表 1
一锅法合成N-错位卟啉的各种工艺
Table 1.
Various synthetic process of N-confused porphyrin by one pot methodology

早在19世纪40年代, 美国著名化学家Linus Pauling就预测了卟啉的同分异构体——N-错位卟啉的存在[1]. 50年后, 科学家们才从实验上确证了N-错位卟啉的结构[2, 3].该发现为卟啉化学的发展开辟了新的研究方向[4].与卟啉类似, N-错位卟啉具有18π电子的共轭体系, 在仿生催化、材料、化学传感器、光动力治疗癌症等领域有广阔的应用前景[5~9], 因此本文主要综述了N-错位卟啉的研究进展.
卟啉(porphyrin), 又称紫质或生命色彩原.它是生命体内血红素等生物大分子的核心结构和活性中心, 扮演着多重且关键性的角色, 例如氧的传递(血红蛋白)、电子传递(细胞色素C)、催化代谢(细胞色素P450)、光合作用(叶绿素)等[10~14].卟啉作为共轭四吡咯大环化合物的代表具有极强的修饰性, 如外围β位、meso位、大环中心或键连方式等修饰.通过改变卟啉大环结构得到的卟啉类似物称为异卟啉[15].异卟啉不仅具有卟啉的一般特性, 还呈现出其它新颖而有趣的性质, 如稳定特殊金属离子、Mobius芳香性、阴离子识别、近红外荧光等性质[16~20].根据对卟啉大环的改变方式可将异卟啉分为四类: (1)改变环的大小, 如扩展卟啉[16~18]等; (2)改变环内的N原子; (3)改变环的饱和状态; (4)改变吡咯单元之间的键接方式, 如N-错位卟啉[2~4]、咔咯[21]等.其中作为异卟啉的典型代表N-错位卟啉、咔咯等已形成了独立的研究领域.
1994年, Furuta[3]和Latos-Grażyński[2]课题组分别独立报道了一种含有N3C核的异卟啉. Furuta课题组[3]将其命名为N-错位卟啉(N-confused porphyrin, NCP).与卟啉1相比, N-错位卟啉2或3有一个N-错位吡咯环, 它的C-21原子位于大环内核, N-2原子位于环外的β位置.因其独特的结构, 表现出很多特殊的光物理和金属配位等性质.
|
|
N-错位卟啉比卟啉分子的对称性更低, 具有独特的互变异构性[5]. N-错位卟啉在二氯甲烷等非极性溶剂中呈红色, 主要以2 (isomer A)形式存在.在N, N-二甲基甲酰胺等极性溶剂中呈绿色, 主要以3 (isomer B)形式存在, 因为环外N—H键可与极性溶剂形成氢键, 从而稳定isomer B的分子构型. N-错位卟啉的互变异构特性赋予其具有两种配位方式, 既可像咔咯一样作为三价阴离子配体, 又可像卟啉一样作为二价阴离子配体.
N-错位卟啉的合成始于20世纪90年代初.直到1999年, Lindsey课题组[22]发现以甲基磺酸为催化剂, 二氯甲烷为溶剂, 苯甲醛与吡咯合成5, 10, 15, 20-四苯基N-错位卟啉(NCTPP)的产率可达40%左右. N-错位卟啉化学的发展才开始进入繁荣阶段.根据起始原料的不同, 将其合成方法分为一锅法、[2+2]法和[3+1]法.一锅法主要用于合成对称N-错位卟啉, [2+2]法和[3+1]法主要用于合成不对称N-错位卟啉及一锅法合成产率较低的对称N-错位卟啉.
一锅法, 也可称为二步法.相应的醛与吡咯在催化剂(甲基磺酸等)作用下, 形成四吡咯甲烷中间体; 其在氧化剂如四氯苯醌(TCQ)或二氯二腈基苯醌(DDQ)和碱(三乙胺, TEA)作用下, 被氧化关环合成N-错位卟啉(Eq. 1).醛与吡咯在酸催化剂的作用下合成两种不同位相的四吡咯甲烷中间体可能是N-错位卟啉生成的机理.因为该中间体的吡咯环上的α位和β位均存在反应活性, 所以氧化关环生成的产物是卟啉与N-错位卟啉的混合物.
N-错位卟啉的合成研究进展最早可追溯到1994年(表 1). Furuta课题组[3]和Latos-Grażyński课题组[2]同年分别以溴化氢和三氟化硼乙醚溶液作为催化剂, 一锅法合成了N-错位卟啉4和5, 产率约4%~7%, 主产物为卟啉.由于N-错位卟啉的合成产率极低限制了其发展.直到1999年Lindsey课题组[22]对这一反应工艺进行优化.改进了反应物原料的配比和甲基磺酸催化剂的浓度(5%~15%), N-错位卟啉4最高产率可达39%.这一重大发现开创了N-错位卟啉化学发展的新时代.此后, 科研工作者投入大量的精力, 在N-错位卟啉合成工艺研究中, 如Lindsey等[23]研究了BF3•Et2O/NaCl等45种催化剂对N-错位卟啉4合成反应的影响.发现33种催化剂均对该反应有催化效果, 四苯基卟啉产率为5%~58%, 四苯基N-错位卟啉4产率为1%~40%.其中, 催化效果最佳的是甲基磺酸. Furuta课题组[24]按经典的Lindsey一锅法以五氟苯甲醛与吡咯为原料甲基磺酸催化合成N-错位卟啉6, 发现产率极低(<0.1%), 难以分离得到. 2017年, Geier课题组[25]探索了影响N-错位卟啉6产率的几个因素如催化剂的种类(甲基磺酸、对甲苯磺酸、三氟甲基磺酸、三氟乙酸和三氟化硼乙醚溶液)、催化剂的用量、氧化剂DDQ的用量和反应时间等, 并对各工艺参数进行优化, 获得了该反应的最佳工艺条件.当三氟甲基磺酸作为催化剂时, N-错位卟啉6的产率可达10%~12%. Kitaoka等[26]探究了溶剂对N-错位卟啉4合成反应的影响.以离子液体替代二氯甲烷作溶剂可以合成N-错位卟啉4, 但产率较低(0%~10%).而采用离子液体与二氯甲烷的混合液作为溶剂时, 不仅可以提高N-错位卟啉4的产率, 还可以减少二氯甲烷的用量.从上述可以看出, 一锅法主要用于对称N-错位卟啉的合成, 催化剂与溶剂的选择及用量对N-错位卟啉的合成具有重要的影响.随着科研工作者的不断探索, 对称取代的四(R-苯基)N-错位卟啉[R=4-氟(35%)、4-氯、4-溴(13%)、4-碘(8%)、4-甲氧基(13%)、4-腈基(6.7%)、4-甲氧基羰基(16%)、4-乙炔基(9.2%)、4-溴甲基(21%)、4-硝基(6.8%)、3-甲氧基(23%)、2', 5'-二甲氧基苯基(26%)和3, 5-二叔丁基(10%)等][27~30]数量越来越多, 种类也越来越丰富.
 下载:
导出CSV
下载:
导出CSV
| 时间/年 | 课题组 | 催化剂 | 产物 | 产率 |
| 1994 | Furuta | 溴化氢 | 4 | 5%~7% |
| 1994 | Latos-Gra|yński | 三氟化硼乙醚溶液 | 5 | 4% |
| 1999 | Lindsey | 甲基磺酸 | 4 | 39% |
| 2003 | Furuta | 甲基磺酸 | 6 | <0.1% |
| 2017 | Geier | 甲基磺酸 | 6 | 6% |
| 对甲苯磺酸 | 6 | 5% | ||
| 三氟甲基磺酸 | 6 | 10%~12% | ||
| 三氟乙酸 | 6 | 4% | ||
| 三氟化硼乙醚溶液 | 6 | 1% |
|
|
(1) |
一锅法不仅可以合成对称N-错位卟啉, 还可以合成不对称N-错位卟啉. Chmielewski课题组[31]以吡咯、α-乙基吡咯和苯甲醛为原料在甲基磺酸催化下一锅法合成了N-错位卟啉4, 7, 8和9 (Eq. 2), 产率均为5%.
|
|
(2) |
Wolff等[32]以3, 5-二叔丁基苯甲醛, 4-硝基(乙炔基、溴)苯甲醛与吡咯合成了三个不对称N-错位卟啉10、11和12 (Eq. 3), 产率均为4%.
|
|
(3) |
虽然一锅法可以合成不对称的N-错位卟啉, 但产物复杂, 分离困难, 产率较低.因此, 一锅法合成不对称N-错位卟啉的研究一直进展不大.一锅法合成对称N-错位卟啉是目前最适用的方法, 但仍存在不足, 如硝基取代N-错位卟啉等的合成产率偏低; 氨基、羧酸和磺酸等取代的N-错位卟啉不能直接通过相应醛为原料合成.
[2+2]法是利用两个二吡咯烷烃化合物在催化剂的作用下合成N-错位卟啉及其衍生物. 2003年, Furuta等[24]发现一锅法合成N-错位卟啉6产率极低(<0.1%), 改用[2+2]法.利用N-错位二吡咯甲烷13与二吡咯甲烷14在甲基磺酸的催化下合成对称N-错位卟啉6 (Eq. 4), 产率为21%.
|
|
(4) |
Dolphin等[33]采用[2+2]法, 由α, β-二吡咯甲烷15与二吡咯烷烃二醛16在混酸(盐酸和醋酸)催化作用下, 合成了不对称N-错位卟啉17 (Eq. 5), 产率为25%.
|
|
(5) |
Narayanan等[34]采用[2+2]法合成了N-错位卟啉4.以二氯甲烷为溶剂, 二吡咯苯基甲烷为原料, 在对甲苯磺酸催化作用下合成N-错位卟啉4, 分离产率为7% (Eq. 6).
2012年, Acharya等[35]采用[2+2]法, 以N-错位二吡咯甲烷18和19、正常的二吡咯甲烷及相应的苯甲醛为原料, 合成了三个不对称N-错位卟啉20, 21和22, 合成产率为3%~5% (Eq. 7).
|
|
(6) |
|
|
(7) |
[2+2]法合成N-错位卟啉及其衍生物, 产率较低, 但可合成一些一锅法无法合成的不对称N-错位卟啉.
[3+1]法是利用三吡咯烷烃化合物与吡咯衍生物在催化剂的作用下合成N-错位卟啉衍生物. 1999年Lash等[36]以CH2Cl2为溶剂, TFA催化吡咯二醛与三吡咯二酸反应, FeCl3氧化关环合成β-烷基取代的N-错位卟啉23 (Eq. 8).
2003年, Furuta课题组[37]利用[3+1]法合成了β-烷基未取代meso位部分取代的N-错位卟啉24 (Eq. 9).以氯仿为溶剂, 在三氟化硼甲醇溶液的催化作用下, 2, 4-二(苯基羟基甲基)吡咯与无取代的三吡咯二烷烃反应, 反应完后加入四氯苯醌氧化剂缩合而成.
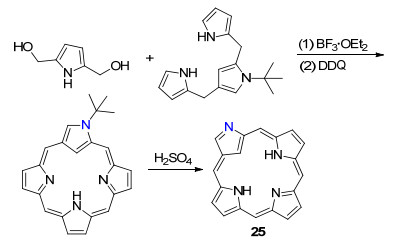
2005年, Furuta课题组[38]利用该法合成了N-错位卟吩25, 主要以三氟化硼乙醚溶液为催化剂, 利用2, 5-二羟甲基吡咯与N-叔丁基N-错位三吡咯二烷烃反应后,加入2, 3-二氯-5, 6-二氰基对苯醌(DDQ)氧化缩合成N-叔丁基N-错位卟吩, 再通过硫酸的酸化作用合成了N-错位卟吩(Scheme 1).
|
|
(8) |
|
|
(9) |
2007年该课题组[39]又利用[3+1]法, 以甲基磺酸或三氟化硼乙醚溶液为催化剂, 利用吡啶基取代的吡咯26与三吡咯二烷烃27 (28)合成了吡啶基取代的N-错位卟啉29 (Eq. 10)和30 (Eq. 11), 产率均为0.5%.
他们课题组[40]还改进了Lash报道的不对称N-错位卟啉合成方法.利用乙醇替代二氯甲烷作为溶剂, 以对甲苯磺酸替代三氟乙酸作为催化剂, 吡咯二醛与三吡咯二酸反应, 对氯苯醌氧化关环合成了N-错位卟啉31(产率: 6%)、32(产率: 3%)和33(产率: 1%) (Eq. 12).
|
|
(10) |
|
|
(11) |
|
|
(12) |
与[2+2]法类似, [3+1]法合成N-错位卟啉的产率较低, 但可合成一些新颖的不对称N-错位卟啉.
随着N-错位卟啉及其衍生物合成方法的不断发展, 其应用研究也越来越受到科研工作者的关注. N-错位卟啉与卟啉分子结构上的相似性导致两者性质方面具有相似之处, N-错位卟啉金属配合物[6, 8]还表现出与金属卟啉类似的催化活性.
卟啉与N-错位卟啉等大环化合物金属配合物在催化领域的应用已经成为卟啉化学的研究热点.例如:在氧化、烯烃的环丙烷化、不对称合成以及氮杂环丙烷化反应等有机合成反应中金属卟啉与金属咔咯等均表现出优异的催化活性[41~43].目前, 关于N-错位卟啉在催化化学中的应用报道较少, 主要是有关催化环丙烷化、氧原子转移、催化氧化和催化环加成等的反应.
2006年, Furuta等[6]报道了四个N-错位卟啉铑配合物34~37 (Eq. 13)在苯乙烯与重氮乙酸乙(叔丁)酯进行环丙烷化的催化应用.研究表明, 它们均有很好的催化活性. 34用于苯乙烯和重氮乙酸乙酯的环丙烷化反应, 产率可达92%, trans/cis为91/9, 转化数(TON)为1840, 明显高于铑卟啉的催化结果(产率71%, trans/cis: 52/48).在N-错位卟啉环上引入更大位阻的均三甲苯基(35), 发现催化产率并没有显著的提高(产率93%), 反式产物的对映选择性反而变差(trans/cis为83/17).当轴向配体换成吡啶基(Py), 发现催化效率下降, 反式产物的对映选择性也下降.然而, 当轴向配体均为吡啶基, 例如36(催化产率60%)与37(催化产率89%), 后者具有吸电基(五氟苯基)可很大程度提高催化活性.另外, 将重氮乙酸乙酯换成重氮乙酸叔丁酯还发现环丙烷化产物的产率变化不大(91%), 但反式产物的对映选择性有明显的提高(trans/cis: 98/2).
|
|
(13) |
2011年, Ziegler等[44]发现N-错位卟啉钴配合物也可催化苯乙烯环丙烷化反应.该课题组合成了两个N-错位卟啉钴配合物38和39, 并研究了它们在催化苯乙烯与重氮乙酸乙酯的环丙烷化反应中的催化性能.结果表明, 它们的催化产率约为80%, 且trans/cis约为98/2, 产物的对映选择性明显优于钴卟啉(trans/cis: 26/74).
|
|
2018年, Furuta等[45]也合成了两个轴向配体为2-硫吡啶的N-错位卟啉钴配合物40和41, 并研究了它们及其还原产物在催化苯乙烯与重氮乙酸乙酯的环丙烷化反应中的催化性能.结果表明, 以40为催化剂(0.5 mol%), 甲苯为溶剂在常温条件下不发生环丙烷化反应.同等条件下, 当温度升高到80 ℃反应6 h时, 其催化产率为70%且trans/cis约为83/17.而将40 (41)与质量分数为0.5%连二亚硫酸钠的二氯甲溶液进行预处理得到其相应的还原产物.意外发现40 (41)的还原产物在常温条件下表现出优异的催化性能, 其催化产率为78% (76%), 且trans/cis为92/8 (90/10).与N-错位卟啉钴配合物39(催化产率为31%且trans/cis约为92/8)相比, 两者的对映选择性相当, 40的还原产物呈现更优异的催化活性.与钴卟啉(催化产率为6%且trans/cis约为72/28)相比, 40的还原产物具有更优异的催化反应活性和对映选择性.这可能与硫吡啶配体向中心金属钴提供电子并稳定N-错位卟啉内核碳原子有关.
N-错位卟啉铑(钴)配合物与铑(钴)卟啉相比, 在催化环丙烷化反应中反应活性更高, 且反式产物的对映选择性也显著提高.
2012年, Futura等[8]合成了两个新的N-错位卟啉铼配合物42和43 (Eq. 14), 并考察了它们在催化三苯基膦与氧化4-苯基吡啶反应的催化性能.
研究结果表明, 4-苯基吡啶的产率可达97%以上, 明显优于三苯基咔咯铼配合物的催化效果(产率9.8%).该催化反应在无氧条件下也可进行, 表明三苯基氧膦上的氧是通过42和43将氧化4-苯基吡啶中的氧转移而得.研究还发现一个有趣的现象, 42和43之间能相互转化. 42与氧化吡啶反应生成43与吡啶, 而43可与三苯基膦反应生成42.
|
|
(14) |
2014年, 我们课题组[46]设计合成了一系列不同推拉电子效应的N-错位卟啉锰配合物44~49, 运用紫外可见光谱、核磁共振氢(碳)谱、质谱、X射线光电子能谱和循环伏安法等手段确证了它们的结构, 研究并比较了44~49对苯乙烯氧化反应的催化活性.
|
|
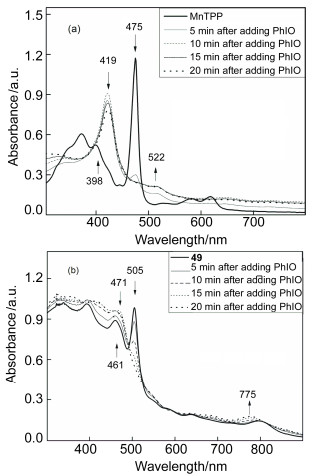
反应时间、溶剂、氧源和取代基等均对催化反应有较大的影响.当亚碘酰苯(PhIO)为氧源, 44~49为催化剂时, 主要产物均为环氧苯乙烷, 且催化活性与四苯基卟啉锰配合物MnTPP相当.表明N-错位卟啉锰配合物具有催化有机底物环氧化的潜力.结合催化实验结果及PhIO存在下的N-错位卟啉锰配合物的紫外可见光谱的变化(图 1), 初步探讨了以PhIO为氧源时催化反应的机理.推测中间体可能为MnV=O, 与锰卟啉的催化机理类似.
2019年Furuta等[47]发现N-错位卟啉钌配合物也可以催化烯烃氧化反应.该课题组合成了三个轴向配体分别为2-硫吡啶、2-吡啶酮和2-亚氨基吡啶的N-错位卟啉钌配合物50、51和52, 并研究了它们(0.076 mol%)对苯乙烯(35 equiv.)氧化反应的催化活性.以双氧水为绿色氧源, 室温条件下, 反应24 h, 50、51和52催化氧化产物主要是BA和SO, 产率分别约为37%~40%和2%~7%.相同条件下, 相比催化剂四苯基卟啉钌配合物(BA: 25%, SO: 3%)与N-错位四苯卟啉钌配合物53 (BA: 26%, SO: 2%)呈现更低的催化活性.无催化剂条件下, BA和SO的产率均低于1%.上述结果表明, 吡啶衍生物等轴向配体的引入可增加N-错位卟啉钌配合物催化苯乙烯氧化反应的催化活性.这可能是由于吡啶衍生物的引入改变了钌与N-错位卟啉配位模式. 53内环中的碳以sp2杂化参与配位, 而50~52内环中的碳以sp3杂化参与配位.该研究成果为催化剂的结构与分子设计提供了新思路, 也为N-错位卟啉在绿色化学领域的发展提供理论与实践基础.
|
|
2019年洪政雄课题组[48]报道了一类具有双功能结构的催化剂N-苯甲酸取代的N-错位卟啉镍54和钯55配合物, 可催化环氧烷烃与二氧化碳的环加成反应, 转化率可达98%, 转化数(TON)为7000.
|
|
其中添加剂2, 6-二甲基吡啶与环氧乙烷上取代基对该环加成反应有重要的影响.不加入该催化剂时无环碳酸酯产物产生, 加入时转化率可达90.4%. 2, 6-二甲基吡啶是该催化反应的关键, 原因可能是具有碱性的2, 6-二甲基吡啶与含有酸性的COOH基团的催化剂生成亲核性更强的COO-基团.从而有利于环加成反应.当环氧乙烷上取代基为吸电子基或空间位阻较大的基团时, 转化率更高.可能是由于吸电基使环氧乙烷中C—C键变长, C—O—C键角变大, 空间位阻较大的基团可增加该基团与N-苯甲酸取代基或meso位苯环之间的范德华作用力, 有利于环氧烷烃开环.该催化反应的机理可能与中心金属离子作为Lewis酸活化环氧烷烃中的氧同时含有更强亲核性的COO-基团进攻环氧乙烷上取代基较少的碳有关.
N-错位卟啉金属配合物可催化烯烃的环丙烷化、氧转移、氧化和环加成反应, 为N-错位卟啉在有机合成反应中的应用提供了理论依据和实践基础.
关于N-错位卟啉在生物化学领域应用的探究一直是卟啉化学的研究热点与重点.目前, 在该领域的应用主要集中在与核酸及蛋白的相互作用和光动力治疗等[7, 9, 49~51].
核酸是生物体遗传信息的载体, 不仅与细胞的生长发育和生命的延续等正常的生命活动有关, 而且还与癌变等异常生命活动息息相关.研究表明端粒、端粒酶与肿瘤细胞的无限增值密切相关.端粒酶在绝大多数的恶性肿瘤细胞中呈阳性表达, 而在正常细胞中呈阴性表达.若能特异性地抑制肿瘤细胞端粒酶的活性, 将抑制肿瘤细胞的无限增殖, 从而使肿瘤细胞停止生长或衰退死亡, 最终达到治愈癌症的目的.核酸的基本单元为核苷酸, 根据组成可以将其分为核糖核酸(RNA)和脱氧核糖核酸(DNA)两类.后者根据结构又可分为DNA双螺旋结构和DNA四螺旋结构(G4-DNA).其中, G4-DNA (G-四链体DNA)的形成可以抑制端粒的延伸和端粒酶的活性.因此, 能稳定G-四链体DNA的药物很有可能作为端粒酶的抑制剂.研究药物分子与DNA的相互作用, 有助于人类从分子水平上了解生命现象的本质, 从而设计出高效低毒的抗癌药物, 最终治愈癌症.随着N-错位卟啉化学的发展, 作为潜在的抗癌药物, N-错位卟啉与DNA相互作用的研究已取得了一定的进步.
Furuta等[49, 50]报道了两个水溶性的N-错位卟啉衍生物56和57, 并采用紫外光谱与圆二色谱(CD)研究了它与双链DNA (dsDNA)和单链(ssDNA)相互作用.
|
|
56与57主要结合在dsDNA的磷酸骨架上或通过沟面相互结合, 且与dsDNA的结合能力顺序为57>56. 56与ssDNA (dsDNA)的CD光谱均呈现正的诱导CD, 但形状不同, 而57与ssDNA (dsDNA)的CD光谱形状类似.表明相较于57, 56的光物理性质受DNA的结构影响较大, 这可能与N-错位卟啉环内的互变异构特性有关.
2010年, 周祥等[51]首次观察到56可以稳定G4-DNA, 并对它具有较高的选择性. CD溶解及光谱与表面等离子共振技术(SPR)实验表明, 56与G4-DNA的亲和能力比58更强, 且能诱导G4-DNA中反平行结构转化为平行与反平行的混合型结构. 58虽可稳定G4-DNA, 但不能导致特定构象的转化.与ds-DNA相比, 56与G4- DNA结合能力更强.同时, 56对平行与反平行结构的G四链体(bcl2)具有较高的选择性, 是ds-DNA的108倍. N-错位卟啉衍生物对G4-DNA具有较强的结合能力和较高的选择性, 为其在癌症的治疗及诊断领域奠定理论基础.
|
|
2015年, 田来进课题组[52, 53]采用紫外与荧光光谱, 研究了两个N-错位卟啉衍生物N-(乙酸三苯锡酯基)- 5, 10, 15, 20-四苯基N-错位卟啉和2-N-甲基-5, 10, 15, 20-四(4-氯苯基)N-错位锡卟啉与小牛胸腺DNA (ct-DNA)相互作用.实验结果表明, 310 K时两者与DNA的结合常数分别为1.92×102和1.23×106 L/mol.他们以吖啶橙(AO)为荧光探针, 考察了不同温度条件下两者对ct-DNA-AO体系的荧光光谱的影响, 发现两者均可猝灭ct-DNA-AO体系的荧光, 且猝灭机理均为静态猝灭.
临床抗癌药物如光动力治疗(PDT)光敏剂主要通过注射方式进入血液, 而转移药物的载体主要是人体血清白蛋白.药物与蛋白结合作用可影响药物在体内的吸收、分布、代谢和排泄.牛血清白蛋白(BSA)与人血清白蛋白(HSA)具有序列同源性, 相似度有76%, 两者的3D结构类似.因此, 研究药物与血清白蛋白的相互作用对药物应用于临床具有指导意义.目前, N-错位卟啉与血清白蛋白(SA)的相互作用的研究已取得了一定成果.
2011年李筱芳等[54]利用紫外光谱和荧光光谱分别测定了N-错位卟啉4、5和59与牛血清白蛋白(BSA)相互作用.实验结果表明, 在温度为293与305 K的条件下, 低浓度条件时, 荧光猝灭机理为静态猝灭; 而在温度为305 K, 且N-错位卟啉的浓度较高时, 其荧光猝灭机理为静态与动态猝灭相互结合. N-错位卟啉与BSA的结合常数约为106 L/mol, 且以疏水相互作用为主, 并发现甲基与甲氧基羰基的取代能提高N-错位卟啉与BSA的结合能力.
|
|
后来, 该课题组[9, 55~57]还设计合成了四个N-错位卟啉衍生物60~63, 并分别研究了它们与BSA的相互作用.研究表明, 60~62与BSA相互作用的荧光猝灭机理为静态与动态猝灭相互结合, 而63与BSA相互作用的荧光猝灭机理为静态猝灭机理. 60和62与BSA的结合常数均约为105 L/mol, 且主要以疏水相互作用为主, 而61和63与BSA的结合常数分别约为106与104 L/mol, 且均主要以静电相互作用为主.根据Föster’s非辐射共振能量转移理论计算得到60~63与BSA结合距离分别为3.63、2.10、3.30和4.37 nm.
|
|
2015年, 田来进课题组[52, 53]采用荧光光谱法还研究了N-(乙酸三苯锡酯基)-5, 10, 15, 20-四苯基N-错位卟啉和2-N-甲基-5, 10, 15, 20-四(4-氯苯基)N-错位锡卟啉与BSA的相互作用. 310 K时两者与BSA的猝灭常数分别为1.30×107和2.60×107 L/mol.前者与BSA相互作用的荧光猝灭机理为动态猝灭, 后者相互作用的猝灭机理为静态猝灭
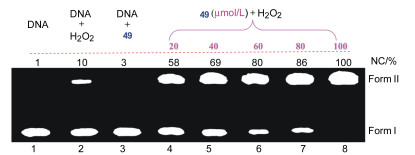
卟啉与咔咯等大环化合物的锰配合物在氧化剂的存在下均表现出较好的核酸酶活性[58~60].我们课题组[61]采用琼脂糖凝胶电泳法研究了在氧化剂双氧水的条件下N-错位卟啉锰配合物49与pBR322质粒DNA的反应. 图 2为不同浓度的49在双氧水存在下断裂pBR322 DNA的电泳图.
49在氧化剂条件下表现出很好的断裂DNA的能力.因此, 可以认为N-错位卟啉锰配合物具有较好的化学核酸酶活性.为了初步探究其氧化断裂DNA的机理, 我们考察了添加剂[如螯合剂乙二胺四乙酸(EDTA)]、羟基自由基(·OH)捕获剂[DMSO、叔丁醇(t-BuOH)]、单线态氧(1O2)捕获剂[组氨酸(L-His)、1, 4-二叠氮双环[2.2.2]辛烷(DABCO)]及超氧阴离子捕获剂[超氧化物歧化酶(SOD)]对49 (60 μmol/L)断裂DNA的影响[59, 60], 结果如图 3所示.
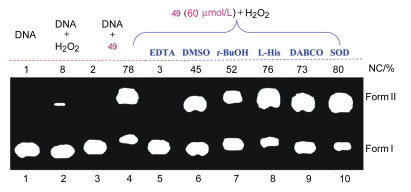
由图 3可见, 与未加添加剂(Lane 4)相比, 加入乙二胺四乙酸(EDTA) (Lane 5)明显减少了Form Ⅱ的含量, 说明EDTA能有效地抑制49的核酸酶活性, 表明49的核酸酶活性与中心金属锰离子密切相关. DMSO (Lane 6)与t-BuOH (Lane 7)同样能使Form Ⅱ的含量减少, 说明羟基自由基可能是造成DNA断裂的活性物中间体.而Lanes 8~10中Form Ⅱ的含量分别为76%、73%和80%, 与未加添加剂Lane 4 (78%)相比无差别.换句话说, L-His、DABCO及SOD对DNA的断裂影响不大.表明49氧化断裂DNA的过程中并未涉及单线态氧和超氧阴离子的产生.因此, 认为N-错位卟啉锰配合物具有氧化断裂DNA的能力, 并且断裂机理与羟基自由基有关[58].
癌症作为威胁人类健康的第一杀手, 是当今最难治愈的疾病之一.治疗癌症主要有手术、放疗、化疗和光动力学疗法(Photodynamic therapy, PDT). PDT是一种正在研究发展中的新技术, 因其独特的优点(与其它治疗手段相比, 创伤小、毒性低、选择性好、适用性好、可重复治疗、可姑息治疗、可协同手术提高疗效和治疗时间短等)在肿瘤治疗中占据了重要地位.光敏剂作为能量的载体和反应的桥梁, 在光动力治疗中有关键的作用, 是PDT的核心.卟啉在光动力治疗癌症领域的应用已取得了较好的研究成果, 例如四间羟基苯基二氢卟吩[Temoporfin, tetra(3-hydroxyphenyl)chlorin, m-THPC, 商品名Foscan]是目前临床上使用的光敏剂.主要用于治疗食管癌、肺癌、胃癌和胰腺癌, 效果令人比较满意, 对基底细胞癌也有疗效, 但有可能会有瘢痕形成[62, 63].早在2001年, Manku等[64]就合成了一个类似于Temoporfin结构的N-错位卟啉——2-N-甲基-5, 10, 15, 20-四-(间羟基)苯基N-错位卟啉64, 并首次研究了其对皮下移植结肠癌细胞株(colo 26)的雌裸鼠的光动力(PDT)治疗效果及皮肤的光毒性.肿瘤的坏死深度及小鼠耳肿胀实验发现, 64在长波处(>650 nm)显示出良好的光动力治疗活性, 而且在皮肤光毒性上明显低于Temoporfin.
|
|
2002年, Dolphin等[29]发现水溶性的季铵盐型N-错位卟啉——N, N'-二甲基四-(烷基)苯基N-错位卟啉盐在700 nm以上的可见光区可有效地产生单线态氧并预测其也可作为潜在的PDT光敏剂. 2012年, Srinivasan等[7]合成了磺酸型水溶性的N-错位卟啉——5, 10, 15, 20-四-(对磺酸基)苯基N-错位卟啉四钠盐65, 并测定了它的光动力治疗活性. 65在747 nm处有较强的吸收(摩尔消光系数为1400 L•mol-1•cm-1), 并能有效地产生单线态氧, 在水和甲醇中的单线态氧的量子产率分别为0.55和0.70.他们利用MTT法测定了65对八种不同癌细胞人体结肠癌细胞(HCT-116)、两种人体乳腺癌细胞(MCF7-ER, PR positive和MDA-MB-231-ER, PR negative)、人体胰腺癌细胞(MIA-PaCa-2)、两种人体子宫颈癌细胞(HeLa和SiHa)和两种人体口腔癌细胞(SCC-172和SCC-131)的光毒性实验.在光照条件下, 均显示出较强的细胞毒性, 对腺癌细胞的光毒性更强, 而暗条件下几乎无细胞毒性. 图 4是以活性氧族的细胞渗透性指示剂CM-H2DCFDA作为探针, 在光照与暗条件下65 (0~12 µmol/L)对MDA-MB-231细胞PDT作用的荧光成像图. CM-H2DCFDA可与单线态氧反应生成绿色荧光的氧化产物.通过荧光成像技术测定该绿色荧光氧化产物的荧光强度, 可间接测定癌细胞体内单线态氧的含量.
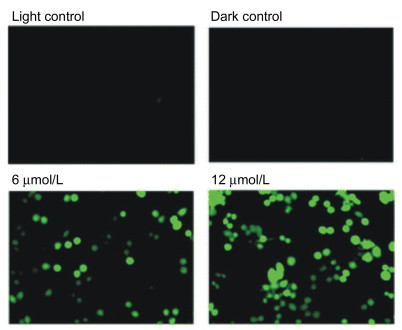
The concentrations are 0 μmol/L (light control), 12 μmol/L in the dark (dark control), 6 μmol/L and 12 μmol/L, respectively
从图 4中可以看出, 随着NCPS浓度的增加, 绿色荧光强度增强, 表明MDA-MB-231细胞中产生了较多的单线态氧.进而, 推测65通过PDT与癌细胞的作用与单线态氧的产生有关.
N-错位卟啉及其衍生物能有效地产生单线态氧, 并在活体小鼠及癌细胞中呈现出显著的PDT活性, 且无皮肤光毒性, 为其在光动力治疗领域的发展提供理论和实践基础.
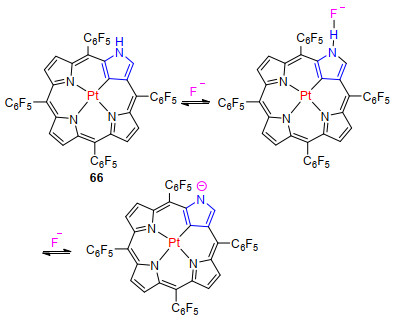
N-错位卟啉及其金属配合物存在外围NH及金属离子, 使这一类物质可作为阴离子或金属离子传感器. Furuta课题组[65]最早发现, N-错位卟啉铂配合物66 (Scheme 2)外围的NH基团作为阴离子的识别单元, 使其可作为阴离子传感器.该配合物的外围NH与Cl-,Br-和I-可通过氢键结合.这一结论可通过核磁共振中外围NH的氢谱信号随着Cl-, Br-和I-的加入向低场移动并伴随紫外吸收光谱中Soret带吸收增强Q带吸收减弱得以证实.然而F-, H2PO4-和OH-将导致外围NH去质子.核磁共振中外围NH的氢谱信号与F-浓度密切相关.当加入0.1 equiv.的F-, 外围NH的氢谱信号向低场移动.当浓度增加到0.4 equiv.时, 外围NH的氢谱信号完全消失.实验结果表明, 少量的F-可与外围NH通过氢键结合, 而大量的F-存在将导致外围NH去质子.
该课题组[66]也报道了一种N-错位卟啉与卟啉共轭连接的二元体阴离子传感器67 (Eq. 15).该二元体中N-错位卟啉作为阴离子识别单元, 卟啉作为接受体.加入F-(氟源:四叔丁基氟化铵), 观察二元体的紫外吸收光谱和荧光光谱变化.发现随着F-的加入, 二元体的紫外光谱在446和702 nm处出现等吸收点, 表明F-与二元体中的N-错位卟啉外围的NH通过氢键1:1结合.随着F-的加入, 二元体的荧光光谱中(以421 nm激发波长)位于730和803 nm处发射峰减弱, 却在703和765 nm处出现两个增强的新发射峰.二元体的荧光量子产率也从0.0053显著增加到0.0153, 表明F-与外围的NH结合加速了二元体中卟啉向N-错位卟啉的能量转移.
Furuta和解永树等[67, 68]还发现N-错位卟啉锡配合物的中心金属锡离子作为阴离子的识别单元赋予其阴离子传感器的性质.
|
|
(15) |
N-错位卟啉锡配合物68 (Eq. 16)具有弱的荧光性质.该配合物68的锡中心轴向可与阴离子(Cl-, Br-和I-)结合(69)且具有较大的结合常数(1.15×105, 2.1×104和6.1×102 L•mol-1), 并能观察到明显的荧光增强现象.基于这一性质可以制备一种化学传感器用于检测特定阴离子.
|
|
(16) |
2018年, Gomes课题组[69]发现N-错位卟啉衍生物还可作为金属离子传感器.该课题组合成了五种N-错位卟啉类材料, 分别为外围N取代丙基NCTPP-Pr+, 负载到二氧化硅NCTPP-Si, 负载到吡啶基修饰的二氧化硅NCTPP-Si-Py+, 负载到MF树脂NCTPP-MF, 负载到吡啶基修饰的MF树脂NCTPP-MF-Py+, 并分别研究了它们对重金属离子Tl(Ⅰ)和Gr(Ⅲ)的敏感性.结果表明五种材料对Gr(Ⅲ)的敏感性均高于Tl(Ⅰ), 还发现阳离子具有调控敏感性的作用.这为N-错位卟啉作为金属离子传感器提供理论和实践基础.
N-错位卟啉与卟啉类似, 均有共轭的大环结构, 具有特殊的光物理和电化学性质.然而基于N-错位卟啉的染料敏化太阳能电池的报道甚少. 2017年Yan课题组[70]报道了4-乙炔基苯甲酸取代(70)和3-C位苯甲酸取代71的N-错位卟啉钯配合物, 并分别研究了它们的光电性质.
|
|
结果表明, 引入苯甲酸等基团增强了N-错位卟啉的光捕获能力.两种类型N-错位卟啉的染料敏化太阳能电池的能量效率均小于0.1%.这可能与电解质不能再生或光电子不能顺利注入到二氧化钛的导带有关.这一推测被理论计算结果证实. N-错位卟啉70和71的最低空分子轨道(LUMO)能级高于TiO2的能级(-3.40 eV), 表明光电子能顺利地注入到TiO2的导带, 但70和71的最高占据分子轨道(HOMO)能级(-4.67和-4.61 eV)与电解质(I-/I3-)的氧化电势(-4.85 eV)不匹配, 换言之两类N-错位卟啉的激发态不能被电解质(I-/I3-)电解还原回基态.该课题组提出对N-错位卟啉外围基团的修饰或中心金属的改变以匹配电解质的电势, 是今后N-错位卟啉染料敏化太阳能电池的研究方向.虽然该报道的N-错位卟啉染料敏化太阳能电池的能量效率较低, 但对N-错位卟啉在染料敏化太阳能电池领域的发展具有重要的指导意义.
扩展卟啉是一类含有4个以上吡咯单元的卟啉[15].与卟啉相比, 扩展卟啉具有更大的π共轭电子结构, 具有独特的光物理性质、特殊的金属配位能力、离子识别和多种构型异构等特性[16~18]. N-错位扩展卟啉是扩展卟啉中存在一个或多个N-错位吡咯环.迄今为止, 已有文献[71~76]报道了5~8个吡咯环的N-错位扩展卟啉.
2003年, Furuta课题组[71, 72]在试图合成N-错位五啉的过程中合成了一个N-错位的双重N-骈环五啉72, 两个双重N-错位五啉衍生物双重N-错位五啉酮73和双重N-错位五啉二酮74. 73通过分子间氢键相互作用以二聚体形式存在, 溶液中不稳定, 在常温条件下氧化为74. 73的紫外光谱显示一个位于544 nm处的Soret带. 74以分子内氢键相互作用以单体形式存在, 其紫外光谱显示其有两个Soret带分别位于542和585 nm.两者均有22个π电子, 符合Huckel规则, 具有显著的芳香性.
|
|
随着环的进一步扩大, N-错位六啉[73~75]呈现更有趣的现象. (1) N-错位六啉存在多种构型.如含一个N-错位吡咯环的六啉(75)、双重N-错位六啉(76)和三重N-错位六啉(77). (2) N-错位六啉可能存在26或28个π电子. N-错位[26]六啉(75)符合Huckel规则, 具有芳香性, 紫外光谱中显示出明显的Soret带和Q带, 而N-错位[28]六啉不符Huckel规则, 无芳香性.例如双重N-错位[26]六啉的紫外光谱呈现出一个位于591 nm处的Soret带和四个Q带位于650~1100 nm, 而双重N-错位[28]六啉的紫外光谱在380~590 nm呈现出三个宽峰. (3) N-错位六啉存在多种对称性.例如双重N-错位六啉为C2对称结构, 空间构型为长方形, 三重N-错位六啉为C3对称结构空间构型为三角形. (4) N-错位六啉作为双核配体可与不同价态的混合金属配位.如N-错位[26]六啉酮可与金(Au3+)和铂(Pt2+)进行配位合成N-错位[26]六啉酮双金属(Au3+, Pt2+)配合物.
|
|
Furuta课题组[76]最近报道了一种目前含吡咯环最多的N-错位扩展卟啉双重N-错位[36]八啉78 (Eq. 17).它具有两种稳定的空间构型, 分别为八字型构型78和哑铃型构型79.两者可通过质子耦合相互转化.它可作为双核配体与铜离子(Cu2+)进行配位形成手性的八字型构型双重N-错位[36]八啉双铜(Ⅱ)配合物.该铜配合物在近红外区呈现出明显的圆二色光谱, 使得该化合物可作为近红外光学手性传感器在分子识别方面有着潜在的应用前景.
|
|
(17) |
近几年来, 关于其它错位异卟啉[77~83]的研究已取得了很大的进展, 如N-错位咔咯(80)[18, 77]、N-错位噻啉(81)[78]、N-错位卟啰啉(82)[79, 80]、双重N-错位卟啉(83, 84)[83]等及其相应的扩展卟啉.它们呈现出独特的光物理性质, 与金属配位形成金属碳键等使得他们在生物医学、光学材料、配位化学、催化化学、阴离子识别等领域具有重要的应用前景.随着越来越多错位异卟啉的不断发现, 越来越多有趣的性质将会被呈现出来.
N-错位卟啉的研究已成为国际上卟啉化学的一个重要领域, 是具有挑战性和创新性的课题. N-错位卟啉的研究目前存在以下问题. (1)采用一锅法合成部分取代的N-错位卟啉产率较低, 甚至难以合成得到.例如:羧基、磺酸基和氨基等取代N-错位卟啉难以用相应的醛与吡咯一锅法制备.一锅法、[2+2]和[3+1]等方法合成N-错位卟啉尤其是不对称N-错位卟啉时, 副产物较多, 且复杂分离较困难; (2) N-错位卟啉在催化化学、生物化学和材料化学等领域的应用研究还有待进一步的拓宽.例如, 目前关于催化领域的报道仅限于催化环丙烷化、氧化和环加成. (3) N-错位扩展卟啉及其它异卟啉的结构和性质及其在各领域的应用少见报道.虽然N-错位卟啉相关研究仍处于初级阶段, 但为今后N-错位卟啉化学的发展提供坚实的理论和实践基础.目前N-错位卟啉研究的主要方向有: (1)优化反应条件, 寻找合适的催化剂和实验方法提高目标化合物的产率和纯度; 深入探讨分子结构与性能之间的关系, 通过分子设计合成不同取代基、不同中心离子、不同对称性的具有功能化的N-错位卟啉衍生物.例如, 将具有抗肿瘤靶向作用的基团引入到N-错位卟啉的结构合成功能化的N-错位卟啉衍生物. (2)拓展各应用领域的研究.如探究新型N-错位卟啉催化剂, 如负载型N-错位卟啉催化剂等; 拓宽催化其它有机反应, 如催化二氧化碳还原, 水光解或电解制氢等绿色反应; 深入生物医学领域的研究, 探索N-错位卟啉的结构与生物活性及产生机理之间的关系; 作为抗肿瘤药物应用于临床研究; 创新多功能材料等领域, 例如传感器、太阳能电池和分子器件等. (3) N-错位扩展卟啉可同时与多个金属离子进行配位, 有望在电子转移等相关领域有潜在的应用前景.关于N-错位扩展卟啉及其它错位异卟啉的结构与性质等有趣的性质有待发现, 例如:探索包括含更多个吡咯环的错位扩展卟啉, 研究其光学和配位性质等.随着科研工作者的不断探索, 越来越多的N-错位卟啉将进一步被合成, 也为其在各领域的应用研究提供理论和实践基础.
Senge, M. O. Angew. Chem., Int. Ed. 2011, 50, 4272. doi: 10.1002/anie.201003660
Chmielewski, P. J.; Latos-Grażyński, L.; Rachlewicz, K.; Glowiak, T. Angew.Chem., Int. Ed. 1994, 33, 779. doi: 10.1002/anie.199407791
Furuta, H.; Asano, T.; Ogawa, T. J. Am. Chem. Soc. 1994, 116, 767. doi: 10.1021/ja00081a047
李筱芳, 刘浩冲, 郑爱庭, 于贤勇, 易平贵, 有机化学, 2011, 31, 166. http://journal15.magtechjournal.com/Jwk3_hxs/yjhx/CN/Y2011/V31/I02/166Li, X. F.; Liu, H. C.; Zhang, A. T.; Yu, X. Y.; Yi, P. G. Chin. J. Org. Chem. 2011, 31, 166 (in Chinese). http://journal15.magtechjournal.com/Jwk3_hxs/yjhx/CN/Y2011/V31/I02/166
Harvey, J. D.; Ziegler, C. J. J. Inorg. Biochem. 2006, 100, 869. doi: 10.1016/j.jinorgbio.2006.01.016
Niino, T.; Toganoh, M.; Andrioletti, B.; Furuta, H. Chem. Commun. 2006, 4335. http://www.ncbi.nlm.nih.gov/pubmed/17047859/
Thomas, A. P.; Saneesh Babu, P. S.; Asha Nair, S.; Ramakrishnan, S.; Ramaiah, D.; Chandrashekar, T. K.; Srinivasan, A.; Radha- krishna-Pillai, M. J. Med. Chem. 2012, 55, 5110. doi: 10.1021/jm300009q
Yamamoto, T.; Toganoh, M.; Furuta, H. Dalton Trans. 2012, 41, 9154. doi: 10.1039/c2dt30885a
Yu, X.; Lu, S.; Yang, Y.; Li, X.; Yi, P. Spectrochim. Acta, Part A 2011, 83, 609. doi: 10.1016/j.saa.2011.09.014
Collman, J. P.; Boulatov, R.; Sunderland, C. J.; Fu, L. Chem. Rev. 2004, 104, 561. doi: 10.1021/cr0206059
Larsen, R. W.; Omdal, D. H.; Jasuja, R.; Niu, S. L.; Jameson, D. M. J. Phys. Chem. B 1997, 101, 8012. doi: 10.1021/jp9640235
Kang, S. A.; Marjavaara, P. J.; Crane, B. R. J. Am. Chem. Soc. 2004, 126, 10836. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM15339156
Yoshioka, S.; Tosha, T.; Takahashi, S.; Ishimori, K.; Hori, H.; Morishima, I. J. Am. Chem. Soc. 2002, 124, 14571. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM12465966
Yang, R. H.; Wang, K. M.; Long, L. P.; Xiao, D.; Yang, X. H.; Tang, W. H. Anal. Chem. 2002, 74, 1088. doi: 10.1021/ac010386b
王全国, 曾凡花, 解永树, 朱为宏, 化学进展, 2009, 21, 1523. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ2009Z2019.htmWang, Q. G.; Zeng, F. H.; Xie, Y. S.; Zhu, W. H. Prog. Chem. 2009, 21, 1523 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HXJZ2009Z2019.htm
Li, Q. Z.; Li, C. J.; Kim, J.; Ishida, M.; Li, X.; Gu, T. T.; Liang, X.; Zhu, W. H.; Ågren, H.; Kim, D. H.; Furuta, H.; Xie, Y. S. J. Am. Chem. Soc. 2019, 141, 5294. doi: 10.1021/jacs.8b13148
Li, Q. Z.; Ishida, M.; Kai, H.; Gu, T. T.; Li, C. J.; Li, X.; Baryshnikov, G.; Liang, X.; Zhu, W. H.; Ågren, H.; Furuta, H.; Xie, Y. S. Angew. Chem., Int. Ed. 2019, 58, 5925. doi: 10.1002/anie.201900010
Maurya, Y. K.; Noda, K.; Yamasumi, K.; Mori, S.; Uchiyama, T.; Kamitani, K.; Hirai, T.; Ninomiya, K.; Nishibori, M.; Hori, Y.; Shiota, Y.; Yoshizawa, K.; Ishida, M.; Furuta, H. J. Am. Chem. Soc. 2018, 140, 6883. doi: 10.1021/jacs.8b01876
Li, C. J.; Zhang, J. L.; Song, J. X.; Xie, Y. S.; Jiang, J. Z. Sci. China, Chem. 2018, 61, 511.
Ding, Y. B.; Tang, Y. Y.; Zhu, W. L.; Xie, Y. S. Chem. Soc. Rev. 2015, 44, 1101. doi: 10.1039/C4CS00436A
Liu, H.-Y.; Mahmood, M. H.; Qiu, S.-X.; Chang, C. K. Coord. Chem. Rev. 2013, 257, 1306. doi: 10.1016/j.ccr.2012.12.017
Geier, G. R.; Haynes, D. M.; Lindsey, J. S. Org. Lett. 1999, 1, 1455. doi: 10.1021/ol9910114
Geier, G. R.; Ciringh, Y.; Li, F.; Haynes, D. M.; Lindsey, J. S. Org. Lett. 2000, 2, 1745. doi: 10.1021/ol005917h
Maeda, H.; Osuka, A.; Ishikawa, Y.; Aritome, I.; Hisaeda, Y.; Furuta, H. Org. Lett. 2003, 5, 1293. doi: 10.1021/ol034227l
Fisher, J. M.; Kensy, V. K.; Geier, G. R. J. Org. Chem. 2017, 82, 4429. doi: 10.1021/acs.joc.7b00195
Kitaoka, S.; Nobuoka, K.; Ishikawa, Y. Tetrahedron 2005, 61, 7678. doi: 10.1016/j.tet.2005.05.097
dela Rosa, M. A. C.; Arco, S. D.; Hung, C.-H. J. Chin. Chem. Soc. (Weinheim, Ger.) 2012, 59, 633. doi: 10.1002/jccs.201100498
Shaw, J. L.; Garrison, S. A.; Alemán, E. A.; Ziegler, C. J.; Modarelli, D. A. J. Org. Chem. 2004, 69, 7423. doi: 10.1021/jo049199e
Xiao, Z.; Dolphin, D. Tetrahedron 2002, 58, 9111. doi: 10.1016/S0040-4020(02)01097-9
Zhu, X.-J.; Jiang, F.-L.; Poon, C.-T.; Wong, W.-K.; Wong, W.-Y. Eur. J. Inorg. Chem. 2008, 2008, 3151. doi: 10.1002/ejic.200800267
Schmidt, I. J.; Chmielewski, P. Tetrahedron Lett. 2001, 42, 6389. doi: 10.1016/S0040-4039(01)01259-X
Wolff, S. A.; Alemán, E. A.; Banerjee, D.; Rinaldi, P. L.; Modarelli, D. A. J. Org. Chem. 2004, 69, 4571. doi: 10.1021/jo049621r
Liu, B. Y.; Bruckner, C.; Dolphin, D. Chem. Commun. 1996, 2141. doi: 10.1002/chin.199708117/full
Narayanan, N. S.; Sridevi, B.; Srinivasan, A.; Chandrashekar, T. K.; Roy, R. Tetrahedron Lett. 1998, 39, 7389. doi: 10.1016/S0040-4039(98)01603-7
Acharya, R.; Paudel, L.; Joseph, J.; McCarthy, C. E.; Dudipala, V. R.; Modarelli, J. M.; Modarelli, D. A. J. Org. Chem. 2012, 77, 6043. doi: 10.1021/jo300810n
Lash, T. D.; Richter, D. T.; Shiner, C. M. J. Org. Chem. 1999, 64, 7973. doi: 10.1021/jo991019r
Furuta, H.; Morimoto, T.; Osuka, A. Org. Lett. 2003, 5, 1427. doi: 10.1021/ol034172n
Morimoto, T.; Taniguchi, S.; Osuka, A.; Furuta, H. Eur. J. Org. Chem. 2005, 2005, 3887. doi: 10.1002/ejoc.200500485
Toganoh, M.; Harada, N.; Morimoto, T.; Furuta, H. Chemistry 2007, 13, 2257. doi: 10.1002/chem.200600776
Furuta, H.; Nanami, H.; Morimoto, T.; Ogawa, T.; Kral, V.; Sessler, J. L.; Lynch, V. Chem.-Asian J. 2008, 3, 592. doi: 10.1002/asia.200700130
Toganoh, M.; Furuta, H. Chem. Commun. 2012, 48, 937. doi: 10.1039/C1CC14633E
Arasasingham, R. D.; He, G. X.; Bruice, T. C. J. Am. Chem. Soc. 1993, 115, 7985. doi: 10.1021/ja00071a008
Brule, E.; de Miguel, Y. R. Tetrahedron Lett. 2002, 43, 8555. doi: 10.1016/S0040-4039(02)02063-4
Fields, K. B.; Engle, J. T.; Sripothongnak, S.; Kim, C.; Zhang, X. P.; Ziegler, C. J. Chem. Commun. 2011, 47, 749. doi: 10.1039/C0CC03894F
Miyazaki, T.; Yamamoto, T.; Mashita, S.; Deguchi, Y.; Fukuyama, K.; Ishida, M.; Mori, S.; Furuta, H. Eur. J. Inorg. Chem. 2018, 203. doi: 10.1002/ejic.201701494/pdf
Peng, S. H.; Mahmood, M. H. R.; Zou, H. B.; Yang, S. B.; Liu, H. Y. J. Mol. Catal. A: Chem. 2014, 395, 180. doi: 10.1016/j.molcata.2014.08.016
Miyazaki, T.; Fukuyama, K.; Mashita, S.; Deguchi, Y.; Yamamoto, T.; Ishida, M.; Mori, S.; Furuta, H. ChemPlusChem 2019, 84, 603. doi: 10.1002/cplu.201800630
dela Cruz, J.; Ruamps, S.; Arco, S.; Hung, C. H. Dalton Trans. 2019, 48, 7527. doi: 10.1039/C9DT00104B
Moriyama, S.; Ikawa, Y.; Furuta, H. Nucleic Acids Symp. Ser. 2007, 207.
Ikawa, Y.; Moriyama, S.; Harada, H.; Furuta, H. Org. Biomol. Chem. 2008, 6, 4157. doi: 10.1039/b810171j
Du, Y. H.; Zhang, D.; Chen, W.; Zhang, M.; Zhou. Y. Y.; Zhou, X. Bioorg. Med. Chem. 2010, 18, 1111. doi: 10.1016/j.bmc.2009.12.049
王晓龙, 硕士论文, 曲阜师范大学, 曲阜, 2015. http://cdmd.cnki.com.cn/Article/CDMD-10446-1015408239.htmWang, X. L. M.S. Thesis, Qufu Normal University, Qufu, 2015 (in Chinese). http://cdmd.cnki.com.cn/Article/CDMD-10446-1015408239.htm
董元赛, 硕士论文, 曲阜师范大学, 曲阜, 2015.Dong, Y. S. M.S. Thesis, Qufu Normal University, Qufu, 2015 (in Chinese).
Yu, X. Y.; Liu, R. H.; Yi, R. Q.; Yang, F. X.; Huang, H. W.; Chen, J.; Ji, D. H.; Yang, Y.; Li, X. F.; Yi, P. G. Spectrochim. Acta, Part A 2011, 78, 1329. doi: 10.1016/j.saa.2011.01.024
Yu, X. Y.; Lu, S. Y.; Yang, Y.; Li, X. F.; Yi, P. G. Spectrochim. Acta, Part A 2012, 91, 113. doi: 10.1016/j.saa.2012.01.068
Lu, S. Y.; Yu, X. Y.; Yang, Y.; Li, X. F. Spectrochim. Acta, Part A 2012, 99, 116. doi: 10.1016/j.saa.2012.09.012
Yu, X. Y.; Liao, Z. X.; Jiang, B. F.; Zheng, L. Y.; Li, X. F. Spectrochim. Acta, Part A 2014, 133, 372. doi: 10.1016/j.saa.2014.05.085
Lu, J.; Liu, H. Y.; Shi, L.; Wang, X. L.; Ying, X.; Zhang, L.; Ji, L. N.; Zang, L. Q.; Chang, C. K. Chin. Chem. Lett. 2011, 22, 101. doi: 10.1016/j.cclet.2010.09.005
Huang, J. T.; Wang, X. L.; Zhang, Y.; Mahmood, M. H. R.; Huang, Y. Y.; Ying, X.; Ji, L.N.; Liu, H. Y. Transition. Met. Chem. 2013, 38, 283. doi: 10.1007/s11243-013-9689-5
Zhang, Y.; Wang, Q.; Wen, J. Y.; Wang, X. L.; Mahmood, M. H. R.; Ji, L. N.; Liu, H. Y. Chin. J. Chem. 2013, 31, 1321. doi: 10.1002/cjoc.201300488
Peng, S. H.; Lü, B. B.; Ali., A.; Wang, J. M.; Ying, X.; Wang, H.; Liu, J. B.; Ji, L. N.; Liu, H. Y. J. Porphyrins. Phthalocyanines 2016, 20, 624. doi: 10.1142/S1088424616500449
Savary, J. F.; Monnier, P.; Fontolliet, C.; Mizeret, J.; Wagnieres, G.; Braichotte, D.; vandenBergh, H. Arch. Otolaryngol. 1997, 123, 162. doi: 10.1001/archotol.1997.01900020042006
Baas, P.; Saarnak, A. E.; Oppelaar, H.; Neering, H.; Stewart, F. A. Br. J. Dermatol. 2001, 145, 75. doi: 10.1046/j.1365-2133.2001.04284.x
Manku, M.; Rice, D.; Milgrom, L. WO 2001/53300, 2001.
Won, D. H.; Toganoh, M.; Uno, H.; Furuta, H. Dalton Trans. 2009, 6151.
Toganoh, M.; Miyachi, H.; Akimaru, H.; Ito, F.; Nagamura, T.; Furuta, H. Org. Biomol. Chem. 2009, 7, 3027. doi: 10.1039/b907775h
Xie, Y. S.; Morimoto, T.; Furuta, H. Angew. Chem., Int. Ed. 2006, 45, 6907. doi: 10.1002/anie.200602481
Ding, Y. B.; Zhu, W. H.; Xie, Y. S. Chem. Rev. 2017, 117, 2203. doi: 10.1021/acs.chemrev.6b00021
Gamelas, S. R. D.; Gomes, A. T. P. C.; Moura, N. M. M.; Faustino, M. A. F.; Cavaleiro, J. A. S.; Lodeiro, C.; Verίssimo, M. I. S.; Fernandes, T.; Daniel-da-Silva, A. L.; Gomes, M. T. S. R.; Neves, M. G. P. M. S. Molecules 2018, 23, 867. doi: 10.3390/molecules23040867
Zhang, N. N.; Chen, J. Q.; Cheng, K.; Li, Y. J.; Wang, L.; Zheng, K. B.; Yang, Q. Q.; Li, D. J.; Yan, J. Y. Res. Chem. Intermed. 2017, 43, 2921. doi: 10.1007/s11164-016-2803-5
Srinivasan, A.; Ishizuka, T.; Furuta, H. Angew. Chem., Int. Ed. 2004, 43, 876. doi: 10.1002/anie.200352946
Srinivasan, A.; Ishizuka, T.; Maeda, H.; Furuta, H. Angew. Chem., Int. Ed. 2004, 43, 2951. doi: 10.1002/anie.200453732
Gokulnath, S.; Yamaguchi, K.; Toganoh, M.; Mori, S.; Uno, H.; Furuta, H. Angew. Chem., Int. Ed. 2011, 50, 2302. doi: 10.1002/anie.201006784
Srinivasan, A.; Ishizuka, T.; Osuka, A.; Furuta, H. J. Am. Chem. Soc. 2003, 125, 878. doi: 10.1021/ja029018v
Xie, Y. S.; Yamaguchi, K.; Toganoh, M.; Uno, H.; Suzuki, M.; Mori, S.; Saito, S.; Osuka, A.; Furuta, H. Angew. Chem., Int. Ed. 2009, 48, 5496. doi: 10.1002/anie.200900596
Mitsuno, K.; Yoshino, T.; Gupta, I.; Mori, S.; Karasawa, S.; Ishida, M.; Furuta, H. Angew. Chem., Int. Ed. 2017, 56, 14252. doi: 10.1002/anie.201708253
Li, M.; Wei, P. C.; Ishida, M.; Li, X.; Savage, M.; Guo, R.; Ou, Z. P.; Yang, S. H.; Furuta, H.; Xie, Y. S. Angew. Chem., Int. Ed. 2016, 55, 3063. doi: 10.1002/anie.201510879
Xie, Y. S.; Wei, P. C.; Li, X.; Hong, T.; Zhang, K.; Furuta, H. J. Am. Chem. Soc. 2013, 135, 19119. doi: 10.1021/ja4112644
Shao, J. W.; Li, C. J.; Kong, J. H.; Jiang, H. R.; Zhao, S. L.; Li, M. Z.; Liang, X.; Zhu, W. L.; Xie, Y. S. Org. Lett. 2018, 20, 1941. doi: 10.1021/acs.orglett.8b00478
Kong, J. H.; Shao, J. W.; Li, C. J.; Qi, D. D.; Li, M. Z.; Liang, X.; Zhu, W. H.; Jiang, J. Z.; Xie, Y. S. Org. Lett. 2017, 19, 650. doi: 10.1021/acs.orglett.6b03816
Zhang, K.; Zhang, J. D.; Li, X.; Guo, R.; Ågren, H.; Ou, Z. P.; Ishida, M.; Furuta, H.; Xie, Y. S. Org. Lett. 2015, 17, 4806. doi: 10.1021/acs.orglett.5b02363
Wei, P. C.; Zhang, K.; Li, X.; Meng, D. Y.; Ågren, H.; Ou, Z. P.; Ng, S.; Furuta, H.; Xie, Y. S. Angew. Chem., Int. Ed. 2014, 53, 14069. doi: 10.1002/anie.201408307
Srinivasan, A.; Furuta, H. Acc. Chem. Res. 2005, 38, 10. doi: 10.1021/ar0302686

图 1 在MnTPP (a)和49 (b)的乙腈溶液中加入PhIO时紫外可见光谱变化图
Figure 1 UV-vis spectra obtained by adding PhIO into the acetonitrile solution of MnTPP (a) and 49 (b)

图 2 不同浓度的49在双氧水存在下断裂pBR322 DNA的电泳图
Figure 2 Agarose gel electrophoresis pattern for the oxidative cleavage of supercoiled pBR322 DNA by various concentrations of manganese N-confused porphyrin 49 in the presence of hydrogen peroxide

图 3 双氧水存在下不同捕获剂对49断裂pBR322 DNA影响的电泳图
Figure 3 Agarose gel electrophoresis pattern for the oxidative cleavage of supercoiled pBR322 DNA by manganese N-confused porphyrin 49 in the presence of hydrogen peroxide and potential inhibitor agents

图 4 以CM-H2DCFDA为探针65对MDA-MB-231细胞PDT作用的荧光成像图
Figure 4 Fluorescence images of MDA-MB-231 cells labeled with the CM-H2DCFDA probe after PDT with 65
The concentrations are 0 μmol/L (light control), 12 μmol/L in the dark (dark control), 6 μmol/L and 12 μmol/L, respectively

图式 2 N-错位卟啉铂配合物66与氟离子结合及去质子化过程
Scheme 2 F- binding and deprotonation process of platinum N-confused porphyrin 66
表 1 一锅法合成N-错位卟啉的各种工艺
Table 1. Various synthetic process of N-confused porphyrin by one pot methodology
| 时间/年 | 课题组 | 催化剂 | 产物 | 产率 |
| 1994 | Furuta | 溴化氢 | 4 | 5%~7% |
| 1994 | Latos-Gra|yński | 三氟化硼乙醚溶液 | 5 | 4% |
| 1999 | Lindsey | 甲基磺酸 | 4 | 39% |
| 2003 | Furuta | 甲基磺酸 | 6 | <0.1% |
| 2017 | Geier | 甲基磺酸 | 6 | 6% |
| 对甲苯磺酸 | 6 | 5% | ||
| 三氟甲基磺酸 | 6 | 10%~12% | ||
| 三氟乙酸 | 6 | 4% | ||
| 三氟化硼乙醚溶液 | 6 | 1% |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们