图式 1.
目标化合物的合成路线
Scheme 1.
Synthesis route of target compounds

脱氢枞胺是天然产物松香的重要改性产品之一[1], 是含有三个手性碳原子的菲结构[2], 其衍生物具有抗菌、抗炎、抗病毒和抗肿瘤等生物活性[3~6].对脱氢枞胺进行改性并拓展其应用领域, 一直是林产化学与工业的前沿研究课题.含N杂环化合物在药物的研究领域举足轻重[7], 例如三嗪类化合物具有抗结核、抗病毒、抗菌和抗炎等活性[8]; 吲哚作为自然界中广泛分布的杂环之一, 具有广泛的药理功能[9]. 2-氨基噻唑及其各种衍生物经常被用来合成各种生物活性分子的前体[10].因此, 根据活性叠加原理, 将脱氢枞胺和上述含氮杂环进行拼接, 可能会获得生物活性良好的化合物.
DNA是抗肿瘤药物的重要作用靶点之一.抗肿瘤药物通过与肿瘤细胞的DNA作用, 抑制DNA复制, 引起DNA损伤, 进而阻断细胞分裂并最终导致细胞死亡.以顺铂为代表的靶向DNA的铂类金属基抗肿瘤药物在临床上获得了极大的成功[11].相比于金属配合物与DNA作用的机理研究取得的巨大进步, 作为抗肿瘤药物重要组成部分的有机小分子与DNA相互作用的研究还没有得到足够的重视, 需要大力地研究以明确其抗肿瘤机理.
据统计, 1940~2014间批准上市的抗肿瘤药物, 质量分数约为37%的抗肿瘤药物来自于天然产物及其衍生物[12].以文卡生物碱、紫杉碱、鬼臼毒素和喜树碱为代表的抗肿瘤药物, 使得植物成为抗肿瘤制剂的重要来源.
综上所述, 本文对脱氢枞胺的环B与C进行了改性, 设计、合成了如Scheme 1所示的3个光学纯含氮脱氢枞基杂环化合物, 乙酰脱氢枞胺-6, 7-(3-巯基)-1, 2, 4-三嗪(5)、乙酰脱氢枞胺-6, 7-吲哚(7)和12-(2-氨基噻唑)-乙酰脱氢枞胺(10).并采用荧光光谱、圆二色光谱及DNA凝胶电泳实验, 探讨了标题脱氢枞胺杂环衍生物5、7、10与DNA作用的模式.通过噻唑蓝(MTT)法测试了它们及其协同氯化铜对MCF-7肿瘤细胞的增殖抑制作用.本研究将为新型脱氢枞胺衍生物的设计、合成及生物活性研究提供理论参考和实验数据.
以提纯后的光学纯脱氢枞胺(1)为母体, 经过乙酰化、氧化、溴代得到乙酰脱氢枞胺-6-溴-7-酮(4), 然后与硫代氨基脲在N, N-二甲基甲酰胺(DMF)中反应得到目标产物乙酰脱氢枞胺-6, 7-(3-巯基)-1, 2, 4-三嗪(5).中间体乙酰脱氢枞胺-7-酮(3)与苯肼盐酸盐在乙醇溶液中经催化、闭环可得到乙酰脱氢枞胺-6, 7-吲哚(7).乙酰脱氢枞胺(2)经催化、溴代、闭环可得到12-(2-氨基噻唑)-乙酰脱氢枞胺(10).最后经柱层析提纯, 得到的产物产率适中.化合物5, 7和10均保留了1的手性中心.
溴化乙锭(EB)是具有共轭芳香环的平面荧光分子, 在溶液中会发生荧光猝灭. DNA本身没有荧光, 当溶液中形成DNA-EB复合物时, EB由于插入了DNA碱基对之间, 受到疏水性保护从而使其荧光增强.因此, EB常作为研究小分子与DNA相互作用模式的荧光探针.在EB-DNA-小分子化合物体系中, 如果小分子也能与DNA发生类似于EB的插入作用, EB与DNA的结合位点会被小分子占据, 就有EB分子游离出来, 体系的荧光强度减弱.化合物5、6、7、10对鲑鱼精DNA-EB体系荧光光谱的影响如图 1所示. DNA-EB体系的荧光强度随着化合物浓度的增加而减弱, 表明化合物与DNA发生了作用[13].根据Stern-Volmer方程F0/F=1+KSVCQ, 可以计算出化合物对DNA-EB体系的荧光猝灭常数KSV; 以F0/F对CQ作图, 其线性拟合斜率即为KSV.化合物5、6、7、10的KSV值分别为1.32×105、9.68×104、1.08×105、1.25×105 mol•L-1, 这些数值提示作用模式可能是插入作用[14], 且作用强度依次是5>10>7>6.

cDNA=1.0×10-4 mol•L-1; cQ=0, 6, 12, 18, 24, 30, 36, 42, 48, 54, 60 μmol•L-1
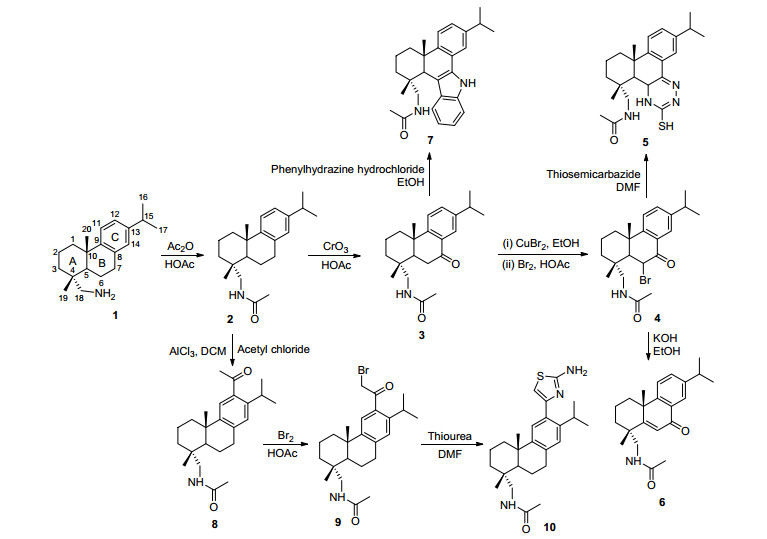
DNA是手性生物大分子, 其CD光谱在278 nm左右的特征正峰是由于DNA碱基对的π-π堆积作用产生的, 在249 nm左右的特征负峰是由DNA的右手螺旋结构引起的[15]. CD光谱是探测DNA构象变化的有效手段, 小分子化合物与DNA发生作用后, 会影响DNA的构象, 从而使得DNA的CD光谱发生变化.一般来说, 静电和沟槽作用对DNA正负吸收峰影响较小或没有影响, 而插入作用对正负吸收峰影响较大[16], CD光谱的变化也可以作为判断化合物与DNA结合模式的依据之一.在cDNA=0.1 mmol•L-1, 结合比率r=c化合物/cDNA=0.12的条件下, 化合物5、6、7、10对鲑鱼精DNA溶液CD光谱的影响如图 2所示.化合物5、6、7、10均能使DNA溶液在278 nm处的正峰强度降低, 且降低幅度5>10>7>6, 其中5和10分别使DNA的正峰发生了2和1 nm的红移. 7、10使负峰的强度降低且变化幅度10>7; 5、6使负峰的强度增强且变化幅度5>6.并且5和7分别使负峰发生了1 nm的蓝移和红移.这些数据表明5、6、7、10都能够使DNA的构型发生一定的变化[16].
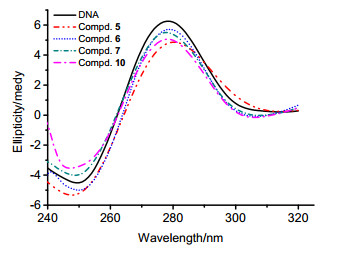
pBR 322质粒DNA通常有三种构型:闭环超螺旋型(Form I), 单链开环缺刻型(Form II), 双链断裂线型(Form III).不同构型的DNA分子量与空间结构紧密程度不同, 导致它们在凝胶电泳中的迁移速率Form I>Form III>Form II[17]. 图 3为不同浓度的化合物5、6、7、10切割pBR 322 DNA的凝胶电泳图.
Lane 1: DNA; Lane 2: DNA+50 µmol•L-1 compounds 5, 6, 7 and 10; Lane 3: DNA+100 µmol•L-1 compounds 5, 6, 7 and 10; Lane 4: DNA+150 µmol• L-1 compounds 5, 6, 7 and 10
从图 3可以看出, 化合物5、7、10都能对DNA进行切割, 特别是化合物5在高浓度时对DNA的切割活性显著增强, 缺刻型的Form II DNA浓度不断增加, 超螺旋型的Form I DNA持续减少[18].对于化合物6而言, 即使增加浓度, 也不会产生任何Form II或Form III DNA.因此, 化合物6不具备切割DNA的活性.凝胶电泳实验结果表明, 化合物5对pBR 322 DNA的切割活性最高, 化合物7与10的切割活性相似, 而化合物6则没有切割活性.这一结果和荧光光谱的结果一致.另外, 芳香杂环的结构及存在与否, 对脱氢枞基衍生物切割pBR 322质粒DNA的活性影响很大.
采用噻唑蓝(MTT)法[19]测定了化合物5, 7, 10及其与CuCl2按1:1的物质的量比进行研磨得到的复配物对人乳腺癌细胞MCF-7增殖的抑制作用(24 h), 以临床用药顺铂(IC50=6.3 µmol•L-1)为阳性对照.结果表明, 化合物5、7和10没有细胞毒活性; 在浓度为32 µmol• L-1时, 化合物1和CuCl2对MCF-7细胞增殖的抑制率分别为86.0%和5.9%, 化合物5和7与铜形成的复配物对MCF-7细胞的抑制率分别为86.7%和34.4%, 而化合物10的铜复配物则没有对MCF-7细胞的增殖产生抑制作用.另外, 化合物1和5的铜复配物对MCF-7细胞的半致死量IC50值分别为3.7和13.8 µmol•L-1.与氯化铜进行复配, 显著地提高了化合物1、5、7的细胞毒活性, 说明金属铜离子能够加强此类化合物杀伤肿瘤细胞的能力, 二者发生协同作用的原因, 很有可能是它们发生了配位作用, 这非常值得进行后续的深入研究.
荧光光谱和圆二色光谱研究表明, 依据活性叠加原理设计、制备的3个新型的脱氢枞胺芳香杂环衍生物5、7和10与鲑鱼精DNA发生了作用, 且作用能力5>10>7.凝胶电泳实验证明化合物5、7、10能够单链切割DNA, 切割能力与荧光光谱研究结果相吻合. MTT实验结果表明, 与氯化铜的协同作用大大增强了化合物5和7对MCF-7细胞的增殖抑制作用; 化合物5的铜复配物对MCF-7细胞的细胞毒活性远高于7的铜复配物.本研究表明在脱氢枞胺B环和C环上引入芳杂环, 可以获得具有良好生物活性的脱氢枞胺衍生物, 并且化合物的生物活性与改性后的结构关系密切.另外, 与铜离子复配能够显著改善化合物的细胞毒活性.
Avance DRX-600核磁共振仪; Ls-55荧光光谱仪; 6310 Ionn Trap LC/MS; Jasco J-810型圆二色谱仪; Elementar Vario Micro型元素分析仪; DYY2-C-核酸电泳仪; PCR梯度仪; Bio-rad Universal Hood II S.N.76S凝胶成像仪; 恒温培养振荡器; Versa max型酶标仪; Thermo-3111二氧化碳培养箱; AllegraX-15R离心机; Olympus倒置荧光显微镜; Esco洁净工作台.
歧化松香胺(广西桂林松泉林化工业有限公司, 含脱氢枞胺70%), 经纯化后使用[20], [α]D20+45 (c 0.10, CHCl3); 乙酸酐(上海阿拉丁试剂有限公司, 分析纯); 氨基硫脲, 硫脲, 苯肼盐酸盐(国药集团化学试剂有限公司); pBR 322质粒DNA、鲑鱼精DNA(南京都莱生物科技有限公司), 其余试剂均为市售分析纯.
在100 mL反应瓶中, 将1 (14.50 g, 50 mmol)溶解在乙酸(30 mL)中, 将乙酸酐(28 mL, 296 mmol)加入, 在室温下搅拌3 h后, 用氯仿(100 mL×3)萃取.萃取液用水洗至中性后, 用无水硫酸钠干燥, 滤液经减压蒸馏后得到粘稠状的2粗品, 经过硅胶柱层析纯化得14.22 g淡黄色油状物, 产率87%. [α]D20+31 (c 0.50, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.03 (s, 3H), 1.31~1.33 (m, 9H), 1.48~1.54 (m, 3H), 1.74~1.87 (m, 3H), 1.99 (t, J=7.8 Hz, 1H), 2.06 (s, 3H), 2.13 (d, J=2.4 Hz, 1H), 2.38 (d, J=12.6 Hz, 1H), 2.90~2.99 (m, 3H), 3.17 (q, 1H), 3.34 (q, 1H), 6.14 (s, 1H), 6.99 (s, 1H), 7.10 (d, J=7.8 Hz, 1H), 7.26~7.27 (d, J=8.4 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.7, 18.8, 19.0, 21.0, 23.4, 24.0, 25.3, 30.2, 33.5, 37.4, 37.5, 38.4, 45.2, 49.9, 123.8, 124.2, 126.9, 134.8, 145.6, 147.2, 170.7; MS (70 eV) m/z: 328.3 [M+H]+. Anal. calcd for C22H33NO: C 80.52, H 10.05, N 4.21; found C 80.68, H 10.16, N 4.28.
在100 mL反应瓶中, 将2溶解在(23.90 g, 70 mmol)乙酸(30 mL)中, 将15 mL三氧化铬溶液(12.00 g, 120 mmol)加入, 在0 ℃条件下搅拌4 h后, 用氯仿(100 mL×3)萃取.萃取液用水洗至中性后, 用无水硫酸钠干燥, 滤液减压蒸馏后得到粘稠状的3粗品.经过硅胶柱层析纯化得14.67 g黄色油状产物, 产率86%. [α]D20+51 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.03 (s, 3H), 1.32 (t, J=7.2 Hz, 9H), 1.48~1.55 (m, 3H), 1.75 (d, J=13.8 Hz, 1H), 1.85 (t, J=13.2 Hz, 1H), 2.04 (s, 3H), 2.11 (s, 1H), 2.38 (d, J=12.6 Hz, 1H), 2.90~2.95 (m, 2H), 3.14 (q, 1H), 3.34 (q, 1H), 6.43~6.45 (t, J=5.4 Hz, 1H), 6.99 (s, 1H), 7.09 (d, J=7.8 Hz, 1H), 7.27 (d, J=8.4 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ: 19.1, 21.0, 23.3, 24.1, 25.4, 33.5, 36.2, 37.4, 37.5, 38.4, 45.2, 50.0, 123.9, 124.2, 126.9, 134.9, 145.6, 147.3, 170.6; IR (KBr) ν: 3373, 3073, 2937, 1721, 1685, 1625, 1562, 1436, 1238 cm-1; MS (70 eV) m/z: 342.19 [M+H]+. Anal. calcd for C22H31NO2: C 77.42, H 9.38, N 4.11; found C 77.54, H 9.46, N 4.02.
方法一:在100 mL反应瓶中, 将3 (1.67 g, 5 mmol)溶解在乙醇(30 mL)中, 再加入溴化铜固体(2.78g, 12.5 mmol), 所得混合物在搅拌条件下回流12 h.然后, 将反应液倒入水(200 mL)中, 用乙酸乙酯(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到红棕色膏状的4.经过硅胶柱层析纯化得0.97 g黄色油状产物, 产率46%.
方法二:在100 mL反应瓶中, 将3 (1.67 g, 5 mmol)溶解在乙酸(40 mL)中, 加入液溴(1.00 g, 6.25 mmol), 所得混合物在室温下搅拌过夜.反应结束后, 将反应液倒入饱和碳酸氢钠水溶液中, 用乙酸乙酯(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到红棕色膏状的4粗品.经过硅胶柱层析纯化得1.53 g黄色油状产物, 产率73%. [α]D20+64 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.15 (s, 3H), 1.21~1.25 (m, 9H), 1.41 (s, 2H), 1.49 (d, J=5.7 Hz, 1H), 1.52 (d, J=5.7 Hz, 1H), 1.67~1.72 (m, 3H), 1.96 (s, 3H), 2.91~2.96 (m, 1H), 3.23~3.27 (m, 2H), 3.54 (q, 1H), 4.72 (d, J=8.7 Hz, 1H), 6.15 (s, 1H), 7.21 (d, J=8.1 Hz, 1H), 7.43 (m, 1H), 7.63 (d, J=2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ: 19.4, 23.0, 23.5, 23.7, 25.0, 33.5, 36.2, 36.4, 38.6, 39.2, 41.8, 51.4, 60.3, 121.8, 125.8, 131.3, 147.0, 147.7, 170.9, 194.4; IR (KBr) ν: 3421, 2960, 1686, 1652, 1558, 1388 cm-1; MS (70 eV) m/z: 420.08 [M]+. Anal. calcd for C22H30BrNO2: C 62.86, H 7.38, N 3.33; found C 62.77, H 7.21, N 3.37.
在50 mL反应瓶中, 将4 (2.10 g, 5 mmol)溶解在DMF (20 mL)中, 加入硫代氨基脲固体(0.55 g, 6 mmol), 在60 ℃下搅拌4 h后, 用乙酸乙酯(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到黄色膏状的5粗品.经过硅胶柱层析纯化得1.12 g黄色固体产物, 产率58%. m.p. 183.1~184.2 ℃; [α]D20-181 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.11 (d, J=5.4 Hz, 6H), 1.25~1.28 (m, 6H), 1.42 (d, J=18.6 Hz, 1H), 1.49 (d, J=2.8 Hz, 1H), 1.54~1.58 (m, 1H), 1.69 (q, 1H), 1.75~1.77 (m, 2H), 1.95 (s, 3H), 2.51 (q, 1H), 3.16 (m, 1H), 3.32 (m, 1H), 5.64 (s, 1H), 6.66 (s, 1H), 7.23~7.28 (m, 3H), 7.45 (s, 1H), 7.76 (d, J=4 Hz, 1H), 9.37 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.6, 23.3, 23.5, 24.1, 24.5, 33.7, 36.4, 36.8, 37.7, 42.3, 45.8, 49.6, 122.8, 123.0, 128.4, 129.9, 146.6, 148.9, 149.8, 170.7; IR (KBr) ν: 3427, 3278, 2957, 2925, 2867, 1649, 1685, 1572, 1494, 1421, 1287 cm-1; MS (70 eV) m/z: 413.2 [M+H]+, 847.2 [2M+Na]+. Anal. calcd for C23H32N4OS: C 66.83, H 8.00, N 13.56; found C 66.69, H 7.91, N 13.37.
在50 mL反应瓶中, 将4 (2.10 g, 5 mmol)溶解在乙醇(20 mL)中, 然后加入氢氧化钾溶液, 控制pH=10, 在60 ℃条件下搅拌4 h后, 用氯仿(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到黄色膏状的6粗品.经过硅胶柱层析纯化得1.42 g黄色油状产物, 产率84%. [α]D20+52 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.28~1.30 (m, 6H), 1.38 (s, 3H), 1.54 (s, 3H), 1.62~1.65 (m, 4H), 1.84~1.87 (m, 1H), 1.95 (s, 3H), 2.51 (d, J=16.2 Hz, 1H); 2.95~2.99 (m, 1H), 3.34 (q, 1H), 3.68 (q, 1H), 5.67 (s, 1H), 6.29 (s, 1H), 7.41 (s, 2H), 7.98 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.7, 23.4, 24.0, 24.1, 25.3, 33.4, 36.1, 37.3, 38.3, 45.2, 49.9, 123.8, 124.1, 126.9, 132.6, 134.8, 145.6, 147.2, 153.5, 170.4, 199.2; IR (KBr) ν: 3360, 2960, 1632, 1607, 1545, 1492, 1276 cm-1; MS (70 eV) m/z: 340.22 [M+H]+, 357.00 (M+NH4)+, 362.28 (M+Na)+. Anal. calcd for C22H29NO2: C 77.65, H 8.82, N 4.12; found C 77.75, H 8.89, N 4.05.
在100 mL反应瓶中, 将3 (1.75 g, 5 mmol)溶解在乙醇(30 mL)中, 然后分别加入苯肼盐酸盐(1.44 g, 10 mmol)和盐酸, 控制pH=1~2, 在80 ℃条件下搅拌6 h后, 用氯仿(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到黄色油状的7粗品.经过硅胶柱层析纯化得1.12 g黄色固体产物, 产率53.7%. m.p.169.1~170.2 ℃; [α]D20+21 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 1.03 (s, 3H), 1.28 (t, J=7.2 Hz, 1H), 1.34 (q, 6H), 1.39 (d, J=3.2 Hz, 1H), 1.50 (s, 3H), 1.69~1.83 (m, 4H), 2.00 (s, 3H), 2.32 (d, J=13.3 Hz, 1H), 2.98 (m, 2H), 3.35 (d, J=18.8 Hz, 1H), 5.76 (s, 1H), 7.18 (m, 3H), 7.30 (d, J=8.1 Hz, 1H), 7.35 (d, J=3.6 Hz, 1H), 7.47 (m, 1H), 8.01 (m, 1H), 8.66 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 17.7, 19.9, 21.3, 23.5, 24.1, 33.8, 36.5, 37.2, 37.4, 40.0, 48.8, 51.2, 111.4, 119.0, 119.7, 121.2, 122.8, 123.6, 126.1, 126.9, 128.4, 129.2, 135.0, 137.2, 146.7, 146.8, 170.7; IR (KBr) ν: 3289, 2959, 2926, 2867, 1656, 1550, 1509, 1440, 1371 cm-1; MS (70 eV) m/z: 415.55 [M+H]+, 432.00 (M+NH4)+, 437.02 (M+Na)+. Anal. calcd for C28H34- N2O: C 80.96, H 8.43, N 6.75; found C 81.01, H 8.51, N 6.71.
在100 mL反应瓶中, 将无水氯化铝(3.60 g, 9 mmol)和乙酰氯(0.70 g, 9 mmol)加入到二氯甲烷(30 mL)中, 室温搅拌1 h后, 向其中滴加含2 (1.96 g, 6 mmol)的二氯甲烷(15 mL)溶液, 在室温继续反应8 h后, 用氯仿(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到粘稠状的8粗品.经过硅胶柱层析纯化得2.26 g淡黄色固体物, 产率68%. m.p. 190.1~193.3 ℃; [α]D20+38 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 0.96 (s, 3H), 1.21~1.24 (m, 9H), 1.28 (q, 1H), 1.43 (q, 4H), 1.71~1.81 (m, 4H), 1.98 (s, 3H), 2.56 (s, 3H), 2.82~2.87 (m, 1H), 2.96 (q, 1H), 3.07 (q, 1H), 3.30 (q, 1H), 3.46~3.48 (m, 1H), 5.52 (s, 1H), 7.07 (s, 1H), 7.40 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.7, 18.8, 23.4, 24.1, 24.2, 25.2, 28.7, 30.1, 30.5, 36.1, 37.3, 37.4, 38.2, 45.0, 49.8, 124.2, 127.0, 136.3, 138.8, 144.8, 146.8, 170.2, 203.4; IR (KBr) ν: 3299, 2966, 2922, 2864, 1683, 1641, 1555, 1460, 1371 cm-1; MS (70 eV) m/z: 370.19 [M+H]+. Anal. calcd for C24H35NO2: C 77.84, H 9.73, N 3.78; found C 77.79, H 9.69, N 3.84.
在100 mL反应瓶中, 将8(1.85 g, 5 mmol)溶解在乙酸(40 mL)中, 然后加入液溴(1.00 g, 6.25 mmol), 在室温条件下搅拌6 h后, 将反应液倒入饱和碳酸氢钠水溶液中, 用氯仿(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到红棕色膏状的9粗品.经过硅胶柱层析纯化得1.42 g淡黄色油状物, 产率63.1%. [α]D20+33 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 0.95 (s, 3H), 1.20~1.24 (m, 9H), 1.25~1.28 (m, 2H), 1.39~1.43 (m, 3H), 1.71~1.78 (m, 4H), 1.99 (s, 3H), 2.85~2.87 (m, 1H), 2.95~2.97 (m, 1H), 3.02~3.06 (m, 1H), 3.29~3.38 (m, 2H), 4.40 (d, J=3.4 Hz, 2H), 5.59 (s, 1H), 7.10 (s, 1H), 7.39 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.7, 18.8, 22.8, 23.0, 24.0, 25.2, 29.6, 30.9, 32.3, 34.4, 36.1, 37.4, 38.1, 45.0, 50.3, 120.3, 127.9, 128.4, 134.5, 143.9, 149.1, 171.4; IR (KBr) ν: 3288, 2966, 2962, 2926, 2867, 1695, 1657, 1549, 1452, 1377 cm-1; MS (70 eV) m/z (%): 450.68 [M+H]+. Anal. calcd for C24H34BrNO2: C 64.29, H 7.81, N 3.12; found C 64.31, H 7.86, N 3.09.
在50 mL反应瓶中, 将9 (2.25 g, 5 mmol)溶解在DMF (20 mL)中, 然后加入硫脲(0.46 g, 6 mmol), 在75 ℃条件下搅拌4 h后, 用乙酸乙酯(100 mL×3)萃取.有机相用无水硫酸钠干燥后, 经减压蒸馏得到黄色膏状的10粗品.经过硅胶柱层析纯化得1.03 g米白色固体产物, 产率48.3%. m.p. 122.5~123.9 ℃; [α]D20+93 (c 0.10, CHCl3); 1H NMR (CDCl3, 600 MHz) δ: 0.95 (s, 3H), 1.18 (q, 6H), 1.23 (s, 3H), 1.26~1.29 (m, 2H), 1.40~1.44 (m, 3H), 1.64~1.67 (m, 1H), 1.73~1.79 (m, 2H), 1.93 (s, 1H), 1.99 (s, 3H); 2.84~2.88 (m, 1H), 2.96 (q, 1H), 3.09 (q, 1H), 3.24~3.29 (m, 2H), 5.28 (s, 2H), 5.51 (s, 1H), 6.34 (s, 1H), 7.02 (s, 1H), 7.23 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ: 18.7, 18.9, 21.0, 23.5, 24.1, 24.2, 25.1, 28.8, 29.9, 36.1, 37.3, 37.4, 38.3, 45.1, 49.8, 104.7, 125.7, 126.0, 131.6, 135.2, 144.1, 146.8, 166.6, 170.2; IR (KBr) ν: 3301, 3184, 2960, 2925, 2866, 1650, 1548, 1517, 1543, 1375 cm-1; MS (70 eV) m/z: 426.29 [M+H]+. Anal. calcd for C25H35N3OS: C 70.42, H 8.45, N 9.86; found C 70.48, H 8.51, N 9.79.
室温条件下, 向样品池中加入3 mL的鲑鱼精DNA-EB(溴化乙锭)混合溶液(cDNA/cEB=10:1)后, 依次向样品池中加入等体积的化合物样品, 使得化合物和DNA的浓度比值梯度地增加, 静置5 min后, 在520 nm的激发波长下测定其荧光发射谱[21].
用Tris缓冲溶液配制鲑鱼精DNA溶液和含化合物的鲑鱼精DNA溶液(结合比率r=c化合物/cDNA), 混匀后室温下静置5 min, 测定其圆二色谱, 测试时扣除Tris缓冲溶液和化合物的吸收[22].
将pBR 322质粒DNA (200 μg)与浓度梯度增加的化合物在pH=7.26的TBE缓冲溶液中活化2.5 h (37 ℃), 用溴酚蓝-EDTA终止反应, 在1%的琼脂糖凝胶上进行电泳, 电泳结束后, 在凝胶成像系统上成像, 观察化合物对DNA的切割情况.
采用MTT法测试了目标化合物对MCF-7肿瘤细胞的体外细胞毒活性, 以临床用药顺铂作为阳性对照.首先将待测化合物溶解于DMSO中, 配制成10 mmol•L-1的溶液, 然后用培养液和牛血清将浓度稀释至1, 2, 4, 8, 16, 32 µmol•L-1 (最终DMSO含量<0.5%).将MCF-7细胞经常规消化、计数, 接种到96孔板中, 置于37 ℃, 5%的CO2培养箱中培养24 h.再将配制好的不同浓度的待测化合物加入96孔板中, 设置对照组, 每孔3个复孔, 于培养箱中培养24 h后, 每孔加入20 μL的MTT溶液, 继续培养4 h.弃去上清液后每孔加入150 μL的DMSO, 摇床振荡10 min混匀, 随后用酶标仪测定570 nm处的吸光值(OD)并计算抑制率, 化合物作用于MCF-7细胞的抑制率达到50%时所对应的浓度定义为半数抑制浓度(IC50).
辅助材料(Supporting Information) 所合成目标化合物的核磁共振氢谱、核磁共振碳谱及高分辨质谱谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
莫启进, 段文贵, 李行任, 黄丹平, 罗湛江, 有机化学, 2011, 31, 1114. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340270.shtmlMo, Q. J.; Duan, W. G.; L, X. R.; Huang, D. P.; Luo, Z. J. Chin. J. Org. Chem. 2011, 31, 1114 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340270.shtml
Fei, B. L.; Li, W.; Xu, W. S.; Long, J. Y.; Liu, Q. B.; Sun, W. Y.; Anson, C. E.; Powell, A. K. Eur. J. Inorg. Chem. 2013, 34, 5919.
Fei, B. L.; Xu, W. S.; Gao, W. L.; Zhang, J.; Zhao, Y.; Long, J. Y.; Anson, C. E.; Powell, A. K. J. Photochem. Photobiol. B 2015, 142, 77. doi: 10.1016/j.jphotobiol.2014.11.008
Jin, X. Y.; Chen, H.; Li, D. D.; Li, A. L.; Wang, W. Y.; Gu, Wen. J. Enzym. Inhib. Med. Chem. 2019, 34, 955.
Wang, Y. C.; Su, C. H.; Li, F. Y.; Liu, L. Z.; Pan, Y. M.; Wu, X. R.; Wang, H. S. Spectrochim. Acta A. 2010, 76, 328. doi: 10.1016/j.saa.2010.03.014
Lin, G. S.; Duan, W. G.; Liu, H. Q.; Ma, Y.; Lei, F. H. Chem. Nat. Compd. 2018, 54, 56. doi: 10.1007/s10600-018-2258-6
Akhtar, J.; Khan, A. A.; Ali, Z.; Haider, R.; Shahar Yar, M. Eur. J. Med. Chem. 2017, 125, 143. doi: 10.1016/j.ejmech.2016.09.023
Marin-Ocampo, L.; Veloza, L. A.; Abonia, R.; Sepulveda-Arias, J. C. Eur. J. Med. Chem. 2019, 162, 435. doi: 10.1016/j.ejmech.2018.11.027
Ali, I.; Mukhtar, S. D.; Hsieh, M. F.; Alothman, Z. A.; Alwarthan, A. RSC Adv. 2018, 8, 37905. doi: 10.1039/C8RA07060A
Khalifa, M. E. Acta Chim. Slov. 2018, 65, 1. doi: 10.17344/acsi.2017.3547
Hassan, A. A.; Aly, A. A.; Mohamed, N. K.; EI Shaieb, K. M.; Makhlouf, M. M.; Abdelhafez, ESMN.; Brase, S.; Nieger, M.; Dalby, K. N.; Kaoud, T. S. Bioorg. Chem. 2019, 85, 585. doi: 10.1016/j.bioorg.2019.02.027
Marques, R. A.; Gomes, A. O. C. V.; de Briton, M. V.; dos Santos, A. L. P.; da Silva, G. S.; de Lima, L. B.; Nunes, F. M.; de Mattos, M. C.; de Oliveira, F. C. E.; Pessoa, C. D.; de Moraes, M. O.; de Fatima, A.; Franco, L. L.; Silva, M. D.; Dantas, M. D. D.; Santos, J. C. C.; Figueiredo, I. M.; da Silva, E. F.; de Aquino, T. M.; de Araujo, J. X.; de Oliveira, M. C. F.; Gunatilaka, A. A. L. J. Photochem. Photobiol. B 2018, 179, 156. doi: 10.1016/j.jphotobiol.2018.01.016
Zhang, F.; Lin, Q. Y.; Li, S. K.; Zhao, Y. L.; Wang, P. P.; Chen, M. M. Spectrochim. Acta, A 2012, 98, 436. doi: 10.1016/j.saa.2012.08.074
Niroomand, S.; Khorasani-Motlagh, M.; Noroozifar, M.; Moodi, A. J. Photochem. Photobiol. B 2012, 117, 132. doi: 10.1016/j.jphotobiol.2012.09.015
边琳, 李连之, 张庆富, 刘后炉, 王大奇, 化学学报, 2011, 69, 1661. doi: 10.3866/PKU.WHXB20110722Bian, L.; Li, L. Z.; Zhang, Q. F.; Liu, H. L; Wang, D. Q. Acta Chim. Sin. 2011, 69, 1661 (in Chinese). doi: 10.3866/PKU.WHXB20110722
Tiwari, A. D.; Mishra, A. K.; Mishra, S. B.; Mamba, B. B.; Maji, B.; Bhattacharya, S. Spectrochim. Acta A 2011, 79, 1050. doi: 10.1016/j.saa.2011.04.018
黄俊腾, 张阳, 王湘利, 计亮年, 刘海洋, 无机化学学报, 2013, 29, 1649.Huang, J. T.; Zhang, Y.; Wang, X. L.; Ji, L. N.; Liu, H. Y. Chin. J Inorg. Chem. 2013, 29, 1649 (in Chinese).
Alizadeh, R.; Afzal, M.; Arjmand, F. Spectrochim. Acta, A 2014, 131, 625. doi: 10.1016/j.saa.2014.04.051
刘娟娟, 卢言菊, 王婧, 毕良武, 赵振东, 有机化学, 2017, 37, 731. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345850.shtmlLiu, J. J.; Lu, Y. J.; Wang, J.; Bi, L. W.; Zhao, Z. D. Chin. J. Org. Chem. 2017, 37, 731 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345850.shtml
陈泳, 博士论文, 南京林业大学, 南京, 2012.Chen, Y. Ph.D. Dissertation, Nanjing Forestry University, Nanjing, 2012 (in Chinese).
冯少波, 雷茜, 张业, 王恒山, 潘英明, 吴强, 童碧海, 有机化学, 2007, 27, 1414. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract330183.shtmlFeng, S. B.; Lei, Q.; Zhang, Y.; Wang, H. S.; Pan, Y. M.; Wu, Q.; Tong, B. H. Chin. J. Org. Chem. 2007, 27, 1414 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract330183.shtml
甘礼社, 周长新, 有机化学, 2009, 29, 849. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328198.shtmlGan, L. S.; Zhou, C. X. Chin. J. Org. Chem. 2009, 29, 849 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328198.shtml

图 1 化合物对鲑鱼精DNA-EB体系的荧光光谱的影响
Figure 1 Effects of compounds on fluorescence spectra of salmon sperm DNA-EB system
cDNA=1.0×10-4 mol•L-1; cQ=0, 6, 12, 18, 24, 30, 36, 42, 48, 54, 60 μmol•L-1

图 2 化合物对鲑鱼精DNA溶液CD光谱的影响
Figure 2 Effects of compounds on CD spectra of salmon Sperm solution

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
