chem. Int. Ed. 2012, 51, 1934.
(g) Ghavtadze, N.; Melkonyan, F. S.; Gulevich, A. V.; Huang, C.; Gevorgyan, V. Nat. Chem. 2014, 6. 122.
(h) Kanyiva, K. S.; Kuninobu, Y.; Kanai, M. Org. Lett. 2014, 16, 1968.
(i) Liu, Y. J.; Liu, Y. H.; Zhang, Z. Z.; Yan, S. Y.; Chen, K.; Shi, B. F. Angew. Chem. Int. Ed. 2016, 55, 13859.
(j) Li, W.; Huang, X.; You, J. Org. Lett. 2016, 18, 666.
(k) Zhao, W.-T.; Lu, Z.-Q.; Zheng, H.; Xue, X.-S.; Zhao, D. ACS Catal. 2018, 8, 7997.
(a) Kakiuchi, F.; Igi, K.; Matsumoto, M.; Chatani, N.; Murai, S. Chem. Lett. 2001, 30, 422.
(b) Murakami, K.; Hirano, K.; Yorimitsu, H.; Oshima, K. Angew. Chem. Int. Ed. 2008, 47, 5833.
(c) Liang, Y.; Zhang, S. G.; Xi, Z. F. J. Am. Chem. Soc. 2011, 133, 9204.
(d) Zhang, Q. W.; An, K.; He, W. Angew. Chem. Int. Ed. 2014, 53, 5667.
(e) Li, L. W.; Zhang, Y. B.; Gao, L.; Song, Z. L. Tetrahedron Lett. 2015, 56, 1466.
(f) Zhang, L.; Hang, Z.; Liu, Z.-Q. Angew. Chem. Int. Ed. 2016, 55, 236.
(g) Xu, Z.; Xu, J. Z.; Zhang, J.; Zheng, Z. J.; Cao, J.; Cui, Y. M.; Xu, L. W. Chem. Asian J. 2017, 12, 1749.
(h) Li, W.; Xiao, G.; Deng, G.; Liang, Y. Org. Chem. Front. 2018, 5, 1488.
(a) Zarate, C.; Martin, R. J. Am. Chem. Soc. 2014, 136, 2236.
(b) Somerville, R. J.; Hale, L. V. A.; Gómez-Bengoa, E.; Burés, J.; Martin, R. J.Am.Chem.Soc. 2018, 140, 8771.
Zarate, C.; Nakajima, M.; Martin, R. J. Am. Chem. Soc. 2017, 139, 1191. doi: 10.1021/jacs.6b10998
Guo, H.; Chen, X.; Zhao, C.; He, W. Chem. Commun. 2015, 51, 17410 doi: 10.1039/C5CC07071F
Cui, B.; Jia, S.; Tokunaga, E.; Shibata, N. Nat. Commun. 2018, 9, 4393. doi: 10.1038/s41467-018-06830-w
Tan, D.-H.; Lin, E.; Ji, W.-W.; Zeng, Y.-F.; Fan, W.-X.; Li, Q.; Gao, H.; Wang, H. Adv. Synth. Cat. 2018, 360, 1032. doi: 10.1002/adsc.201701497
Liu, X. W.; Zarate, C.; Martin, R. Angew. Chem. Int. Ed. 2019, 58, 2064. doi: 10.1002/anie.201813294
Guo, L.; Chatupheeraphat, A.; Rueping, M. Angew. Chem. Int. Ed. 2016, 55, 11810. doi: 10.1002/anie.201604696
Pu, X.; Hu, J.; Zhao, Y.; Shi, Z. ACS Catal. 2016, 6, 6692. doi: 10.1021/acscatal.6b01956
Huang, Z.-D.; Ding, R.; Wang, P.; Xu, Y.-H.; Loh, T.-P. Chem. Commun. 2016, 52, 5609. doi: 10.1039/C6CC00713A
Scharfbier, J.; Oestreich, M. Synlett 2016, 27, 1274. doi: 10.1055/s-0035-1561407
Xue, W.; Qu, Z.-W.; Grimme, S.; Oestreich, M. J. Am. Chem. Soc. 2016, 138, 14222. doi: 10.1021/jacs.6b09596
Xue, W.; Oestreich, M. Angew. Chem. Int. Ed. 2017, 56, 11649. doi: 10.1002/anie.201706611
Scharfbier, J.; Hazrati, H.; Irran, E.; Oestreich, M. Org. Lett. 2017, 19, 6562. doi: 10.1021/acs.orglett.7b03279
Okuda, Y.; Sato, M.; Oshima, K.; Nozaki, H. Tetrahedron Lett. 1983, 24, 2015. doi: 10.1016/S0040-4039(00)81831-6
Xue, W.; Shishido, R.; Oestreich, M. Angew. Chem. Int. Ed. 2018, 57, 12141. doi: 10.1002/anie.201807640
Oestreich, M.; Auer, G. Adv. Synth. Catal. 2005, 347, 637. doi: 10.1002/adsc.200404381
Chu, C. K.; Liang, Y.; Fu, G. C. J. Am. Chem. Soc. 2016, 138, 6404. doi: 10.1021/jacs.6b03465
Zhang, L.; Oestreich, M. Org. Lett. 2018, 20, 8061. doi: 10.1021/acs.orglett.8b03714
(a) Awell, A.; Bokermann, G. C. US 3772347, 1973.
(b) Matsumoto, H.; Nagashima, S.; Yoshihiro, K.; Nagai, Y. J. Organomet. Chem. 1975, 88, C1.
(c) Tobisu, M.; Kita, Y.; Ano, Y.; Chatani, N. J. Am. Chem. Soc., 2008, 130, 15982.
(d) Yamamoto, Y.; Matsubara, H.; Murakami, K.; Yorimitsu, H.; Osuka, A. Chem. Asian J. 2015, 10, 219.
Trost, B. M.; Yoshida, J. Tetrahedron Lett. 1983, 24, 4895. doi: 10.1016/S0040-4039(01)99804-1
Yamamoto, E.; Ukigai, S.; Ito, H. Synlett 2017, 28, 2460. doi: 10.1055/s-0036-1590835

图式 2 Ni/Cu催化特戊酸酯碳氧键活化硅化反应
Scheme 2 Ni/Cu-catalyzed silylation of pivalate via C—O activation

图式 4 镍催化芳甲醚与有机硅硼试剂交叉偶联
Scheme 4 Ni-catalyzed cross-coupling of methoxyl aromatics and silyl boranes

图式 5 Pd催化卤代芳烃与有机硅硼试剂交叉偶联
Scheme 5 Pd-catalyzed cross-coupling of aromatic halides and silyl boranes

图式 8 碱促进的氟代烃脱氟硅化反应
Scheme 8 Base promoted defluorosilylation of fluoroarenes and fluoroalkanes

图式 10 Pd催化卤代苄与有机硅硼试剂交叉偶联
Scheme 10 Pd-catalyzed cross-coupling of benzylic halides and silyl boranes

图式 11 铜催化烷基三氟甲磺酸酯与有机硅硼试剂交叉偶联
Scheme 11 Cu-catalyzed cross-coupling of alkyl aliphatic triflates and silyl boranes

图式 12 铜催化烷基碘代物与有机硅硼试剂交叉偶联
Scheme 12 Cu-catalyzed cross-coupling of alkyl iodides and silyl boranes

图式 13 铜催化烷基羧酸衍生物与有机硅硼试剂交叉偶联
Scheme 13 Cu-catalyzed cross-coupling of aliphatic carboxylic acid derivatives and silyl boranes

图式 14 铜催化烷α-三氟甲磺酰氧取代脂肪腈或酸酯与有机硅硼试剂交叉偶联
Scheme 14 Cu-catalyzed cross-coupling of α-triflyloxy nitriles or esters and silyl boranes

图式 15 钯催化烯基或烯丙基磷酸酯与有机硅镁试剂交叉偶联
Scheme 15 Pd-catalyzed cross-coupling of allylic or vinyl phosphates and organosilyl magnesium reagents

图式 16 铁或钴催化溴代烷烃与有机硅镁试剂交叉偶联
Scheme 16 Fe/Co-catalyzed cross-coupling of alkyl bromides and organosilyl magnesium reagents

图式 17 铜催化烯丙基碳酸酯或氨基甲酸酯与有机硅锌试剂交叉偶联
Scheme 17 Cu-catalyzed cross-coupling of allylic esters or carbamates and organosilyl zinc reagents

图式 18 镍催化烷基溴代物与有机硅锌试剂交叉偶联
Scheme 18 Ni-catalyzed cross-coupling of alkyl bromides and organosilyl zinc reagents

图式 19 铜催化烷基溴代物与有机硅锌试剂交叉偶联
Scheme 19 Cu-catalyzed cross-coupling of vinyliodonium salts and organosilyl zinc reagents

图式 20 铑催化芳基氰化物与联硅烷交叉偶联
Scheme 20 Rh-catalyzed cross-coupling of nitriles and disilanes

图式 21 钯催化氯代芳烃与不对称联硅试剂交叉偶联
Scheme 21 Pd-catalyzed cross-coupling of aromatic chlorides and silylsilatranes

图式 22 钯催化磷酸酯与有机硅铝试剂交叉偶联
Scheme 22 Pd-catalyzed cross-coupling of vinyl or allyl phosphates and organosilyl aluminum reagents

图式 23 钯催化溴代芳烃或碘代烯烃与有机硅铝试剂交叉偶联
Scheme 23 Pd-catalyzed cross-coupling of aromatic bromides or vinyl iodides and organosilyl aluminum reagents

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:




