图式 1.
有机硅烷化合物的合成方法
Scheme 1.
Synthetic methods for organosilicon compounds

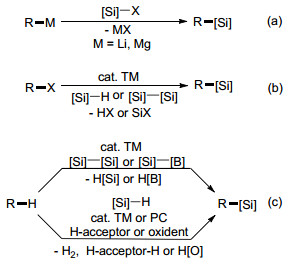
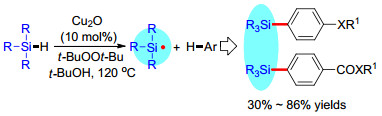
有机硅烷化合物是分子中含有C—Si键的有机化合物, 被广泛应用于制造有机硅光电材料、硅橡胶、涂层、成型植入物、压敏粘合剂、硅表面活性剂等含硅材料[1].而且, 因为有机硅烷化合物具有低毒性和易被代谢的特点, 它们在药物化学领域亦有重要地位[2].此外, 由于C—Si键不稳定, 很容易转化为各种C—C键或碳-杂键, 许多有机硅烷还是有机合成中重要的反应中间体[3].因此, 发展简便高效的有机硅烷合成方法受到科学家们的广泛关注和重视, 是有机化学前沿的热点领域.
传统合成有机硅烷化合物的方法主要通过氯硅烷或环氧硅烷与有机金属试剂的反应来构建C—Si键(Scheme 1a).这些方法不仅需要用到等物质的量的有机金属试剂, 官能团容忍性差, 还会产生大量对环境不友好的金属盐副产物, 有明显的局限性.随后, 一系列通过卤代烃与硅烷或双硅烷的C—Si交叉偶联反应合成有机硅烷的方法被开发出来(Scheme 1b), 但它们以卤代烃作为原料, 反应前需要对反应底物进行预卤化, 其大规模应用同样受到限制.
C—H键是有机化合物中最基本也是分布最广泛的化学键之一, 通过它们的直接硅烷化来合成有机硅烷避免了底物的预活化, 与传统方法相比, 具有良好的步骤经济性和原子经济性(Scheme 1c). C—H键的硅烷化拥有较长的历史, 1982年, Curtis和Epstein报道了[4]首例过渡金属催化苯的不定向硅烷化反应.此后, 过渡金属催化C—H键的硅烷化反应构建C—Si键的方法不断被报道, 得到蓬勃发展, 成果斐然.经过科学家们近四十年的努力, 包括钯、铑、铱、钌、镍、铜、钴、铁等在内的许多过渡金属被成功应用于催化C—H键的硅烷化反应, 为有机硅烷化合物的合成提供了大量简便、高效和实用的技术手段.
2015年, Hartwig[5], Sharma[6]和王从洋[7]课题组分别对过渡金属催化C—H键硅烷化的方法进行了系统的综述. 2016年, 尚筱洁和柳忠全[8]总结了涉及C—H/Si—H键官能团化的自由基反应.不过, 随着该领域的快速发展, 近年来又有许多突破性研究成果涌现出来, 其中不乏一些概念全新的反应体系和活化策略.在此背景下, 本文将系统地综述2015年以来通过过渡金属催化C—H键的直接硅烷化反应来合成有机硅烷的新方法和新策略.需要指出的是, 鉴于最近张扬会课题组[9]在他们自己工作的基础上综述了以双硅烷作为硅源的钯催化C—H键硅烷化反应, 本文将不再赘述相关内容.
本节主要讨论通过碳-金属键的形成来实现C—H键断裂/C—Si键偶联的过渡金属催化反应.根据C—H键不同的活化机制和硅烷化反应类型, 过渡金属催化的C—H键活化/硅烷化反应可以分为以下三大类: (1)导向基团导向的C—H键活化/硅烷化反应; (2)非导向的C—H键活化/硅烷化反应; (3)分子内C—H键活化/硅烷化反应.
2016年, Pilarski课题组[10]报道了钌催化杂芳烃C(sp2)—H键的硅烷化反应(Eq. 1).该反应可以在烷基保护的氨基或无保护的伯氨基团导向下选择性地进行邻位C(sp2)—H键的硅烷化反应, 而且, 在没有导向基团的情况下反应仍然可以顺利进行, 其硅烷化反应主发生在杂芳烃的2号位.作者发现, 对于导向的邻位C(sp2)—H键硅烷化反应, 产率受导向基团的电性影响较大, 太长链或σ-给电子效应弱的导向基团反应效率较低.
游劲松课题组[11]报道了首例钌催化的一级C(sp3)—H键分子间硅基化反应(Eq. 2).该方法具有催化剂负载低、底物范围广、官能团耐受性良好且对空气不敏感的特点, 为C(sp3)—Si键的构建提供了一种方便实用的途径. Murata课题组[12]则在180 ℃的条件下实现了噁唑啉导向、钌催化的选择性γ位一级C(sp3)—H键的硅烷化反应(Eq. 3).除噁唑啉外的吡啶和吡唑等氮杂环也可以作为该反应的导向基团.
|
|
(1) |
|
|
(2) |
|
|
(3) |
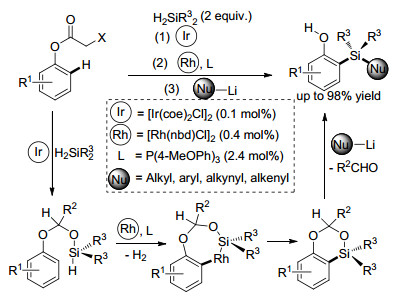
Jeon课题组[13]发展了一种缩醛基团无痕导向、铱/铑双金属协同催化的苯酚邻位C(sp2)—H键硅烷化的方法(Scheme 2).作者利用两种过渡金属催化剂的协同作用, 以原位生成且容易脱除的缩醛作为导向基团, 成功实现了苯酚邻位C(sp2)—H键的硅烷化反应.该反应可以应用于多取代芳烃的制备, 在合成上具有较高的潜在应用价值.
Tobita课题组[14, 15]先后报道了钌催化苯乙炔的氢化/反式氢硅化、邻位C(sp2)—H键硅烷化串联反应(Eq. 4).该反应条件温和, 以一种刚性的双硅烷作为配体, 以硅氢作为硅源, 芳基和烯基C(sp2)—H键均可以顺利地进行硅烷化反应.作者发现, 氢硅烷的电性和取代基对反应的产率和选择性有较大的影响, 当使用体积较大的HSi(OSiMe3)3时, 仅能得到炔烃氢硅烷化的产物, 而无C(sp2)—H键硅烷化反应发生; 而带有吸电子基团、中等体积的HSiMe(OSiMe3)2和HSi(OEt)3, 能够顺利参与反应, 生成相应的双硅烷化产物.当使用体积较小的HSi(OMe)3进行反应时, 会导致催化剂分解而没有任何产物生成.
|
|
(4) |
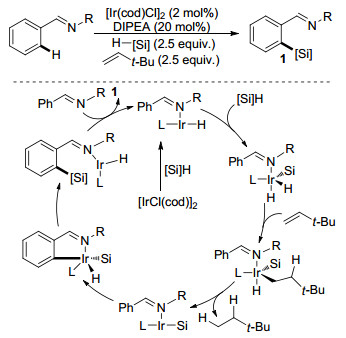
2017年, 李鹏飞课题组[16]报道了铱催化亚胺导向的芳基邻位C(sp2)—H键硅烷化反应(Scheme 3).该反应以HSiMe(OSiMe3)2作为硅烷化试剂, 使用二异丙基乙胺(DIPEA)作为添加剂, 叔丁基乙烯作为氢受体, 以良好的区域选择性和产率合成了多种芳基硅烷化合物.在该反应中, 电中性或富电子的底物其硅烷化效率相对较高, 而带有吸电子基的底物只能得到中等偏下的产率.当邻位上带有取代基时, 产率也有所降低.此外, 该催化体系还适用于未活化C(sp3)—H键的硅烷化.
2018年, Takai课题组[17]报道了铱催化喹啉的选择性C(sp2)—H键活化/硅烷化反应(Eq. 5).作者以叔丁基乙烯作为氢受体, [Ir(OMe)(cod)]2作为催化剂, 在不需要额外配体的情况下, 选择性地实现了喹啉8位C(sp2)—H键的硅烷化反应.实验发现, 当喹啉2位上有取代基或喹啉环上存在供电子基团时, 反应效果更好.机理研究表明, C(sp2)—H键的断裂选择性地发生在8位上, 是不可逆的过程, 而C—Si键的形成是迅速发生的.该反应为靶点药物的结构修饰提供了一种简便高效的合成途径.
Yoshikai课题组[18]报道了一种亚胺导向、铁催化的芳基C(sp2)—H键硅烷化反应(Eq. 6).该反应以降冰片烯作为氢受体, 以廉价的Fe3(CO)12作为催化剂, 而[Ru3(CO)12]则无法催化该反应.反应所得到的产物可以在过氧化物和光照条件下, 转化为相应的氰基化合物, 但该反应效率不高, 有待进一步改进.
|
|
(5) |
|
|
(6) |
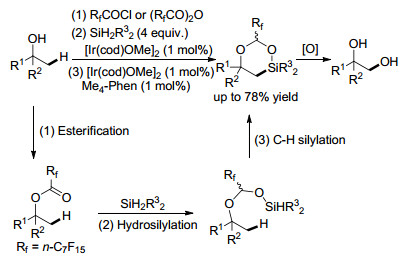
Hartwig课题组[19]报道了全氟缩醛无痕导向的脂肪醇β-C(sp3)—H键的硅烷化反应, 在一锅反应中合成了各种取代的1, 2-二醇化合物(Scheme 4).反应经历了两个铱催化硅烷化反应过程:酯羰基的氢硅化反应生成缩醛和分子内C(sp3)—H键硅烷化.二乙基硅烷在整个反应过程中发挥双重作用, 在酯的氢硅化反应中充当还原剂以及作为C(sp3)—H键硅烷化的导向基团兼硅源.
2019年, 李滨课题组[20]报道了首例钌(Ⅱ)催化噁唑导向的邻位C(sp2)—H键选择性硅烷化反应(Eq. 7).在该催化体系下, 各种噁唑啉或苯并[d]噁唑均可以成功导向单硅烷化反应, 收率中等至良好, 具有良好的官能团耐受性和区域选择性, 为多种含硅噁唑的合成提供了一条方便实用的途径.
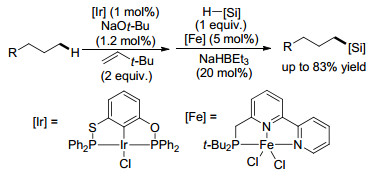
2015年, 黄正课题组[21]报道了一种通过铱、铁联合催化的脱氢/异构化/硅氢化串联反应将简单烷烃转化为直链烷基硅烷的方法(Scheme 5), 在形式上实现了烷烃末端一级C(sp3)—H键的直接硅烷化反应.在该催化体系中, 钳形配合的铱催化剂的作用是催化烷烃脱氢, 而铁催化剂则主要起到催化烯烃异构化/氢硅烷化的作用, 从而提供了一种简单高效的合成直链烷基硅烷的方法.
Minami和Hiyama等[22]通过C(sp2)—H键的直接硅烷化反应实现了保护的2-(羟甲基)苯基二甲基-杂芳基/烯基硅烷(杂芳基/烯基-HOMSi试剂)的合成(Eq. 8).作者发现, 杂芳烃和烯烃的直接硅烷化反应为杂芳基/烯基HOMSi试剂的合成提供了简便有效的途径, 各种杂芳烃和末端烯烃均可以通过C—Si偶联实现硅烷化.此外, 该反应体系还可以应用于杂芳烃的双硅烷化反应.
|
|
(8) |
2016年, Jeon课题组[23]报道了钌卡宾催化的芳基乙烯C(sp2)—H键的硅烷化反应(Eq. 9).实验发现, 以顺- 1, 5-环辛二烯作为氢受体、二环己基磷氯化苯基钌卡宾作为催化剂时, 以良好的官能团耐受性和较高的E/Z选择性合成了芳乙烯基硅烷化合物. 2017年, Mankad课题组[24]以硅基硼烷作为硅源, 实现了铜催化烯烃的脱氢硅烷化反应(Eq. 10).在该反应体系下, 无论是贫电子还是富电子烯烃均可以实现硅烷化.作者发现, 该反应能够选择性地进行烯烃C(sp2)—H键硅烷化而不发生C=C氢硅烷化反应的关键就在于使用了二正戊基酮(6-undecanone)作为HBpin受体.此外, Marciniec等[25]以烯基硅醚作为硅源经过钌催化的脱乙烯/C—H键硅烷化合成了一系列取代的氮杂硅醚化合物(Eq. 11).
|
|
(9) |
|
|
(10) |
|
|
(11) |
2017年, Mitsudo和Suga等[26]通过铑催化的分子内Si—H/C(sp2)—H偶联反应, 发展了一种合成多并环苯并噻咯衍生物的简便方法(Eq. 12).在该反应中, 贫电子的双膦配体dppe-F20效果最好.该催化体系可适用于各种2位硅氢取代的联芳基化合物的分子内环化反应, 且能耐受一系列常见的取代基团, 还可应用于高度π共轭的加长梯形硅杂多芳环体系的构建.
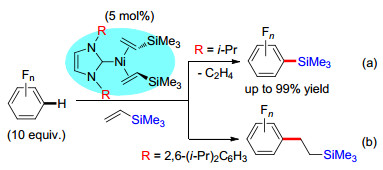
2017年, Johnson课题组[27]报道了镍催化多氟芳烃C(sp2)—H键的硅烷化或硅乙基化反应.该反应使用多氟苯与乙烯基硅烷反应, 在不同的氮杂环卡宾/镍催化体系下, 分别得到不同产物.当氮杂环卡宾(NHC)氮原子上的取代基为体积较小的异丙基时, 反应得到C(sp2)—H键硅烷化的产物(Scheme 6a).而当NHC氮原子上的取代基为体积较大的2, 6-二异丙基苯基时, 反应得到多氟苯基乙基硅烷(Scheme 6b). 2019年, 该课题组[28]报道了对该反应机理的研究结果, 发现NHC取代基的体积对多氟芳烃C(sp2)—H键官能团化的选择性有较大影响.当NHC取代基的体积较小时, 镍催化剂与乙烯基硅烷络合之后容易脱去乙烯, 生成硅基镍物种, 因而可以发生C(sp2)—H键硅烷化反应.而当NHC取代基的体积较大时, 乙烯分子难以释放, 故而发生乙烯基硅烷的氢芳基化反应.
|
|
(12) |
2018年, 侯召民课题组[29]报道了钇催化甲基硫醚α位C(sp3)—H键的硅烷化反应(Eq. 13).该方法具有原子经济性高、底物范围广、产率高和反应条件简单等优点.在钇催化体系下, 硅烷化反应选择性地发生在α-硫甲基的C(sp3)—H键上, 以较高的产率合成了一系列很难通过其他方法合成的α-硅烷硫醚衍生物.
|
|
(13) |
硅杂环化合物是一类重要的有机杂环化合物, 在药物化学和材料科学领域具有独特的应用价值, 其合成一直以来备受科学家的关注.最近, 赵东兵课题组[30]对过渡金属催化的硅杂环化合物的合成方法进行了系统的综述, 主要概括了通过C—H键硅基化、C—Si键活化以及成环反应等策略来构建硅杂环化合物的方法.其中, 分子内的C—H键活化/硅烷化反应是构建各种硅杂环化合物最直接高效的方法.
2014年, 陈润锋和黄维等[31]报道了铑催化的分子内Si—H/C—H偶联反应, 发展了一种高原子经济性地构建二苯并氮杂环硅烷的方法(Eq. 14).该反应以叔丁基乙烯作为氢受体, 在135 ℃下反应24 h, 即能以高达92%的产率得到相应的氮杂环硅烷产物.同时, 作者还重点研究了所合成的二苯并氮杂环硅烷化合物的光电性能.
|
|
(14) |
2016年, Hartwig课题组[32]报道了一种铑催化环丙烷C(sp3)—H键的对映选择性分子内硅烷化反应(Eq. 15).该反应使用金属Rh作为催化剂, 以手性双膦(S)-DTBM-Segphos为配体, 环丙烯作为氢受体, 具有较高的收率、对映选择性和官能团耐受性.所得到的氧杂环硅烷化合物经过简单的氧化即可得到手性保留的2-羟甲基环丙醇衍生物.
|
|
(15) |
Murata等[33]发展了一种用于2-(二烷氢硅基)联苯的分子内C(sp2)—H键活化/硅烷化反应的PtCl2/三(3, 5-二甲基-1-吡唑基)硼氢化钾(TpMe2K)催化体系, 以良好的产率合成了一系列二苯并噻咯衍生物(Eq. 16). 2017年, Takai课题组[34]利用[RhCl(cod)]2和PPh3催化体系实现了类似的分子内C(sp2)—H键活化/硅烷化反应(Eq. 17).该反应既不需要氧化剂, 也不需要外加氢受体.
2016年, Takai课题组[35]报道了一种通过铑催化的分子内脱氢硅基化反应对映选择性地合成手性硅杂[6]螺烯的方法(Eq. 18).作者以金属铑与(R)-(S)-BPPFA配体作为催化剂, 从2-(硅基)菲联萘出发, 经历分子内C(sp2)—H键的脱氢硅基化反应, 合成了罕见的单纯对映体:硅杂[6]螺烯衍生物, 且不需要外加氧化剂.
|
|
(16) |
|
|
(17) |
|
|
(18) |
何伟课题组[36]报道了铑催化硅杂环丁烷的开环/分子内C(sp2)—H硅烷基化反应.得到了一系列高产率、高区域选择性的π-共轭噻咯衍生物(Eq. 19).研究证明该反应经历了一个罕见的五元环钌金属物种的β-氢消除/分子内C(sp2)—H硅烷基化的反应过程.值得一提的是, 该反应体系还适用于分子内C(sp3)—H硅烷基化反应.
|
|
(19) |
2017年, 何伟题组[37]在以上反应的基础上, 发展了一种构建含有硅手性中心的二苯并噻咯的合成方法(Eq. 20).该反应首先通过钌催化的硅杂环丁烷开环/分子内不对称C(sp2)—H硅基化, 生成手性氢噻咯中间体, 继而与芳烃发生手性保留的分子间C(sp2)—H硅烷化反应, 高对映选择性、高产率地合成多取代二苯并噻咯化合物.
同年, 施章杰等[38]报道了铱与手性二氮配体催化的高对映选择性分子内C—H键硅烷化反应, 用于合成手性苯并环硅醚(Eq. 21).所使用的二氮配体包含一个四氢喹啉联噁唑啉结构, 可以与[Ir(cod)OMe]2很好地协同催化分子内硅烷化反应.底物探索发现, 所有对称的无取代、间位取代和对位取代二芳基甲氧基硅氢都能以较高的收率和良好的对映选择性(>90%)获得相应的环硅醚产物, 且不对称的双芳基甲氧硅烷亦能适用于该反应.机理研究表明, 芳基C(sp2)—H键的断裂既不是可逆过程, 也不是反应的决速步骤.随后, Hartwig课题组[39]利用同样的催化体系实现了分子内C(sp3)—H键的对映选择性硅烷化反应, 高选择性地构建了手性季碳中心(Eq. 22).与上述C(sp2)—H键的硅烷化反应不同, C(sp3)—H键活化是该反应的决速步.
|
|
(20) |
|
|
徐利文课题组[40]报道了一种铱催化硅氧烷基芳烃邻位C(sp2)—H键硅烷化的方法.在该反应中邻位的硅氧基硅氢可以在形式上看做是分子内C(sp2)—H键活化的导向基团(Eq. 23).该反应具有良好的官能团耐受性和区域选择性, 可用于合成含有两个潜在可修饰硅原子的不对称环状氧杂环二硅烷类化合物.
|
|
(23) |
黄正课题组[41]报道了钌催化选择性一级C(sp3)—H键的分子内硅烷化反应, 用于合成含硅杂环化合物(Eq. 24).该反应最低仅需0.1 mol%的钌催化剂, 以顺式环辛烯作为氢受体, 具有良好的官能团耐受性, 成功地实现了C、N、Si、Ge原子的α位置上C—H键的分子内硅烷化.机理实验证明C(sp3)—H键的断裂是该反应的决速步骤.随后, 该课题组[42]还报道了钌催化的苄氧硅烷分子内C(sp2)—H键的硅烷化反应(Eq. 25).
|
|
(24) |
|
|
(25) |
谭启涛和许斌等[43]报道了通过三步铑催化的分子内C—H/Si—H脱氢偶联环化反应来合成硅杂π-碗状sumanene衍生物的方法(Eq. 26).作者利用单晶X射线衍射首次确定了该类化合物的平面几何构型, 并用紫外-可见光谱和荧光光谱研究了它们的光学性质.
|
|
(26) |
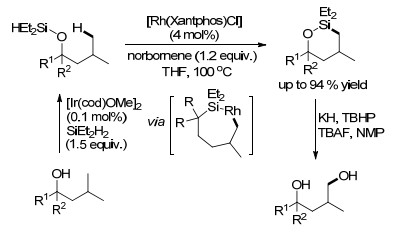
2018年, Hartwig课题组[44]报道了铑催化烷烃C(sp3)—H键的分子内选择性硅烷化反应(Scheme 7).作者以降冰片烯作为氢受体, 使用[Rh(Xantphos)Cl]来催化一级C(sp3)—H键的选择性硅烷化反应, 来合成六元环氧杂硅烷, 该氧杂环硅烷化产物经过氧化后可以得到重要的1, 4-二醇衍生物, 且具有良好官能团耐受性, 还可以应用于复杂分子的合成与官能化.机理研究表明, C(sp3)—H键活化是该反应的决速步骤.
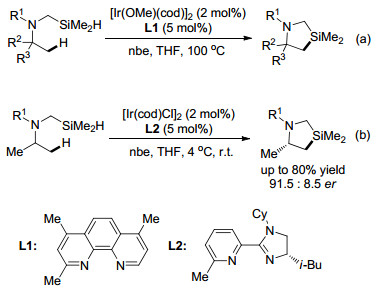
随后, Hartwig课题组[45]还报道了铱催化脂肪族胺β-位C(sp3)—H键的选择性硅烷化反应.该反应使用2, 4, 7-三甲基菲啰啉L1作为配体, 在温和的条件下即可选择性地实现脂肪胺一级C(sp3)—H键的硅烷化反应, 且具有良好的官能团耐受性.反应所得到的硅杂吡咯烷化合物可以很容易被过氧化物氧化为相应的1, 2-醇胺(Scheme 8a).除此之外, 作者还制备了一系列极少用于手性催化的手性吡啶基咪唑配体.当使用吡啶基咪唑L2作为配体时, 在室温下即能以较高的产率和对映选择性获得目标硅杂吡咯烷衍生物(Scheme 8b), 为手性1, 2-醇胺的合成提供了一种实用方法.
赵东兵课题组[46]报道了铑催化2-芳基酚衍生物的六元硅环化反应, 合成了二苯并噻咯和平面手性的硅氧杂并环茂金属衍生物(Eq. 27).作者通过分子内的C(sp2)—H键硅烷化成功地构建了六元二苯并氧硅杂环.此外, 作者在手性膦配体和钌催化体系下, 实现了对映选择性的C(sp2)—H键分子内硅烷化, 以较高的对映选择性合成了一系列平面手性硅氧杂环并茂金属化合物, 所得到的产物被成功应用于各种转化反应.
Shibata课题组[47]发展了一种铱催化的分子内C(sp2)—H/Si—H脱氢偶联合成取代三苯并硅杂环庚烷衍生物的方法(Eq. 28).反应从2', 6'-二芳基-2-(氢硅基)联苯出发, 以叔丁基乙烯作为氢受体, 首次合成了多取代三苯并硅杂环庚烷, 通过手性HPLC拆分得到其纯对映体, 并研究测定了它们消旋化反应的能垒.
|
|
(27) |
|
|
(28) |
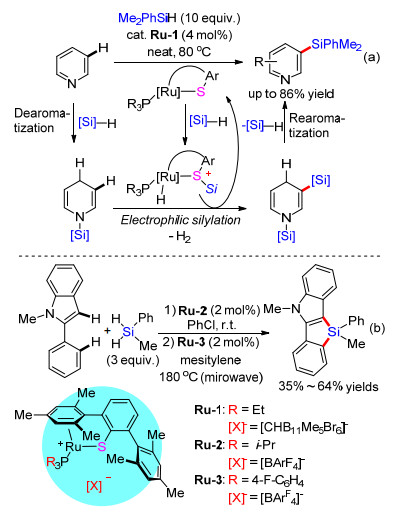
本节主要概括经由硅基正离子或类Friedel-Crafts反应过程而实现的过渡金属催化C—H键硅烷化反应. 2015年, Oestreich课题组[48]报道了钌催化吡啶间位C(sp2)—H键的亲电硅烷化反应(Scheme 9a).该反应经历了氢化去芳构化/亲电硅烷化/芳构化的反应历程.作者使用硫酚/膦配位金属钌Ru-1作为催化剂, 通过钌与硫的协同作用活化Si—H, 生成硫原子稳定的正离子硅烷中间体, 该中间体与去芳构化后的二氢吡啶发生亲电硅烷化, 再芳构化得到间位硅烷化的吡啶化合物. 2017年[49]该课题组将该催化体系拓展到两步C(sp2)—H键的Friedel-Crafts硅烷化反应(Scheme 9b).作者使用两种配体配合的钌催化剂Ru-2和Ru-3分别催化分子间和分子内C(sp2)—H键硅烷化反应, 在两步、一锅反应中构建了硅杂多并环吲哚骨架.虽然该方法的底物范围有一定的局限性, 但它显著地扩展了有机硅烷化合物的多样性.
2016年, Oestreich课题组[50]还报道了非贵金属铁催化的富电子芳烃的C(sp2)—H键亲电硅烷化反应(Eq. 29).作者使用NaBArF4来活化FeCl2作为催化剂, 由此生成的路易斯酸可以促进富电子芳烃与氢硅烷的亲电取代反应, 反应条件简单, 不需要添加额外的氢受体, 与典型的Friedel-Crafts反应比较相似.
随后, 黄正课题组[51]利用一种钳形钌配合物作为催化剂, 催化杂芳烃的区域选择性硅烷化反应(Eq. 30).作者以(TMSO)2MeSiH和Et3SiH作为硅源, 与各种氧、硫杂环芳烃反应, 以出色的C-2选择性、良好的官能团耐受性得到了一系列杂环芳基硅烷化合物, 并成功将它们用于钯催化的Hiyama-Denmark偶联反应.
|
|
(29) |
|
|
(30) |
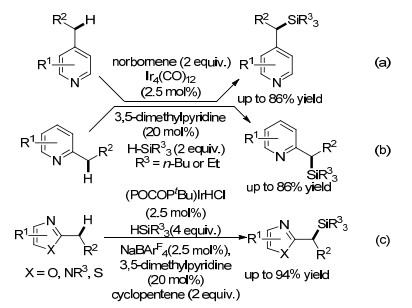
2017年, Fukumoto课题组[52, 53]先后报道了铱催化4-烷基吡啶和2-烷基吡啶的苄位烷基C(sp3)—H键的硅烷化反应(Scheme 10a, b), 作者使用Ir4(CO)12作为催化剂, 降冰片烯作为氢受体, 3, 5-二甲基吡啶作为添加剂, 可以选择性地得到多种4-(1-硅基)吡啶与2-(1-硅基)吡啶衍生物, 在反应体系中并没有检测到吡啶C(sp2)—H键硅烷化副反应产物.当使用[Ir(OMe)(cod)]2作为催化剂时不发生反应, 但在一氧化碳(CO)气氛下反应能够正常进行, 证明了CO配体对该反应的重要性.作者发现该反应对硅基化试剂有严格的要求, 仅有三乙基硅烷反应效果较好.实验证明吡啶环的3, 5号位上的取代基对反应没有影响, 2, 6号位上的取代基由于位阻关系, 会导致低的反应收率, 而2, 4, 6-三甲基吡啶则不反应. 2018年, 该课题组[54]还实现了铱催化各种唑类杂芳烃苄基位C(sp3)—H的硅烷化反应(Scheme 10c).
Tsuchimoto课题组[55]报道了一种锌催化吲哚3号位C(sp2)—H键的亲电硅基化反应(Eq. 31).作者在锌-吡啶体系下, 以硅氢作为硅源, 得到了一系列3号位硅基化的吲哚衍生物, 并释放出氢气.机理研究表明该反应经历了锌促进硅氢对富电子芳烃的亲电取代反应过程, C(sp2)—H键的断裂是该反应的决速步骤.
|
|
(31) |
2018年, Nakajima课题组[56]以镍作为催化剂, 以氯硅烷作为硅源实现了烯烃的硅基Heck反应(Eq. 32).无论是三氯硅烷、二氯硅烷还是单氯硅烷均可以适用于该反应体系, 并可以在一锅反应中直接醇解, 以中等到良好的产率得到相应的烯基硅酯化合物.
|
|
(32) |
Oestreich课题组[57]报道了铜催化胺α位C(sp3)—H的亲电硅烷化反应, 提供了一种合成α-硅烷化胺衍生物的合成方法(Eq. 33).作者对氮原子上的不同保护基进行了探索, 发现磺酰胺取代基的电性对反应有明显的影响, 富电子比缺电子的反应效果好, 且磷酰胺与苯甲酰胺也适用于该反应, 表现出较好的底物耐受性.该反应的原料N-卤代酰胺还可以直接原位生成, 无需纯化即可应用于后续反应.
|
|
(33) |
通过C—H键自由基反应来合成有机硅烷的报道相对较少. 2016年, 蔡春课题组[58]报道了铜催化烯烃和富电子杂芳烃的硅烷化反应(Eq. 34).作者使用醋酸铜作为催化剂, 过氧叔丁醚作为氧化剂, 经过铜催化过氧化物产生叔丁氧自由基, 随后发生自由基攫氢过程, 产生硅基自由基, 继而经由自由基脱氢偶联反应, 选择性地得到一系列反式烯基硅烷或杂芳基硅烷化合物.在该反应中, 供电子基取代苯乙烯的反应效率要优于吸电子基取代苯乙烯.机理研究证明该反应经历了硅基自由基机理.同年, 该课题组还报道了铁催化的烯烃自由基硅烷化反应[59].
|
|
(34) |
2017年, 柳忠全课题组[60]报道了一种铜催化的芳烃C(sp2)—H键自由基选择性硅烷化方法(Scheme 11).作者以氢硅烷作为自由基前体, 在铜/过氧化物体系下产生硅基自由基, 继而发生芳烃/杂芳烃C(sp2)—H键的自由基脱氢取代反应.该反应适用于各种富电子和缺电子芳烃的对位选择性硅烷化, 这与Friedel-Crafts亲电C(sp2)—H键硅烷化或过渡金属催化脱氢硅烷化反应显著不同, 扩大了该反应的实用范围.
最近, 张永强课题组[61]报道了可见光促进的杂芳烃C(sp2)—H键硅烷化反应(Eq. 35).该反应使用三烷基硅氢(HSiR3)作为硅自由基前体, 在可见光照射的条件下, 以铱作为光催化剂, 实现了缺/富电子杂环芳烃和芳基腈的自由基C—Si偶联反应, 获得中等到良好的产率和优秀的区域选择性.该方法操作简单、反应条件温和、使用安全且容易获得的Na2S2O8、双(三甲基硅基)过氧化物(BTMSPO)或iPr3SiSH作为自由基引发剂, 为含硅有机小分子的合成提供了一种成本低廉、环境友好的合成方法.
本文综述了2015年以来过渡金属催化C—H键硅烷化反应的最新研究进展.根据C—H键的不同活化机制, 主要分为以下几类: (1)过渡金属催化的C—H键活化/硅烷化反应; (2) C—H键的亲电硅烷化反应; (3) C—H键的自由基硅烷化反应.其中, 过渡金属催化的C—H键活化/硅烷化反应是发展最早也是最成熟的策略, 具有丰富的反应多样性和强大的选择性控制能力.而其他两类C—H键硅烷化反应的发展相对较晚, 它们普遍具有反应条件温和、操作简便的优点, 为C—H键的硅烷化提供了许多新颖的催化体系和反应机制.总之, 近年来科学家发展了许多类型各异的C—H键硅烷化反应, 已为结构多样的有机硅化合物的合成提供了一些实用价值较高的合成方法.
然而, 该领域仍存在许多亟待解决的问题和挑战.首先, 催化剂诱导的对映选择性C—H键硅烷化的方法仍然较少, 且主要局限于分子内反应, 发展分子间C—H键、尤其是C(sp3)—H键的对映选择性硅烷化反应是目前面临的主要挑战.其次, 对于许多近年来发现的C—H键硅烷化反应其机理还不够明确, 研究并弄清楚它们的反应历程和选择性调控机制, 是目前需要解决的问题.此外, 设计和发展新的催化体系和拓展C—H键硅烷化反应类型, 尤其是发展可回收重复利用的高效非均相催化体系, 拓展过渡金属催化C—H键硅烷化反应在有机合成、新材料创制、药物设计与研发等领域中的实际应用范围, 是该领域今后的重点发展方向.
(a) Elbing, M.; Bazan, G. C. Angew. Chem., Int. Ed. 2008, 47, 834.
(b) Hill, R. M. In Silicone Surfactants, Surfactants Science Series, Ed.: Hill, R. M., Marcel Dekker, New York, 1999, Vol. 86, pp. 1~48.
(c) Muzafarov, A. M. Silicon Polymers, Springer, Heidelberg, 2011.
(d) Jones, R. G.; Ando, W.; Chojnowski, J. Silicon-Containing Polymers, Kluwer Academic, Dordrecht, 2000.
(e) You, Y.; An, C.-G.; Kim, J.-J.; Park, S. Y. J. Org. Chem. 2007, 72, 6241.
(f) Bai, D.; Han, S.; Lu, Z.-H.; Wang, S. Can. J. Chem. 2008, 86, 230.
(g) Barroso, G.; Li, Q.; Bordia, R. K.; Motz, G. J. Mater. Chem. A 2019, 7, 1936.
(h) Sims, R. A.; Harmer-Bassell, S. L.; Quinton, J. S. Appl. Surf. Sci. 2019, 475, 999.
(i) Itami, K.; Ito, H.; Yano, Y.; Miyauchi, Y.; Mitoma, N. WO 2017131190, 2017.
(a) Mortensen, M.; Husmann, R.; Veri, E.; Bolm, C. Chem. Soc. Rev. 2009, 38, 1002.
(b) Franz, A. K.; Wilson, S. O. J. Med. Chem. 2013, 56, 388.
(c) Gluyas, J. B. G.; Burschka, C.; Doerrich, S.; Vallet, J.; Gronemeyer, H.; Tacke, R. Org. Biomol. Chem. 2012, 10, 6914.
(a) Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063.
(b) Rappoport, Z.; Apeloig, Y. Chemistry of Organosilicon Compounds, Wiley-VCH, New York, 2001.
(c) Li, L.; Zhang, Y.; Gao, L.; Song, Z. Tetrahedron Lett. 2015, 56, 1466.
Gustavson, W. A.; Epstein, P. S.; Curtis, M. D. Organometallics 1982, 1, 884. doi: 10.1021/om00066a028
Cheng, C.; Hartwig, J. F. Chem. Rev. 2015, 115, 8946. doi: 10.1021/cr5006414
Sharma, R.; Kumar, R.; Kumar, I.; Singh, B.; Sharma, U. Synthesis 2015, 47, 2347. doi: 10.1055/s-00000084
Yang, Y.; Wang, C. Sci. China Chem. 2015, 58, 1266. doi: 10.1007/s11426-015-5375-0
Shang, X.; Liu, Z.-Q. Org. Biomol. Chem. 2016, 14, 7829. doi: 10.1039/C6OB00797J
Zhou, B.; Lu, A.; Zhang, Y. Synlett 2019, 30, 685. doi: 10.1055/s-0037-1610339
Devaraj, K.; Sollert, C.; Juds, C.; Gates, P. J.; Pilarski, L. T. Chem. Commun. 2016, 52, 5868. doi: 10.1039/C6CC00803H
Li, W.; Huang, X.; You, J. Org. Lett. 2016, 18, 666. doi: 10.1021/acs.orglett.5b03593
Kon, K.; Suzuki, H.; Takada, K.; Kohari, Y.; Namikoshi, T.; Watanabe, S.; Murata, M. ChemCatChem 2016, 8, 2202. doi: 10.1002/cctc.v8.13
Hua, Y.; Asgari, P.; Avullala, T.; Jeon, J. J. Am. Chem. Soc. 2016, 138, 7982. doi: 10.1021/jacs.6b04018
Komuro, T.; Kitano, T.; Yamahira, N.; Ohta, K.; Okawara, S.; Mager, N.; Okazaki, M.; Tobita, H. Organometallics 2016, 35, 1209. doi: 10.1021/acs.organomet.5b01013
Kitano, T.; Komuro, T.; Ono, R.; Tobita, H. Organometallics 2017, 36, 2710. doi: 10.1021/acs.organomet.7b00528
Wang, H.; Wang, G.; Li, P. Org. Chem. Front. 2017, 4, 1943. doi: 10.1039/C7QO00340D
Murai, M.; Nishinaka, N.; Takai, K. Angew. Chem., Int. Ed. 2018, 57, 5843. doi: 10.1002/anie.201801229
Xu, W.; Pek, J. H.; Yoshikai, N. Asian J. Org. Chem. 2018, 7, 1351. doi: 10.1002/ajoc.v7.7
Bunescu, A.; Butcher, T. W.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 1502. doi: 10.1021/jacs.7b12150
Liu, S.; Zhang, S.; Lin, Q.; Huang, Y.; Li, B. Org. Lett. 2019, 21, 1134. doi: 10.1021/acs.orglett.9b00085
Jia, X.; Huang, Z. Nat. Chem. 2015, 8, 157.
Minami, Y.; Komiyama, T.; Hiyama, T. Chem. Lett. 2015, 44, 1065. doi: 10.1246/cl.150359
Bokka, A.; Jeon, J. Org. Lett. 2016, 18, 5324. doi: 10.1021/acs.orglett.6b02642
Mazzacano, T. J.; Mankad, N. P. ACS Catal. 2017, 7, 146. doi: 10.1021/acscatal.6b02594
Sztorch, B.; Frackowiak, D.; Pyziak, J.; Czapik, A.; Hoffmann, M.; Marciniec, B. Dalton Trans. 2017, 46, 4975. doi: 10.1039/C7DT00201G
Mitsudo, K.; Tanaka, S.; Isobuchi, R.; Inada, T.; Mandai, H.; Korenaga, T.; Wakamiya, Atsushi; Murata, Y.; Suga, S. Org. Lett. 2017, 19, 2564. doi: 10.1021/acs.orglett.7b00878
Elsby, M. R.; Johnson, S. A. J. Am. Chem. Soc. 2017, 139, 9401. doi: 10.1021/jacs.7b05574
Elsby, M. R.; Liu, J.; Zhu, S.; Hu, L.; Huang, G.; Johnson, S. A. Organometallics 2019 38, 436. doi: 10.1021/acs.organomet.8b00786
Y. Luo, H.-L. Teng, C. Xue, M. Nishiura, Z. Hou, ACS Catal. 2018, 8, 8027. doi: 10.1021/acscatal.8b02405
Han, J. L.; Qin, Y.; Sun, Y.; Zhao, D. Sci. Sin. Chim. 2019, 49, 672. doi: 10.1360/N032018-00240
Li, H.; Wang, Y.; Yuan, K.; Tao, Y.; Chen, R.; Zheng, C.; Zhou, X.; Li, Ju.; Huang, W. Chem. Commun. 2014, 50, 15760. doi: 10.1039/C4CC06636G
Lee, T.; Hartwig, J. F. Angew. Chem., Int. Ed. 2016, 55, 8723. doi: 10.1002/anie.201603153
Murata, M.; Takizawa, M.; Sasaki, H.; Kohari, Y.; Sakagami, H.; Namikoshi, T.; Watanabe, S. Chem. Lett. 2016, 45, 857. doi: 10.1246/cl.160415
Murai, M.; Okada, R.; Asako, S.; Takai, K. Chem.-Eur. J. 2017, 23, 10861. doi: 10.1002/chem.v23.45
Murai, M.; Okada, R.; Nishiyama, A.; Takai, K. Org. Lett. 2016, 18, 4380. doi: 10.1021/acs.orglett.6b02134
Zhang, Q.-W.; An, K.; Liu, L.-C.; Guo, S.; Jiang, C.; Guo, H.; He, W. Angew. Chem., Int. Ed. 2016, 55, 6319. doi: 10.1002/anie.201602376
Zhang, Q.-W.; An, K.; Liu, L.-C.; Zhang, Q.; Guo, H.; He, W. Angew. Chem., Int. Ed. 2017, 56, 1125. doi: 10.1002/anie.201609022
Su, B.; Zhou, T.-G.; Li, X.-W.; Shao, X.-R.; Xu, P.-L.; Wu, W.-L.; Hartwig, J. F.; Shi, Z.-J. Angew. Chem., Int. Ed. 2017, 56, 1092. doi: 10.1002/anie.201609939
Su, B.; Hartwig, J. F. J. Am. Chem. Soc. 2017, 139, 12137. doi: 10.1021/jacs.7b06679
Lin, Y.; Jiang, K.-Z.; Cao, J.; Zheng, Z.-J.; Xu, Z.; Cui, Y.; Xu, L.-W. Adv. Synth. Catal. 2017, 359, 2247. doi: 10.1002/adsc.v359.13
Fang, H; Hou, W.; Liu, G.; Huang, Z. J. Am. Chem. Soc. 2017, 139, 11601. doi: 10.1021/jacs.7b06798
Fang, H.; He, Q.; Liu, G.; Huang, Z. Synlett 2017, 28, 2468. doi: 10.1055/s-0036-1590982
Zhou, D.; Gao, Y.; Liu, B.; Tan, Q.; Xu, B. Org. Lett. 2017, 19, 4628. doi: 10.1021/acs.orglett.7b02254
Karmel, C.; Li, B.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 1460. doi: 10.1021/jacs.7b11964
Su, B.; Lee, T.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 18032. doi: 10.1021/jacs.8b10428
Zhao, W.-T.; Lu, Z.-Q.; Zheng, H.; Xue, X.-S.; Zhao, D. ACS Catal. 2018, 8, 7997. doi: 10.1021/acscatal.8b01992
Shibata, T.; Uno, N.; Sasaki, T.; Takano, H.; Sato, T.; Kanyiva, K. S. J. Org. Chem. 2018, 83, 3426. doi: 10.1021/acs.joc.8b00233
Wbbolt, S; Oestreich, M. Angew. Chem., Int. Ed. 2015, 54, 15876. doi: 10.1002/anie.201508181
Omann, L.; Oestreich, M. Organometallics 2017, 36, 767. doi: 10.1021/acs.organomet.6b00801
Yin, Q.; Klare, H. F. T.; Oestreich, M. Angew. Chem., Int. Ed. 2016, 55, 3204. doi: 10.1002/anie.201510469
Fang, H.; Guo, L.; Zhang, Y.; Yao, W.; Huang, Z. Org. Lett. 2016, 18, 5624. doi: 10.1021/acs.orglett.6b02857
Fukumoto, Y.; Hirano, M.; Chatani, N. ACS Catal. 2017, 7, 3152. doi: 10.1021/acscatal.7b00539
Fukumoto, Y.; Hirano, M.; Matsubara, N.; Chatani, N. J. Org. Chem. 2017, 82, 13649. doi: 10.1021/acs.joc.7b02375
Hirano, M.; Fukumoto, Y.; Matsubara, N.; Chatani, N. Chem. Lett. 2018, 47, 385. doi: 10.1246/cl.171137
Yonekura, K.; Iketani, Y.; Sekine, M.; Tani, T.; Matsui, F.; Kamakura, D.; Tsuchimoto, T. Organometallics 2017, 36, 3234. doi: 10.1021/acs.organomet.7b00382
Matsumoto, K.; Huang, J.; Naganawa, Y.; Guo, H.; Beppu, T.; Sato, K.; Shimada, S.; Nakajima, Y. Org. Lett. 2018, 20, 2481. doi: 10.1021/acs.orglett.8b00847
Feng, J.-J.; Oestreich, M. Org. Lett. 2018, 20, 4273. doi: 10.1021/acs.orglett.8b01698
Gu, J.; Cai, C. Chem. Commun. 2016, 52, 10779. doi: 10.1039/C6CC05509E
Xu, R.; Cai, C. Catal. Commun. 2018, 107, 5. doi: 10.1016/j.catcom.2017.12.023
Xu, Z.; Chai, L.; Liu, Z.-Q. Org. Lett. 2017, 19, 5573. doi: 10.1021/acs.orglett.7b02717
Liu, S.; Pan, P.; Fan, H.; Li, H.; Wang, W.; Zhang, Y. Chem. Sci. 2019, 3817.

图式 2 缩醛无痕导向Ir/Rh催化的苯酚C(sp2)—H键硅烷化反应
Scheme 2 Ir/Rh-catalyzed ortho-C(sp2)—H silylation of phenols with traceless acetal directing groups

图式 3 铱催化的亚胺导向C(sp2)—H键硅烷化反应
Scheme 3 Imine directed Ir-catalyzed C(sp2)—H bonds silylation

图式 4 全氟缩醛无痕导向的醇β-C(sp3)—H键硅烷化反应
Scheme 4 Traceless silylation of β-C(sp3)—H bonds of alcohols via perfluorinated acetals

图式 5 铱/铁串联催化烷烃末端C(sp3)—H键的硅烷化反应
Scheme 5 Iridium-iron-catalyzed tandem silylation of terminal β-C(sp3)—H bonds of alkanes

图式 6 镍催化的C(sp2)—H键硅烷化/烷基化反应
Scheme 6 Ni-catalyzed silylation/alkylation of C(sp2)—H bonds

图式 8 铱催化脂肪胺的β-选择性C(sp3)—H键的硅烷化反应
Scheme 8 Ir-catalyzed β-selective C(sp3)—H silylation of aliphatic amines

图式 10 铱催化氮杂芳烃苄位C(sp3)—H键的硅烷化反应
Scheme 10 Ir-catalyzed benzylic C(sp3)—H silylation of N-heteroarenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

































 下载:
下载:


