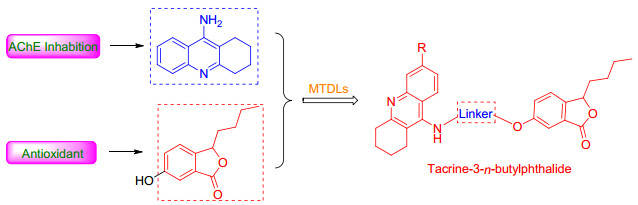
Figure 1.
Structures of tacrine, NBP and tacrine hybrids
Design, Synthesis and Evaluation of Novel Tacrine-3-n-butylphthalide Hybrids as Multifunctional Cholinesterase Inhibitors
Wandong Liu , Yu Yang , Jiaming Li , Yanyan Guo , Fan Jin , Bin Zhang

Alzheimer's disease (AD), a multifaceted neurodegenerative disease is primarily associated with dementia, which often leads to cognitive decline, psychological and behavioral abnormalities, and ultimately death.[1] At present, the number of AD patients is on the rise. It is estimated that in 2050, more than 100 million individuals will be affected by AD worldwide.[2] Pathogenic background of AD is extremely complex, and many factors, such as decreased level of acetylcholine (ACh), [3] β-amyloid (Aβ) deposits, [4] tau protein aggregation, [5] oxidative stress, neuroinflammation, are thought to play significant roles in the pathogenesis of the disease. Based on the cholinergic hypothesis, the current pharmacotherapy for AD is to maintain the levels of acetylcholine (ACh) by inhibiting cholinesterase.
There are two types of cholinesterase, i.e., acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE).[5, 6] The AChE consists of two binding sites connected by a 2.0 nm deep narrow gorge. One is catalytic anionic site (CAS) at the bottom of the gorge and the other is peripheral anionic site (PAS) near the entrance.[7, 8] In general, inhibitors that bind to either site could inhibit the AChE. Various studies have confirmed that PAS plays a crucial role in Aβ pro-aggregating action of AChE. Thus, dual-site inhibitors that interact with both CAS and PAS appear to be more beneficial for AD treatment.[9] Moreover, in healthy brains, AChE hydrolyzes the major ACh while BuChE only plays a secondary role. However, in terms of AD, the activity of AChE gradually decreases, while that of BuChE significantly increases. In the later stage of the treatment for the disease, BuChE becomes more important in regulating the ACh level in cholinergic neurons. Therefore, both AChE and BuChE are valuable therapeutic targets for the treatment of AD.[10, 11]
In contrast to cholinergic hypothesis, the recent amyloid hypothesis suggests that accumulation of Aβ peptide in the brain plays a central role in the pathogenesis of AD.[12] The antioxidant system in brain progressively decays during aging. Especially in AD brain, it declines more rapidly than normal brain. An increasing body of evidence indicates that the oxidative damage occurs in earliest stage of AD pathogenesis and promotes the formation of other pathological hallmarks of the disease, such as amyloid plaques and neurofibrillary tangles. Thus, antioxidants are also beneficial for AD treatment.
At present, the AChE inhibitors for the clinical management of AD include donepezil, rivastigmine, and galantamine. But the current treatment strategies for AD can only alleviate the symptoms rather than stop or reverse the progression of disease.[13] Hence, developing multitargetdirected ligands (MTDLs) to act as multifunctional agents to treat this disease is the mainstream for AD treatment. Based on the importance of BuChE, designing multifunctional molecules to modify a known ChE inhibitor with additional biological properties such as lowering the Aβ aggregation, oxidative stress and chelating metals, is the common approach.
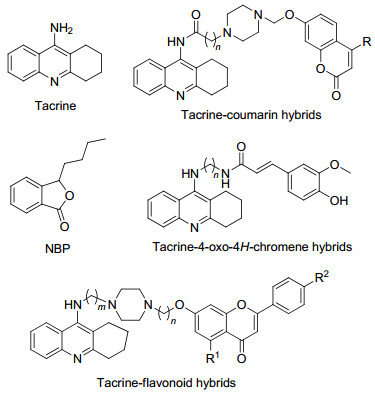
Tacrine (Figure 1), the first ChE inhibitor approved by the FDA (Food and Drug Administration) for AD treatment, was withdrawn due to severe liver toxicity.[14] However, the high potency in ChE inhibition and low molecular weight suitable for modification still make it a widely used scaffold for developing multi-target agents. A number of multi-target molecules designed by connecting tacrine with another fragment with additional activities have been developed, such as tacrine-coumarin, [15] tacrine-4-oxo-4H- chromene[9] and tacrine-flavonoid hybrids.[16] Among these compounds, most of them exhibited enhanced biological profile than tacrine, and several compounds with fewer side effects.
3-n-Butylphthalide (NBP, Figure 1) was approved by the State Food and Drug Administration (SFDA) of China for clinical use in stroke patients in 2002. Recently, they have attracted more attention due to a variety of biological activities related to AD. Study on bone marrow of rat confirmed that NBP reduced accumulation of reactive oxygen species (ROS) induced by hydrogen peroxide (H2O2).[18] In addition, a double transgenic study on L-NBP indicated a significant improvement in spatial learning and memory deficits and also an inhibition of tau hyperphosphorylation by regulation of key kinase activity.[19]
Given the activities of tacrine and NBP, in an attempt to obtain new multi-target molecules with both ChE inhibitory and antioxidants activity for the AD treatment, in this paper, a series of novel tacrine-NBP hybrids (Figure 2) have been synthesized, pharmacologically evaluated and calculated by molecular-simulation. Firstly, the AChE and BuChE inhibition and antioxidant activity of these novel compounds were evaluated. Then, molecular docking studies were carried out to study the binding mode against AChE.
The designed compounds 10a~10h, 14a~14c and 17a~17h were illustrated in Scheme 1. Compound 1 was obtained by reacting 2-carboxybenzaldehyde with diethylamine, then compound 2 was obtained by reacting compound 1 with n-butylmagnesium bromide, and com- pound 3 was obtained by the catalytic cyclization reaction of compound 2 by p-toluenesulfonic acid (PTS).
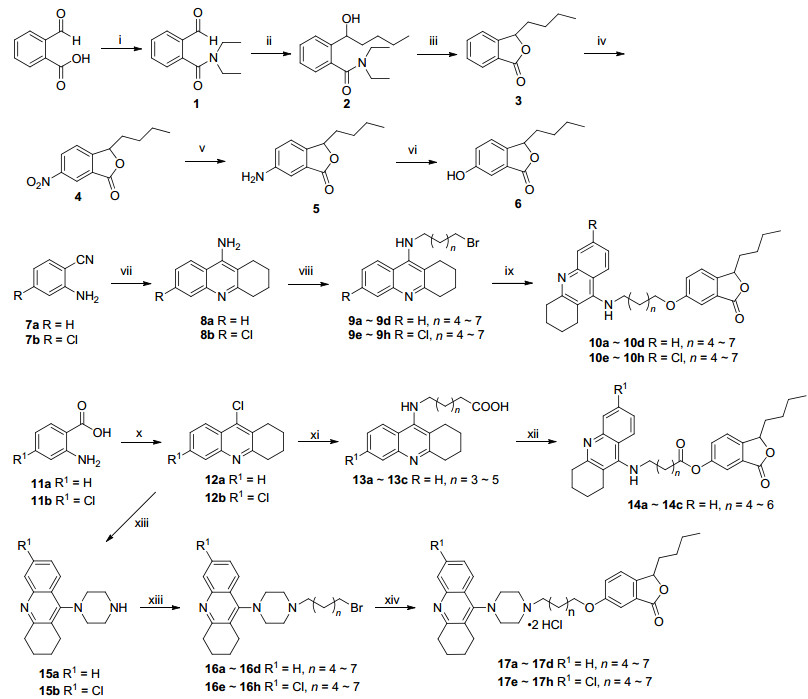
Reagents and conditions: (ⅰ) (1) SOCl2, toluene, 80 ℃; (2) diethylamine, DCM, 0 ℃; (ⅱ) (1) CH3(CH2)3Br, Mg, I2, THF, 60 ℃; (2) Grignard reagent, THF, -20 ℃; (ⅲ) p-TsOH•HCl, toluene, 80 ℃; (ⅳ) KNO3, H2SO4, r.t.; (ⅴ) Fe, AcOH, THF, H2O, 70 ℃; (ⅵ) (1) NaNO2, 30% H2SO4, urea, 0 ℃; (2) Cu(NO3)2•3H2O, Cu2O; (ⅷ) cyclohexanone, ZnCl2, 100 ℃; (ⅷ) different dibromo-alkanes, Cs2CO3, KI, CH3CN, 55 ℃; (ⅸ) Compound 6, Cs2CO3, KI, CH3CN, 60 ℃; (ⅹ) cyclohexanone, POCl3, 100 ℃; (ⅹⅰ) different amino-alkanoic acid, phenol, KI, 180 ℃; (ⅹⅱ) Compound 6, DCC, DMAP, r.t.; (ⅹⅲ) piperazine, 1-pentanol, N2, 140 ℃; (ⅹⅳ) (1) different dibromoalkanes, Cs2CO3, KI, CH3CN, 60 ℃ (2) CH3COCH3, HCl (conc.)
Compound 4 was obtained by the nitration of compound 3 under H2SO4 and KNO3. After the reduction reaction of the nitro of compound 4 with Fe/NH4Cl, compound 5 was obtained. Finally, after the diazotization of compound 5 with NaNO2 and 30% H2SO4, followed by hydrolysis reaction, the key intermediate 6 was obtained.
On the one hand, compounds 8a and 8b were obtained by the Lewis acid mediated cyclodehydration reaction of compounds 7a~7b and cyclohexanone. Then other key intermediates 9a~9h were obtained by the nucleophilic substitution of compounds 8a and 8b in the presence of potassium iodide. Finally compounds 10a~10h were obtained by the nucleophilic substitution of compounds 9a~9h with NBP derivatives 6.
Intermediates 12a~12b were obtained by reacting anthranilic acid with cyclohexanone [20]. Other key intermediates 13a~13c were obtained by nucleophilic substitution of diffierent amino alkanoic acid in the presence of potassium iodide, and compounds 14a~14c were obtained by reacting 13a~13c with NBP derivative 6. On the other hand, other key intermediates 15a~15b were obtained by reacting compounds 12a~12b with excess anhydrous piperazine. Compounds 16a~16h were obtained by reacting different dibromoalkanes with intermediates 15a~15b. Finally, compounds 17a~17h were obtained by reacting compounds 16a~16h with NBP derivative 6.
The inhibitory activities of the test compounds 10a~10h, 14a~14c, 17a~17h against hAChE (from human erythrocytes) and hBuChE (from human erythrocytes) were evaluated according to the spectrophotometric method described by Ellman et al.[21], while tacrine and donepezil were used as reference compounds. The IC50 values and selectivity index (the ratio of hAChE and hBuChE) of all test compounds were summarized in Table 1. The results indicated that all new target compounds showed moderate inhibitory activities to both ChEs with IC50 values ranging from sub-micromolar to nanomolar. Most of compounds exhibited more potent inhibitory activity for AChE than that for BuChE, indicating that these compounds were selective inhibitors for AChE. Compared with compounds 10a~10d, compounds 10e~10h exhibited an increase in AChE inhibition and a decrease in BuChE inhibition, indicating that a chlorine group played a crucial role in inhibitory activity of cholinesterase. In addition, the results showed that the piperazine ring did not have significant influence on the inhibition activity of these compounds for AChE and BuChE.
 下载:
导出CSV
下载:
导出CSV
| Compd. | R | R1 | n | IC50a/(nmol•L-1) | SId | |
| hAchEb | hBchEc | |||||
| 10a | H | — | 4 | 247.30±25.17 | 6109±378 | 24.70 |
| 10b | H | — | 5 | 38.65±3.91 | 5227±461 | 135.24 |
| 10c | H | — | 6 | 156.20±13.46 | 354±36 | 2.27 |
| 10d | H | — | 7 | 342.70±32.43 | 4639±397 | 13.54 |
| 10e | Cl | — | 4 | 549.80±55.39 | 51.31±5.09 | 0.09 |
| 10f | Cl | — | 5 | 659.30±67.27 | 85.18±7.28 | 0.13 |
| 10g | Cl | — | 6 | 363.40±33.56 | 33.69±3.49 | 0.09 |
| 10h | Cl | — | 7 | 174.20±15.64 | 165.5±15.6 | 0.95 |
| 14a | — | H | 4 | 45.27±3.57 | 5074±283 | 112.08 |
| 14b | — | H | 5 | 144.60±10.21 | 358±18.45 | 2.48 |
| 14c | — | H | 6 | 330.00±18.45 | 4708±256 | 14.27 |
| 17a | — | H | 4 | 195.00±14.64 | 766±38 | 3.93 |
| 17b | — | H | 5 | 441.00±521.47 | 2905±164 | 6.59 |
| 17c | — | H | 6 | 244.50±14.41 | 3947±206 | 16.14 |
| 17d | — | H | 7 | 72.77±4.17 | 6410±394 | 88.09 |
| 17e | — | Cl | 4 | 148.90±8.24 | 446.8±276 | 3.00 |
| 17f | — | Cl | 5 | 268.60±14.27 | 87.48±6.49 | 0.33 |
| 17g | — | Cl | 6 | 44.81±2.49 | 5867±312 | 130.93 |
| 17h | — | Cl | 7 | 270.20±16.14 | 7908±578 | 29.27 |
| Donepezil | — | — | — | 7.11±0.46 | 7530±474 | 1059.07 |
| Tacrine | — | — | — | 200.70±11.21 | 27.12±1.85 | 0.14 |
| a IC50 values represent the concentration of inhibitor required to decrease enzyme activity by 50% and are the mean of five independent experiments, each performed in triplicate (SD=standard deviation). b From human erythrocytes. c BuChE from human serum. d AChE selectivity index=IC50(hBuChE)/IC50(hAChE) | ||||||
The length of alkyl spacer between tacrine and 3-n-butylphthalide moiety played a crucial role in the ChE inhibitory activity, and when the length of the alkyl spacer was 7 or 8, the compounds exhibited the most potent AChE and BuChE inhibitory activity. Compound 10b with a seven-carbon spacer exhibited the most potent AChE inhibitory activity with IC50 value of 38.65 nmol•L-1, which was 5-fold stronger than that of tacrine. Compound 10g with an eight-carbon spacer exhibited the most potent BuChE inhibitory activity with IC50 value of 33.69 nmol• L-1, which was close to tacrine and 220-fold greater than that of donepezil.
To understand the possible binding mode of the potent compounds with AChE or BuChE, molecular modeling studies were carried out using AutoDock Tools software suit and the results using PyMOL were visualized. The binding mode of compound 10b in AChE was investigated based on the X-ray crystal structure of donepezil bound to recombinant human acetylcholinesterase (hAChE, PDB code 4EY7). As shown in Figure 3A, the tacrine moiety of 10b was located at the CAS of AChE, which showed π-π stacking interactions with the indole ring of Trp 86 (0.42 nm). Besides, the tacrine moiety could also interact with Tyr 124 via a π-donor hydrogen bond (0.30 nm). The isobenzofuranone ring adopted an appropriate orientation to stack against the indole ring of Trp 286 in PAS with the ring-to-ring distance of 0.49 nm. In addition, the oxygen atoms in the carbonyl and ether linkages of isobenzofuranone were engaged in hydrogen bond contacts with residues Ser 293 (0.19 nm) and Arg 296 (0.26 nm), respectively. The simulated results indicated that 10b was a dual site binding inhibitor for AChE.
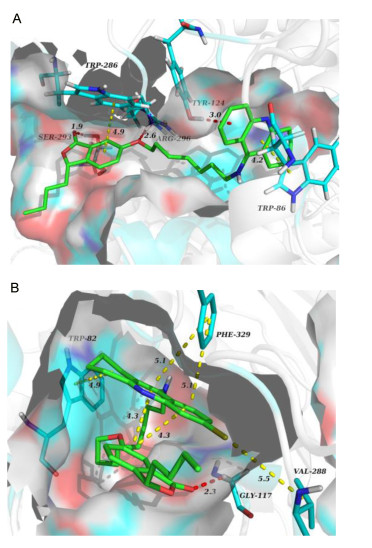
Hydrogen bonds are shown by red dashed lines and π-π stacking interactions are shown by yellow dashed lines
Similarly, the molecular modeling analysis of 10g was performed to elucidate its possible binding modes with BuChE (PDB code 1P0I) active site (Figure 3B). Since the narrow active site of BuChE, the conformation of 10g was folded and intramolecular interactions were formed between tacrine moiety and isobenzofuranone ring. Two pivotal residues of the binding site Phe 329 and Trp 82 were involved in binding with tacrine moiety through π-π stacking interaction, and the ring-to-ring distances of 0.49 and 0.51 nm respectively. Moreover, a key hydrogen bond (0.23 nm) was observed between Gly 117 and isobenzofuranone ring. The chlorine atom of the tacrine moiety made π-alkyl interaction with Val 288. These features may account for why 10g strongly inhibits BuChE.
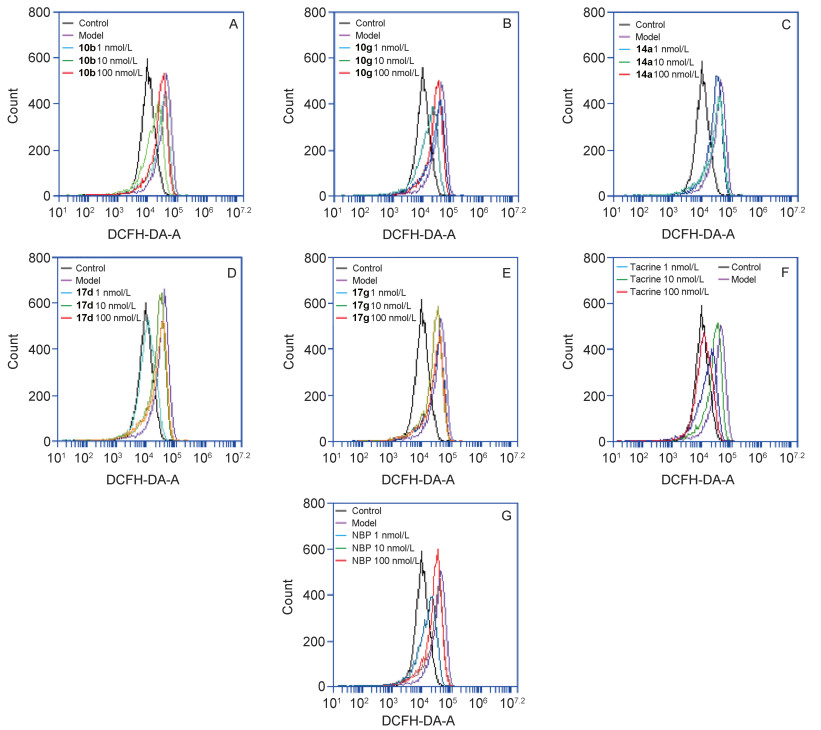
In rat Aβ-induced injury model of hippocampal neurons, intracellular ROS of target compounds 10b, 10g, 14a, 17d and 17g were evaluated using the DCFH-DA with 3-n-butylphthalide and tacrine as reference compound.[22] The results indicated that all compounds retained the potent inhibitory activity for Aβ1-42, inducing that rat hippocampal neuronal cells secrete ROS of the parent compound 3-n-butylphthalide (Table 2). Connecting 3-n-butylphtha- lide with tacrine moiety by varying the linker between the two fragments did not significantly affect the antioxidant activity of 3-n-butylphthalide, which indicated that hybridizing these two scaffolds was reasonable.
下载:
导出CSV
| Compd. | ROS levels (folds of control) | ||||
| Control | Model | 1 nmol•L-1 | 10 nmol•L-1 | 100 nmol•L-1 | |
| NBP | 1±0.225 | 3.397±0.265 | 2.547±0.463 | 2.437±0.065* | 1.503±0.343** |
| Tacrine | 1±0.293 | 3.183±0.245 | 2.468±0.419 | 1.628±0.161** | 1.270±0.170** |
| 10b | 1±0.539 | 3.676±0.405 | 2.758±0.655 | 2.550±0.276 | 1.675±0.194** |
| 10g | 1±0.320 | 3.105±0.233 | 2.490±0.352 | 2.169±0.201* | 1.433±0.174** |
| 14a | 1±0.232 | 3.357±0.377 | 2.637±0.178 | 2.553±0.482 | 2.474±0.287 |
| 17d | 1±0.569 | 3.544±0.264 | 2.779±0.104 | 2.190±0.300** | 1.096±0.144** |
| 17g | 1±0.592 | 2.839±0.081 | 2.892±0.340 | 2.684±0.481 | 2.455±0.380 |
| a Compared with model *p < 0.05, **p < 0.01. | |||||
A series of novel tacrine-3-n-butylphthalide hybrids as multi-target agents have been designed, synthesized and evaluated for ChE inhibition and antioxidation activity. The biological screening results indicated that all the target compounds exhibited potent ChE inhibitory activity with IC50 value in nanomolar range, and also retained antioxidant activity close to NBP. Among these compounds, 10b possessed the highest inhibitory activity for AChE, which was 5-fold more active than that of the reference com-pound tacrine, and with a moderate BuChE inhibition. The molecular simulation further confirmed that 10b was a dual site binding inhibitor for AChE. In vitro antioxidant activities assay, 10b retained the potent inhibitory activity for Aβ1-42 inducing rat hippocampal neuronal cells secreting ROS. Finally, all these results confirmed 10b as a potential multifunctional agent for the treatment of AD.
Rat hippocampal neurons were treated with different concentrations of compounds in the presence or absence of Aβ1-42 (2 µmol•L-1). After 48 h incubation, the ROS levels were measured with DCFH-DA probe. Three independent experiments, for each condition, were performed in triplicate. The abscissa indicates the value of the relative intensity of the scattered light signal (unit: channel), and the ordinate relative to the number of cells (unit: Zahl)
The reagents and solvents for this reaction were purchased from common commercial suppliers. If necessary, purification was carried out prior to use. Melting points were measured using melting point apparatus (RDCSY-I) and were uncorrected. 1H NMR and 13C NMR spectra were recorded on 400 MHz and 101 MHz instruments (Bruker, Fallanden, Switzerland), respectively, with tetramethylsilane (TMS) as internal standard. HRMS spectra were measured with a Q-Tof Ultima Global mass spectrometer. Column chromatography was performed on silica gel (90~150 mm, Qingdao Marine Chemical Inc.). Compounds 5, 8a~8b, 12a~12b and 13a~13c were prepared following previously published methods.[23~25]
A mixture of compound 5 (6.0 g, 29.2 mmol) and 30% H2SO4 (50 mL) was stirred at ice bath. A solution of NaNO2 (3.2 g, 46.8 mmol) in water (10 mL) was then slowly added dropwise to the reaction mixture under the condition of ice bath, and then the mixture was stirred further for 0.5 h and kept at 0~5 ℃. After adding to a few grains of urea, a solution of Cu(NO3)2•3H2O (105.9 g, 438.5 mmol) in water (200 mL) was then added to the reaction mixture. Then Cu2O (4.2 g, 29.2 mmol) was added to the above reaction mixture, and the mixture was stirred for 10 min. Afterwards, the reaction mixture was extracted with CH2Cl2. The combined organic layer was washed with water, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography using petroleum ether/ethyl acetate (V:V=1:1) as eluent to obtain target compounds 6 as yellow solid, yield 54.4%. m.p. 105.4~107.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.41 (d, J=2.0 Hz, 1H), 7.32 (d, J=8.8 Hz, 1H), 7.24 (dd, J=8.4, 2.4 Hz, 1H), 6.50 (s, 1H), 5.46 (dd, J=7.6, 4.0 Hz, 1H), 2.07~1.99 (m, 1H), 1.80~1.73 (m, 1H), 1.53~1.35 (m, 4H), 0.92 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 171.63, 157.34, 142.22, 127.31, 122.89, 122.80, 111.07, 82.05, 34.53, 26.82, 22.43, 13.85; HRMS calcd for C12H13O3 [M-H]- 205.0865, found 205.0865.
Compounds 8a or 8b (4.3 mmol), Cs2CO3 (12.8 mmol) and KI (0.85mmol) were dissolved in acetonitrile (30 mL). The solution was stirred for 1 h under argon at room temperature. Then, different dibromoalkanes (8.6 mmol) were added to the mixture. After stirred at 55 ℃ for 3.5 h, the mixture was filtered and evaporated under reduced pressure. The residue was washed successively with H2O and brine, dried over Na2SO4 and concentrated in vacuo to afford the compounds 9a~9h as a yellow oil. The crude product was used directly in the next alkylation reaction without further purification.
A mixture of 6 (0.5 g, 2.6 mmol), Cs2CO3 (2.5 g, 7.7 mmol), compounds 9a~9h (0.9 g, 2.6 mmol) and KI (0.09 g, 0.5 mmol) was dissolved in acetonitrile and refluxed at 60 ℃ for 2 h. The mixture was filtered and filtrate was evaporated to remove the solvent. The residue was purified by column chromatography (petroleum ether/ethyl acetate, V:V=5:1, plus 10 mL of triethylamine per 1000 mL).
3-Butyl-6-((6-((1, 2, 3, 4-tetrahydroanthracen-9-yl)amino)-hexyl)oxy)isobenzofuran-1(3H)-one (10a): Yellow oil, 72.1% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.43 (d, J=8.0 Hz, 1H), 8.02 (d, J=8.0 Hz, 1H), 7.88~7.83 (m, 2H), 7.58~7.54 (m, 2H), 7.29 (dd, J=8.4, 2.4 Hz, 1H), 7.24 (s, 1H), 5.56 (dd, J=7.2, 3.6 Hz, 1H), 4.03 (t, J=6.4 Hz, 2H), 3.87 (s, 2H), 3.02 (s, 2H), 2.67 (s, 2H), 2.07~1.99 (m, 1H), 1.82~1.61 (m, 9H), 1.50~1.22 (m, 8H), 0.86 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.28, 159.94, 155.98, 150.93, 142.85, 138.32, 132.86, 127.07, 125.52, 125.38, 124.04, 123.30, 119.54, 115.94, 111.40, 108.05, 81.35, 68.42, 47.51, 34.11, 30.21, 28.78, 28.31, 26.84, 26.17, 25.50, 24.49, 22.34, 21.91, 20.70, 14.28; HRMS calcd for C31H39N2O3 [M+H]+ 487.2961, found 487.2946.
3-Butyl-6-((7-((1, 2, 3, 4-tetrahydroacridin-9-yl)amino)-heptyl)oxy)isobenzofuran-1(3H)-one (10b): Yellow oil, 78.2% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.40 (d, J=8.4 Hz, 1H), 7.97 (d, J=8.4 Hz, 1H), 7.83 (t, J=7.6 Hz, 1H), 7.70 (s, 1H), 7.58~7.54 (m, 2H), 7.31 (dd, J=8.4, 2.4 Hz, 1H), 7.24 (s, 1H), 5.56 (dd, J=7.2, 3.6 Hz, 1H), 4.03 (t, J=6.4 Hz, 2H), 3.83 (q, J=6.8 Hz, 2H), 3.01 (s, 2H), 2.67 (s, 2H), 2.06~1.99 (m, 1H), 1.83~1.61 (m, 9H), 1.43~1.22 (m, 10H), 0.86 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.27, 159.95, 155.68, 151.42, 142.84, 138.90, 132.62, 127.09, 125.37, 125.29, 124.02, 123.31, 120.15, 116.24, 111.73, 108.09, 81.33, 68.52, 47.61, 34.13, 30.30, 28.85, 28.76, 28.68, 26.84, 26.49, 25.80, 24.54, 22.35, 22.00, 20.87, 14.27; HRMS calcd for C32H41N2O3 [M+H]+ 501.3117, found 501.3128.
3-Butyl-6-((8-((1, 2, 3, 4-tetrahydroacridin-9-yl)amino)-heptyl)oxy)isobenzofuran-1(3H)-one (10c): Yellow oil, 75.6% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.40 (d, J=8.4 Hz, 1H), 7.98 (d, J=8.4 Hz, 1H), 7.83 (t, J=7.6 Hz, 1H), 7.70 (s, 1H), 7.57~7.53 (m, 2H), 7.31 (d, J=8.4 Hz, 1H), 7.25 (s, 1H), 5.56 (dd, J=7.2, 3.6 Hz, 1H), 4.03 (t, J=6.4 Hz, 2H), 3.83 (q, J=6.8 Hz, 2H), 3.01 (s, 2H), 2.67 (s, 2H), 2.06~1.99 (m, 1H), 1.83~1.61 (m, 9H), 1.39~1.22 (m, 12H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.26, 159.94, 155.59, 151.36, 142.80, 138.88, 132.52, 127.06, 125.35, 125.23, 124.01, 123.29, 120.12, 116.22, 111.63, 108.04, 81.32, 68.52, 47.54, 34.13, 30.33, 29.06, 29.02, 28.93, 28.67, 26.85, 26.47, 25.82, 24.59, 22.35, 22.01, 20.85, 14.27; HRMS calcd for C33H43N2O3 [M+H]+ 515.3274, found 515.3254.
3-Butyl-6-((9-((1, 2, 3, 4-tetrahydroacridin-9-yl)amino)-heptyl)oxy)isobenzofuran-1(3H)-one (10d): Yellow oil, 68.3% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.28 (d, J=8.4 Hz, 1H), 7.83 (d, J=8.4 Hz, 1H), 7.71 (t, J=8.0 Hz, 1H), 7.55 (d, J=8.4 Hz, 1H), 7.47 (t, J=8.0 Hz, 1H), 7.31 (d, J=8.4 Hz, 1H), 7.25 (s, 1H), 6.79 (s, 1H), 5.56 (dd, J=7.2, 3.6 Hz, 1H), 4.03 (t, J=6.4 Hz, 2H), 3.65 (q, J=6.8 Hz, 2H), 2.96 (s, 2H), 2.68 (s, 2H), 2.05~1.96 (m, 1H), 1.82~1.61 (m, 9H), 1.39~1.22 (m, 14H), 0.86 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.27, 159.96, 153.98, 153.84, 142.80, 142.11, 130.97, 127.08, 124.65, 124.62, 123.99, 123.42, 123.30, 117.92, 113.40, 108.06, 81.31, 68.52, 47.90, 34.14, 30.69, 30.58, 29.32, 29.07, 29.05, 28.95, 26.84, 26.57, 25.87, 24.92, 22.46, 22.35, 21.65, 14.27; HRMS calcd for C34H45N2O3[M+H]+ 529.3430, found 529.3410.
3-Butyl-6-((6-((6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)-amino)heptyl)oxy)isobenzofuran-1(3H)-one (10e): Pale yellow oil, 50.2% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.15 (d, J=9.2 Hz, 1H), 7.70 (s, 1H), 7.54 (d, J=8.4 Hz, 1H), 7.34 (d, J=8.8 Hz, 1H), 7.31~7.27 (m, 1H), 7.24 (s, 1H), 5.61 (q, J=6.8 Hz, 1H), 5.56 (dd, J=7.6, 4.0 Hz, 1H), 4.00 (t, J=6.8 Hz, 1H), 3.58 (t, J=6.8 Hz, 1H), 3.50~3.43 (m, 2H), 2.89 (s, 2H), 2.67 (t, J=6.0 Hz, 2H), 2.06~1.98 (m, 1H), 1.83~1.63 (m, 9H), 1.41~1.22 (m, 8H), 0.86 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.32, 159.96, 159.10, 151.41, 147.31, 142.86, 133.27, 127.10, 126.41, 126.11, 124.01, 123.34, 118.68, 116.06, 108.08, 81.35, 68.40, 48.20, 35.54, 34.10, 33.51, 32.55, 32.36, 30.74, 28.81, 27.65, 26.82, 26.36, 25.41, 22.94, 22.57, 22.35, 14.29; HRMS calcd for C31H38ClN2O3 [M+H]+ 521.2571, found 521.2546.
3-Butyl-6-((7-((6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- amino)heptyl)oxy)isobenzofuran-1(3H)-one (10f): Pale yellow oil, 49.3% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.13 (d, J=9.2 Hz, 1H), 7.70 (s, 1H), 7.53 (d, J=8.4 Hz, 1H), 7.34 (d, J=8.8 Hz, 1H), 7.33~7.27 (m, 2H), 7.23 (s, 1H), 5.58~5.52 (m, 2H), 3.99 (t, J=6.4 Hz, 1H), 3.43~3.42 (m, 2H), 2.87 (t, J=6.0 Hz, 2H), 2.67 (t, J=6.0 Hz, 2H), 2.04~1.97 (m, 1H), 1.79~1.53 (m, 9H), 1.34~1.21 (m, 10H), 0.85 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.28, 159.96, 159.71, 151.00, 148.00, 142.80, 132.89, 127.10, 125.89, 123.95, 123.79, 123.29, 119.02, 116.28, 108.07, 81.31, 68.47, 48.28, 34.13, 33.95, 30.91, 28.84, 26.84, 26.65, 25.81, 25.52, 23.05, 22.75, 22.35, 14.27; HRMS calcd for C32H40ClN2O3 [M+H]+ 535.2727, found 535.2694.
3-Butyl-6-((8-((6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- amino)heptyl)oxy)isobenzofuran-1(3H)-one (10g): Pale yellow oil, 51.6% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.14 (d, J=9.2 Hz, 1H), 7.71 (s, 1H), 7.54 (d, J=8.0 Hz, 1H), 7.33~7.29 (m, 2H), 7.25 (s, 1H), 5.59~5.53 (m, 2H), 4.00 (t, J=6.4 Hz, 1H), 3.43~3.42 (m, 2H), 2.88 (t, J=6.0 Hz, 2H), 2.67 (m, 2H), 2.05~1.98 (m, 1H), 1.81~1.50 (m, 9H), 1.37~1.24 (m, 12H), 0.86 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.17, 159.93, 159.59, 150.91, 148.05, 142.66, 132.84, 127.16, 127.09, 125.76, 123.75, 123.66, 123.09, 118.99, 116.13, 108.01, 81.22, 68.45, 48.32, 34.21, 33.97, 30.99, 29.14, 28.94, 26.88, 26.67, 25.80, 25.50, 23.06, 22.76, 22.35, 14.19; HRMS calcd for C33H42ClN2O3 [M+H]+ 549.2884, found 549.2851.
3-Butyl-6-((9-((6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)-amino)heptyl)oxy)isobenzofuran-1(3H)-one (10h): Pale yellow oil, 53.3% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.14 (d, J=9.2 Hz, 1H), 7.71 (s, 1H), 7.54 (d, J=8.0 Hz, 1H), 7.34~7.29 (m, 2H), 7.25 (s, 1H), 5.59~5.53 (m, 2H), 4.02 (t, J=6.4 Hz, 1H), 3.44~3.42 (m, 2H), 2.88 (t, J=6.0 Hz, 2H), 2.67 (m, 2H), 2.05~1.98 (m, 1H), 1.81~1.50 (m, 9H), 1.39~1.22 (m, 14H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.29, 159.96, 159.22, 151.29, 147.54, 142.76, 133.16, 127.10, 126.60, 126.03, 123.89, 123.30, 123.23, 118.76, 116.00, 108.06, 81.31, 68.50, 48.25, 34.13, 33.63, 30.88, 29.31, 29.05, 28.94, 26.84, 26.63, 26.42, 25.45, 22.98, 22.63, 22.35, 14.25; HRMS calcd for C34H44ClN2O3 [M+H]+ 563.3040, found 563.3022.
A mixture of compounds 13a~13c (1.7 mmol), 6 (1.87 mmol) and CH2Cl2 (15 mL) was stirred at 0 ℃. A solution of DCC (2.55 mmol) in CH2Cl2 (10 mL) was then slowly added dropwise to the reaction mixture at 0 ℃. The mixture was added to DMAP (0.85 mmol) and stirred at 0 ℃ for 0.5 h. Then, the reaction mixture was moved to the condition of room temperature and stirred overnight. After the reaction, it was diluted with water and extracted in CH2Cl2. The combined organic layer was washed with water, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography using CH2Cl2/CH3OH (V:V=10:1) as eluent to obtain target compounds 14a~14c.
1-Butyl-3-oxo-1, 3-dihydroisobenzofuran-5-yl-6-((1, 2, 3, 4- tetrahydroacridin-9-yl)amino)hexanoate (14a): Brown oil, 41.8% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.40 (d, J=8.4 Hz, 1H), 7.71~7.55 (m, 2H), 7.41 (t, J=8.0 Hz, 2H), 7.25 (t, J=7.2 Hz, 1H), 7.07 (d, J=8.0 Hz, 2H), 5.76~5.74 (m, 1H), 3.89~3.84 (m, 2H), 2.98 (s, 2H), 2.68 (s, 2H), 2.58 (t, J=7.2 Hz, 1H), 2.26 (t, J=7.2 Hz, 1H), 1.83~1.24 (m, 16H), 0.88 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 175.12, 170.48, 158.89, 157.57, 151.38, 149.21, 146.40, 141.15, 128.81, 127.73, 127.05, 123.90, 123.02, 120.18, 115.75, 109.89, 81.37, 48.23, 34.30, 33.74, 33.35, 30.74, 26.87, 26.38, 25.42, 24.80, 23.07, 22.68, 22.37, 21.19, 14.28; HRMS calcd for C31H37N2O4 [M+H]+ 501.2753, found 501.2745.
1-Butyl-3-oxo-1, 3-dihydroisobenzofuran-5-yl-7-((1, 2, 3, 4- tetrahydroacridin-9-yl)amino)heptanoate (14b): Brown oil, 52.6% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.19 (d, J=8.8 Hz, 1H), 7.76 (s, 1H), 7.58 (t, J=8.0 Hz, 1H), .42~7.36 (m, 2H), 7.25 (t, J=8.0 Hz, 1H), 7.07 (d, J=8.0 Hz, 1H), 5.91~5.90 (m, 1H), 3.56~3.49 (m, 2H), 2.92 (t, J=6.4 Hz, 2H), 2.70 (t, J=6.4 Hz, 2H), 2.55~2.53 (m, 2H), 1.83~0.82 (m, 21H); 13C NMR (101 MHz, DMSO-d6) δ: 175.07, 173.77, 157.84, 157.24, 151.57, 146.02, 129.79, 128.95, 127.34, 123.87, 122.22, 120.04, 119.18, 115.68, 115.54, 109.89, 81.35, 48.24, 34.19, 33.59, 33.16, 30.83, 30.77, 28.77, 28.62, 26.50, 26.40, 25.42, 24.94, 24.78, 23.03, 22.61, 14.27. HRMS calcd for C32H38- N2O4 [M+H]+ 515.2910, found 515.2901.
1-Butyl-3-oxo-1, 3-dihydroisobenzofuran-5-yl-8-((1, 2, 3, 4- tetrahydroacridin-9-yl)amino)heptanoate (14c): Brown oil, 45.3% yield. 1H NMR (400 MHz, DMSO-d6) δ: 8.40 (d, J=8.4 Hz, 1H), 7.99 (d, J=8.4 Hz, 1H), 7.82 (t, J=7.6 Hz, 1H), 7.65 (s, H), 7.54 (t, J=8.0 Hz, 1H), 7.41 (t, J=8.0 Hz, 1H), 7.25 (t, J=8.0 Hz, 1H), 7.09 (d, J=8.0 Hz, 1H), 5.76~5.75 (m, 1H), 3.85~3.80 (m, 2H), 3.01 (s, 2H), 2.67 (s, 2H), 2.55 (t, J=7.2 Hz, H), 1.81~0.83 (m, 23H); 13C NMR (101 MHz, DMSO-d6) δ: 172.20, 169.56, 158.21, 150.91, 147.23, 129.93, 128.38, 127.08, 126.18, 124.30, 123.65, 123.55, 122.24, 120.65, 118.42, 116.18, 81.65, 48.34, 33.88, 30.99, 28.87, 26.98, 26.61, 25.93, 25.59, 25.08, 24.69, 24.53, 23.21, 22.90, 22.33, 14.26; HRMS calcd for C33H41N2O4 [M+H]+ 529.3066, found 529.3083.
A mixture of compound 12a or 12b (4.2 mmol) and piperazine (21.0 mmol) in pentanol (10 mL) was stirred at 140 ℃ under nitrogen for 12 h. After cooled, the mixture was diluted with water (20 mL) and extracted with CH2Cl2. The organic phase was dried (MgSO4), then filtered off, evaporated and purified by flash column chromatography (CH2Cl2/CH3OH, V:V=10:1) to obtain the title compounds 15a~15b.
9-(Piperazin-1-yl)-1, 2, 3, 4-tetrahydroacridine (15a): Yellow solid, 67.7% yield. m.p. 148.8~150.9 ℃ (lit.[26] m.p. 148.8~150.9 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.14 (d, J=8.4 Hz, 1H), 7.97 (d, J=8.4 Hz, 1H), 7.62~7.58 (m, 1H), 7.46~7.42 (m, 1H), 3.40~3.33 (m, 4H), 3.16~3.12 (m, 6H), 2.97 (t, J=6.4 Hz, 2H), 1.99~1.85 (m, 4H); 13C NMR (101 MHz, CDCl3) δ: 160.44, 152.96, 147.92, 128.83, 128.27, 127.69, 126.00, 124.92, 123.92, 51.59, 47.04, 34.13, 26.85, 23.04, 22.85; ESI-MS m/z: 268.20 [M+H]+.
6-Chloro-9-(piperazin-1-yl)-1, 2, 3, 4-tetrahydroacridine(15b): White solid, 65.4% yield. m.p. 119.3~121.1 ℃ (lit.[26] m.p. 119.3~121.11 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.17 (d, J=9.0 Hz, 1H), 7.85 (d, J=8.4 Hz, 1H), 7.48 (dd, J=9.0, 2.2 Hz), 3.7 (s, 4H), 3.01~2.98 (m, 2H), 2.93~2.87 (m, 6H), 1.87~1.79 (m, 4H); 13C NMR (101 MHz, CDCl3) δ: 161.94, 153.28, 148.22, 133.10, 127.88, 127.42, 126.63, 125.77, 124.40, 51.80, 46.92, 34.02, 26.64, 22.88, 22.60; ESI-MS m/z: 304.15 [M+H]+.
Compound 15a or 15b (3.4 mmol), Cs2CO3 (10.2 mmol) and KI (0.68 mmol) were dissolved in acetonitrile (15 mL). The solution was stirred for 1 h under argon at room temperature. Then, different dibromoalkanes (6.8 mmol) were added to the mixture. After stirred at 60 ℃ for 3 h, the mixture was filtered and evaporated under reduced pressure. The residue was washed successively with H2O, brine, dried over Na2SO4 and concentrated in vacuo to afford compounds 16a~16h as a yellow oil. The crude product was used directly in the next alkylation reaction without further purification.
A mixture of 6 (0.5 g, 2.6 mmol), Cs2CO3 (2.5 g, 7.7 mmol), compounds 16a~16h (0.9 g, 2.6 mmol) and KI (0.09 g, 0.5 mmol) was dissolved in acetonitrile and reflux stirred at 60 ℃ for 2 h. The mixture was filtered and evaporated to remove the solvent. The residue was purified by column chromatography (petroleum ether/ethyl acetate, V:V=5:1, plus 10 mL of triethylamine per 1000 mL) as a yellow oil, which was dissolved in acetone (5 mL), and then 5 drops of concentrated hydrochloric acid was added dropwise. The crystal precipitation was filtered, and the filter cake was dried to obtain a yellow solid.
3-Butyl-6-((6-(4-(1, 2, 3, 4-tetrahydroacridin-9-yl)pipera- zin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17a): Yellow solid, 78.0% yield. m.p. 207.9~210.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.25 (d, J=8.4 Hz, 1H), 8.20 (d, J=8.4 Hz, 1H), 7.99 (t, J=7.6 Hz, 1H), 7.77 (t, J=7.6 Hz, 1H), 7.58 (d, J=7.6 Hz, 1H), 7.35 (dd, J=8.4, 2.0 Hz, 1H), 7.29 (d, J=2.0 Hz, 1H), 5.57 (dd, J=7.6, 4.0 Hz, 1H), 4.14~4.06 (m, 4H), 3.78 (d, J=13.6 Hz, 2H), 3.61 (d, J=12.0 Hz, 2H), 3.30~3.17 (m, 6H), 2.89 (t, J=6.0 Hz, 2H), 2.09~1.22 (m, 18H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.30, 160.13, 160.00, 156.68, 142.91, 133.29, 127.85, 127.14, 125.98, 125.73, 124.08, 123.45, 123.38, 120.58, 108.20, 81.36, 68.50, 55.97, 51.58, 48.50, 34.12, 29.16, 28.73, 26.84, 26.32, 26.27, 25.49, 23.29, 22.35, 22.13, 20.64, 14.28; HRMS calcd for C35H46N3O3 [M+H]+ 556.3539, found 556.3523.
3-Butyl-6-((7-(4-(1, 2, 3, 4-tetrahydroacridin-9-yl)pipera- zin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17b): Yellow solid, 77.5% yield. m.p. 196.6~198.5 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.27 (d, J=8.4 Hz, 1H), 8.20 (d, J=8.8 Hz, 1H), 7.98 (t, J=7.6 Hz, 1H), 7.77 (t, J=7.6 Hz, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.35 (dd, J=8.4, 2.4 Hz, 1H), 7.28 (d, J=2.4 Hz, 1H), 5.56 (dd, J=7.6, 3.6 Hz, 1H), 4.13~4.08 (m, 4H), 3.78 (d, J=13.2 Hz, 2H), 3.61 (d, J=11.6 Hz, 2H), 3.38~3.19 (m, 6H), 2.89 (t, J=5.6 Hz, 2H), 2.09~1.23 (m, 20H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.32, 160.13, 159.98, 156.56, 142.88, 138.29, 133.27, 127.81, 127.09, 126.00, 125.59, 124.08, 123.40, 123.33, 120.45, 108.11, 81.37, 68.55, 55.98, 51.54, 48.52, 34.12, 29.10, 28.88, 28.72, 26.85, 26.60, 26.29, 25.74, 23.30, 22.36, 22.12, 20.62, 14.30; HRMS calcd for C36H48N3O3 [M+H]+ 570.3696, found 570.3683.
3-Butyl-6-((8-(4-(1, 2, 3, 4-tetrahydroacridin-9-yl)pipera- zin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17c): Yellow solid, 69.9% yield. m.p. 204.6~206.5 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.19 (d, J=8.4 Hz, 2H), 7.94 (t, J=8.0 Hz, 1H), 7.74 (t, J=8.0 Hz, 1H), 7.57 (d, J=8.8 Hz, 1H), 7.34 (dd, J=8.4, 2.4 Hz, H), 7.27 (d, J=2.4 Hz, 1H), 5.56 (dd, J=7.6, 4.0 Hz, 1H), 4.11~4.04 (m, 4H), 3.70 (d, J=13.6 Hz, 2H), 3.59 (d, J=12.0 Hz, 2H), 3.30~3.17 (m, 6H), 2.89 (t, J=6.0 Hz, 2H), 2.09~1.22 (m, 22H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.31, 160.00, 157.14, 142.87, 132.70, 127.54, 127.11, 126.01, 125.74, 124.07, 123.68, 123.40, 81.35, 68.59, 56.01, 51.64, 48.30, 34.12, 28.97, 28.95, 26.84, 26.58, 26.28, 23.32, 22.35, 20.92, 14.30; HRMS calcd for C37H50N3O3 [M+H]+ 584.3852, found 584.3840.
3-Butyl-6-((7-(4-(1, 2, 3, 4-tetrahydroacridin-9-yl)pipera- zin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17d): Yellow solid, 67.9% yield. m.p. 175.1~177.3 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.29 (d, J=8.4 Hz, 1H), 8.19 (d, J=8.4 Hz, 1H), 7.98 (t, J=7.6 Hz, 1H), 7.77 (t, J=7.6 Hz, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.34 (dd, J=8.4, 2.4 Hz, 1H), 7.26 (d, J=2.4 Hz, 1H), 5.56 (dd, J=7.6, 4.0 Hz, 1H), 4.15~4.04 (m, 4H), 3.79 (d, J=13.6 Hz, 2H), 3.61 (d, J=12.0 Hz, 2H), 3.28 (t, J=6.0 Hz, 2H), 3.36~3.17 (m, 6H), 2.89 (t, J=6.0 Hz, 2H), 2.09~1.24 (m, 24H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.31, 160.12, 159.98, 156.54, 142.84, 138.25, 133.27, 127.80, 127.07, 126.01, 125.57, 124.07, 123.38, 123.28, 120.40, 108.09, 81.36, 68.59, 56.00, 51.54, 48.54, 34.12, 32.50, 31.19, 29.25, 29.14, 28.98, 26.84, 26.62, 26.29, 25.92, 23.35, 22.36, 22.12, 20.61, 14.30; HRMS calcd for C38H52N3O3 [M+H]+ 598.4009, found 598.3986.
3-Butyl-6-((6-(4-(6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- piperazin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17e): Yellow solid, 52.2% yield. m.p. 165.2~168.1 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.33 (s, 1H), 8.21 (d, J=9.2 Hz, 1H), 7.76 (d, J=8.8 Hz, 1H), 7.58 (d, J=8.4 Hz, 1H), 7.36 (dd, J=8.4, 2.4 Hz, 1H), 7.29 (d, J=2.4 Hz, 1H), 5.57 (dd, J=7.6, 4.0 Hz, 1H), 4.13~4.06 (m, 4H), 3.77 (d, J=13.6 Hz, 2H), 3.61 (d, J=12.0 Hz, 2H), 3.43~3.35 (m, 2H), 3.27~3.18 (m, 4H), 2.88 (t, J=6.4 Hz, 2H), 2.09~1.24 (m, 18H), 0.87 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.30, 159.98, 157.71, 142.89, 139.36, 137.30, 128.41, 127.88, 127.10, 125.91, 124.07, 123.38, 122.06, 119.63, 108.13, 81.36, 68.48, 55.92, 51.49, 48.57, 34.12, 31.18, 28.73, 26.85, 26.32, 25.49, 23.29, 22.36, 22.12, 20.65, 14.30; HRMS calcd for C35H45ClN3O3 [M+H]+ 590.3149, found 590.3111.
3-Butyl-6-((7-(4-(6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- piperazin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17f): Yellow solid, 62.5% yield. m.p. 133.7~135.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.33 (s, 1H), 8.22 (d, J=9.2 Hz, 1H), 7.75 (d, J=8.8 Hz, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.36 (dd, J=8.4, 2.4 Hz, 1H), 7.27 (d, J=2.4 Hz, 1H), 5.57 (dd, J=7.6, 4.0 Hz, 1H), 4.12~4.06 (m, 4H), 3.76 (d, J=13.6 Hz, 2H), 3.60 (d, J=12.0 Hz, 2H), 3.43~3.35 (m, 2H), 3.26~3.18 (m, 4H), 2.87 (t, J=6.0 Hz, 2H), 2.10~1.25 (m, 20H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.31, 159.97, 157.75, 142.86, 139.49, 137.18, 128.36, 127.81, 127.07, 125.94, 124.07, 123.39, 122.06, 119.84, 108.09, 81.37, 68.55, 55.98, 51.51, 48.54, 34.12, 31.18, 28.88, 28.71, 26.85, 26.58, 25.74, 23.31, 22.36, 22.14, 20.70, 14.30; HRMS calcd for C36H47ClN3O3 [M+H]+ 604.3306, found 604.3280.
3-Butyl-6-((8-(4-(6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- piperazin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17g): Yellow solid, 52.7% yield. m.p. 159.8~161.2 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.33 (s, 1H), 8.21 (d, J=9.2 Hz, 1H), 7.76 (d, J=8.8 Hz, 1H), 7.57 (d, J=8.4 Hz, 1H), 7.34 (dd, J=8.4, 2.4 Hz, 1H), 7.27 (d, J=2.4 Hz, 1H), 5.56 (dd, J=7.6, 4.0 Hz, 1H), 4.13~4.06 (m, 4H), 3.79 (d, J=13.6 Hz, 2H), 3.60 (d, J=12.0 Hz, 2H), 3.42~3.35 (m, 2H), 3.27~3.18 (m, 4H), 2.87 (t, J=6.0 Hz, 2H), 2.09~1.22 (m, 22H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.30, 159.99, 157.70, 142.86, 139.40, 137.28, 128.41, 127.87, 127.09, 125.89, 124.06, 123.38, 122.04, 119.60, 108.12, 81.36, 68.59, 55.96, 51.48, 48.58, 34.13, 31.18, 29.50, 28.95, 26.85, 26.58, 26.32, 25.86, 23.31, 22.36, 22.12, 20.64, 14.30; HRMS calcd for C37H49ClN3O3 [M+H]+ 618.3462, found 618.3440.
3-Butyl-6-((9-(4-(6-chloro-1, 2, 3, 4-tetrahydroacridin-9-yl)- piperazin-1-yl)hexyl)oxy)isobenzofuran-1(3H)-one (17h): Yellow solid, 70.0% yield. m.p. 109.6~110.7 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.38 (s, 1H), 8.20 (d, J=8.0 Hz, 1H), 7.75 (d, J=7.6 Hz, 1H), 7.57 (d, J=7.6 Hz, 1H), 7.34 (d, J=6.8 Hz, 1H), 7.26 (s, 1H), 5.56 (dd, J=7.6, 4.0 Hz, 1H), 4.13~4.06 (m, 4H), 3.79 (d, J=13.6 Hz, 2H), 3.60 (d, J=12.0 Hz, 2H), 3.42~3.35 (m, 2H), 3.27~3.18 (m, 4H), 2.87 (t, J=6.0 Hz, 2H), 2.09~1.24 (m, 24H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 172.51, 162.20, 159.77, 145.05, 141.30, 139.63, 130.69, 130.13, 129.29, 128.03, 126.27, 125.58, 124.18, 121.64, 110.32, 83.57, 70.81, 58.21, 53.68, 50.84, 36.34, 31.58, 31.45, 31.35, 31.19, 29.05, 28.83, 28.53, 28.13, 25.56, 24.57, 24.31, 22.80, 16.50; HRMS calcd for C38H51ClN3O3 [M+H]+ 632.3619, found 632.3623.
Acetylcholinesterase (AChE, E.C.3.1.1.7, from human erythrocytes), butyrylcholinesterase (BuChE, E.C.3.1.1.8, from human serum), acetylthiocholine iodide (ATCI), S-butylthiocholine iodide (BTCI), donepezil and tacrine hydrochloride were purchased from Sigmae Aldrich (St. Louis, MO, USA), 5, 50-dithiobis-(2-nitrobenzoic acid) (Ellman's reagent, DTNB) was purchased from Beijing China-Ocean SciTec CoLtd. The inhibitory activities of test compounds 10a~10h, 14a~14c and 17a~17h was evaluated by Ellman's method. Stock solution of test compounds was dissolved in a minimum volume of DMSO (The volume fraction is 1%) and then diluted using the buffer solution (50 mmol•L-1 TriseHCl, pH=8.0, 0.1 mol•L-1 NaCl, 0.02 mol•L-1 MgCl2•6H2O). In each well of the plate, 50 μL of 0.01 MDTNB, 10 μL of AChE or 10 μL of BuChE were incubated with 50 μL of different concentrations of test compounds (1~1000 nmol•L-1) at 37 ℃ for 15 min, followed by addition of (50 μL) acetyl- thiocholine iodide (10 mmol•L-1) or S-butyrylthiocholine iodide (20 mmol•L-1), and the absorbance was measured at different time intervals (0, 60, 120, and 180 s) at wavelength of 412 nm. The concentration of compound producing 50% enzyme activity inhibition (IC50) was calculated by nonlinear regression analysis of the response concentration (log) curve, using the Graph-Pad Prism program package (GraphPad Software, San Diego, CA). Results are expressed as the mean±SEM of at least three different experiments performed in triplicate.
The molecular docking simulation was performed using ANTODOCK 4.2.6 imbedded into AutoDockTools-1.5.6. The active sites were generated from the co-crystallized ligands (Donepezil)[27, 28] within AChE protein structures (PDB codes: 4EY7), respectively. The active site of BChE (PDB codes: 1P0I) was determained according to the report by Edgar Sawatzky energy minimized.[29] Selected compounds 10b and 10g were sketched and energy minimized using Chem3D 16.0, and they were docked into the above prepared proteins active sites. Polar hydrogens were added to all ligands and proteins with the AutoDock Tools (ADT) program prior to docking with Autodock 4.2.6 program.[30] A grid box 4.0 nm in size was centered on the active site of protein. The number of operations in the Genetic Algorithm is set to 100, and other parameters remain the default. All graphical representations in Figure 3 were rendered using PyMOL.[31]
Brain cell cultures were obtained from the cerebral hippocampi of newborn male Sprague-Dawley rats (from Beijing Vital River Laboratory Animal Technology Co., Ltd.) within 24 h. Hippocampal neurons were removed from the brain, and then were dissected in ice-cold D-Hank's medium. They were digested with trypsin at 37 ℃, dissociated by pipetting repeatedly, filtered through a 200 mesh screen. Then the filtrate was centrifuged (800 r/min, 10 min), the supernatant was discarded, and the appropriate amount of the culture solution was suspended and precipitated in a 6-well plastic culture plate. Then cells incubated with 5% CO2 at 37 ℃.
Cells, which have grown for 7d, were selected to establish the Aβ induction model. The cells were plated in 12-well plates at a concentration of 4×104 cells/well. After 24 h of routine culture, the medium was changed to a complete DMEM (Gibco, Grand Island, NY, USA) medium containing Aβ1-42 (2 μmol•L-1), and cells incubated with 5% CO2 at 37 ℃ for 48 h. The induction process of model group was completed, and then the drug-adminis- tered group was changed to a complete DMEM medium containing Aβ1-42 (2 μmol•L-1) and a corresponding concentration of the compound, cells incubated with 5% CO2 at 37 ℃ for 48 h. However, the control group was incubated with conventional complete DMEM medium and incubated with 5% CO2 at 37 ℃ for 48 h.
Intracellular reactive oxygen species (ROS) generation was estimated by flow cytometry (FCM) using DCFH-DA fluorescent probe. Cells were washed three times with PBS and DCFH-DA, diluted to a final concentration of 10 μmol•L-1, incubated at 37 ℃ for 30 min in the dark, and subsequently cells were washed twice with PBS. They were digested with trypsin, the medium was added to terminate the digestion, the cells were centrifuged at 1000 r/min for 5 min to collect the cells, washed twice with PBS, and the cell pellet was collected by centrifugation. The collected cells were resuspended in PBS to make a suspension of 1×106 cells/mL. Intracellular ROS generation was estimated by FCM.
Supporting Information The 1H NMR, 13C NMR and HRMS spectra of compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
Roberson, E. D.; Mucke, L. Science 2006, 314, 781. doi: 10.1126/science.1132813
Ulep, M. G.; Saraon, S. K.; Mclea, S. J. Nurse Pract. 2018, 14, 129. doi: 10.1016/j.nurpra.2017.10.014
Sun, Y.; Chen, J.; Chen, X.; Huang, L.; Li, X. S. Bioorg. Med. Chem. 2013, 21, 7406. doi: 10.1016/j.bmc.2013.09.050
Ulus, R.; Zengin, K. B.; Gazioğlu, I.; Kaya, M. Bioorg. Chem 2017, 70, 245. doi: 10.1016/j.bioorg.2017.01.005
Xie, Q.; Wang, H.; Xia, Z.; Liu, M. Y.; Zhang, W. W.; Wang, X. H.; Fu, W. J. Med. Chem. 2008, 51, 2027. doi: 10.1021/jm070154q
Bolognesi, M.; Minarini, A.; Rosini, M.; Melchiorre, C. Mini-Rev. Med. Chem. 2008, 8, 960. doi: 10.2174/138955708785740652
Sussman, J. L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Science 1991, 253, 872. doi: 10.1126/science.1678899
Chen, Y.; Sun, J.; Fang, L.; Liu, M.; Peng, S.; Liao, H.; Lehmann, J. J. Med. Chem. 2012, 55, 4309. doi: 10.1021/jm300106z
Fernández-Bachiller, M.; Pérez, C.; Monjas, L.; Rademann, J.; Rodríguez-Franco, M. I. J. Med. Chem. 2012, 55, 1303. doi: 10.1021/jm201460y
Roger, M. L; Steven, G. P.; Albert, E. INT. J. Neuropsychoph. 2006, 9, 101.
Shanks, M.; Kivipelto, M.; Bullock, R.; Lane, R. Curr. Med. Res. Opin. 2009, 25, 2439. doi: 10.1185/03007990903209332
Hardy, J.; Selkoe, D. J. Science 2002, 297, 353. doi: 10.1126/science.1072994
Luo, Z.; Sheng, J.; Sun, Y.; Lu, C. J.; Yan, J.; Liu, A. Q.; Luo, H. B.; Huang, L. J. Med. Chem. 2013, 56, 9089. doi: 10.1021/jm401047q
Jarrott, B. Pharmacol. Res. 2017, 116, 29 doi: 10.1016/j.phrs.2016.12.033
Xie, S. S.; Wang, X. B.; Li, J. Y.; Yang, L.; Kong, L. Y. Eur. J. Med. Chem. 2013, 64, 540. doi: 10.1016/j.ejmech.2013.03.051
Li, S. Y.; Wang, X. B.; Xie, S. S.; Jiang, N.; Wang, K. D. G.; Yao, H. Q.; Sun, H. B. Eur. J. Med. Chem. 2013, 69, 632. doi: 10.1016/j.ejmech.2013.09.024
Zhang, P.; Guo, Z. F.; Xu, Y. M.; Li, Y. S.; Song, J. G. Biomed. Pharmacother. 2016, 83, 658. doi: 10.1016/j.biopha.2016.07.040
Sun, B.; Feng, M.; Tian, X.; Liu, X. W.; Zhang, Y. Y.; Ke, X. J.; Huang, S. S. Neurosci. Lett. 2012, 516; 247. doi: 10.1016/j.neulet.2012.04.003
Peng, Y.; Hu, Y.; Xu, S.; Li, P.; Li, J.; Lu, L.; Yang, H. J. Alzheimers. Dis. 2012, 29, 379. doi: 10.3233/JAD-2011-111577
Carlier, P. R.; Han, Y. F.; Chow, E. S. H.; Li, C. P-L.; Wang, H.; Lieu, T. X.; Wong, H. S. Bioorg. Med. Chem. 1999, 7, 351. http://d.old.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ028219189/
Ellaman, G. Biochem. Pharmacol. 1961, 7, 88. doi: 10.1016/0006-2952(61)90145-9
Lawler, J. M.; Song, W.; Demaree, S. R. Free. Radic. Biol. Med. 2003, 35, 9. doi: 10.1016/S0891-5849(03)00186-2
Wang, X.; Wang, L.; Huang, Z.; Huang, Z. J.; Sheng, X.; Li, T. T.; Ji, H. Bioorg. Med. Chem. Lett. 2013, 23, 198. doi: 10.1016/j.bmcl.2012.10.115
McKenna, M. T.; Proctor, G. R.; Young, L. C.; Harvey, A. L. J. Med. Chem. 1997, 40, 3516. doi: 10.1021/jm970150t
María, I. R.; María, I. F.-B.; Concepción, P. J. Med. Chem. 2006, 49, 459. doi: 10.1021/jm050746d
Li, X. K.; Wang, H.; Xu, Y. X.; Liu, W. W.; Gong, Q.; Wang, W.; Qiu, X. X. ACS Chem. Neurosci. 2017, 8, 2708. doi: 10.1021/acschemneuro.7b00259
Kulmacz, R. J.; Lands, W. E. M. Prostaglandins 1983, 25, 531. doi: 10.1016/0090-6980(83)90025-4
Kurumbail, R. G.; Stevens, A. M.; Gierse, J. K.; McDonald, J. J.; Stegeman, R. A.; Pak, J. A.; Gildehaus, D.; Iyashiro, J. M. Nature 1996, 384, 644. doi: 10.1038/384644a0
Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Sotriffer, C. A.; Heilmann, J.; Decker, M. J. Med. Chem. 2016, 59, 2067 doi: 10.1021/acs.jmedchem.5b01674
Morris, G. M.; Huey, R.; Olson, A. J. Curr. Protoc. Bioinformatics. 2008, 8, 8.
Lill, M. A.; Danielson, M. L. J. Comput.-Aided Mol. Des. 2011, 25, 13. doi: 10.1007/s10822-010-9395-8

Scheme 1 Synthetic route of target compounds
Reagents and conditions: (ⅰ) (1) SOCl2, toluene, 80 ℃; (2) diethylamine, DCM, 0 ℃; (ⅱ) (1) CH3(CH2)3Br, Mg, I2, THF, 60 ℃; (2) Grignard reagent, THF, -20 ℃; (ⅲ) p-TsOH•HCl, toluene, 80 ℃; (ⅳ) KNO3, H2SO4, r.t.; (ⅴ) Fe, AcOH, THF, H2O, 70 ℃; (ⅵ) (1) NaNO2, 30% H2SO4, urea, 0 ℃; (2) Cu(NO3)2•3H2O, Cu2O; (ⅷ) cyclohexanone, ZnCl2, 100 ℃; (ⅷ) different dibromo-alkanes, Cs2CO3, KI, CH3CN, 55 ℃; (ⅸ) Compound 6, Cs2CO3, KI, CH3CN, 60 ℃; (ⅹ) cyclohexanone, POCl3, 100 ℃; (ⅹⅰ) different amino-alkanoic acid, phenol, KI, 180 ℃; (ⅹⅱ) Compound 6, DCC, DMAP, r.t.; (ⅹⅲ) piperazine, 1-pentanol, N2, 140 ℃; (ⅹⅳ) (1) different dibromoalkanes, Cs2CO3, KI, CH3CN, 60 ℃ (2) CH3COCH3, HCl (conc.)

Figure 3 (A) 3D docking model of compound 10b with AChE, and (B) 3D docking model of compound 10g with BuChE
Hydrogen bonds are shown by red dashed lines and π-π stacking interactions are shown by yellow dashed lines

Figure 4 Effect of different compounds on Aβ1-42 inducing ROS produced in rat hippocampal neurons
Rat hippocampal neurons were treated with different concentrations of compounds in the presence or absence of Aβ1-42 (2 µmol•L-1). After 48 h incubation, the ROS levels were measured with DCFH-DA probe. Three independent experiments, for each condition, were performed in triplicate. The abscissa indicates the value of the relative intensity of the scattered light signal (unit: channel), and the ordinate relative to the number of cells (unit: Zahl)
Table 1. The AChE and BuChE inhibitory activities of tacrine, donepezil and compounds.
| Compd. | R | R1 | n | IC50a/(nmol•L-1) | SId | |
| hAchEb | hBchEc | |||||
| 10a | H | — | 4 | 247.30±25.17 | 6109±378 | 24.70 |
| 10b | H | — | 5 | 38.65±3.91 | 5227±461 | 135.24 |
| 10c | H | — | 6 | 156.20±13.46 | 354±36 | 2.27 |
| 10d | H | — | 7 | 342.70±32.43 | 4639±397 | 13.54 |
| 10e | Cl | — | 4 | 549.80±55.39 | 51.31±5.09 | 0.09 |
| 10f | Cl | — | 5 | 659.30±67.27 | 85.18±7.28 | 0.13 |
| 10g | Cl | — | 6 | 363.40±33.56 | 33.69±3.49 | 0.09 |
| 10h | Cl | — | 7 | 174.20±15.64 | 165.5±15.6 | 0.95 |
| 14a | — | H | 4 | 45.27±3.57 | 5074±283 | 112.08 |
| 14b | — | H | 5 | 144.60±10.21 | 358±18.45 | 2.48 |
| 14c | — | H | 6 | 330.00±18.45 | 4708±256 | 14.27 |
| 17a | — | H | 4 | 195.00±14.64 | 766±38 | 3.93 |
| 17b | — | H | 5 | 441.00±521.47 | 2905±164 | 6.59 |
| 17c | — | H | 6 | 244.50±14.41 | 3947±206 | 16.14 |
| 17d | — | H | 7 | 72.77±4.17 | 6410±394 | 88.09 |
| 17e | — | Cl | 4 | 148.90±8.24 | 446.8±276 | 3.00 |
| 17f | — | Cl | 5 | 268.60±14.27 | 87.48±6.49 | 0.33 |
| 17g | — | Cl | 6 | 44.81±2.49 | 5867±312 | 130.93 |
| 17h | — | Cl | 7 | 270.20±16.14 | 7908±578 | 29.27 |
| Donepezil | — | — | — | 7.11±0.46 | 7530±474 | 1059.07 |
| Tacrine | — | — | — | 200.70±11.21 | 27.12±1.85 | 0.14 |
| a IC50 values represent the concentration of inhibitor required to decrease enzyme activity by 50% and are the mean of five independent experiments, each performed in triplicate (SD=standard deviation). b From human erythrocytes. c BuChE from human serum. d AChE selectivity index=IC50(hBuChE)/IC50(hAChE) | ||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Effect of different concentrations compounds on Aβ1-42 inducing ROS produced in rat hippocampal neuronsa
| Compd. | ROS levels (folds of control) | ||||
| Control | Model | 1 nmol•L-1 | 10 nmol•L-1 | 100 nmol•L-1 | |
| NBP | 1±0.225 | 3.397±0.265 | 2.547±0.463 | 2.437±0.065* | 1.503±0.343** |
| Tacrine | 1±0.293 | 3.183±0.245 | 2.468±0.419 | 1.628±0.161** | 1.270±0.170** |
| 10b | 1±0.539 | 3.676±0.405 | 2.758±0.655 | 2.550±0.276 | 1.675±0.194** |
| 10g | 1±0.320 | 3.105±0.233 | 2.490±0.352 | 2.169±0.201* | 1.433±0.174** |
| 14a | 1±0.232 | 3.357±0.377 | 2.637±0.178 | 2.553±0.482 | 2.474±0.287 |
| 17d | 1±0.569 | 3.544±0.264 | 2.779±0.104 | 2.190±0.300** | 1.096±0.144** |
| 17g | 1±0.592 | 2.839±0.081 | 2.892±0.340 | 2.684±0.481 | 2.455±0.380 |
| a Compared with model *p < 0.05, **p < 0.01. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们