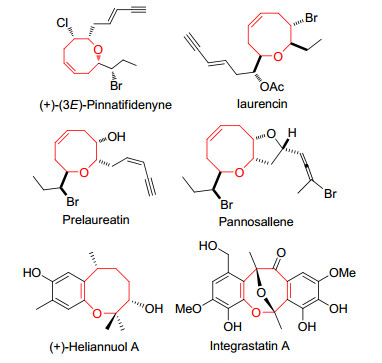
图 1.
具有八元环醚结构的天然产物分子和药物活性分子
Figure 1.
Representative examples of eight-membered ethers possessing biological activities

具有八元环醚结构的有机化合物在有机合成化学、生物化学和药物开发研究领域具有广泛应用[1], 在已经分离得到的天然产物, 特别是海洋天然产物中往往存在八元环醚骨架结构.例如: (+)-(3E)-Pinnatifidenyne[2], Iaurencin[3], Prelaureatin[4]和Pannosallene[5]就是海洋中红藻的代谢产物, (+)-Heliannuol A[6]是一种天然的倍半萜类化合物, 具有优越的抗病毒药物活性, Integrastatin A[7]具有抑制HIV-1整合酶的活性, 目前已经上市作为艾滋病治疗药物(图 1).正是基于八元环醚化合物所具有的生物和药物活性, 发展方便高效合成八元环醚的有效方法越来越引起有机化学家的研究兴趣[8].本文综述近年来出现的合成八元环醚化合物的最新研究进展.
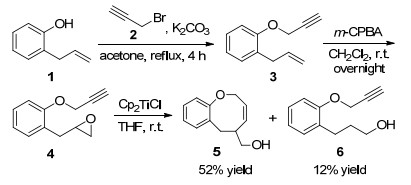
过渡金属催化是有机合成化学常见的反应形式, 因为过渡金属存在d轨道电子或者具有空的d轨道, 容易失去电子或夺取电子, 具有较强的氧化还原能力, 因此不同种类的过渡金属催化剂在有机合成中得到广泛应用. 2006年Roy课题组[9]报道了Cp2TiCl促进的8-endo自由基环加成反应构建八元环醚化合物的新方法, 反应首先利用2-烯丙基苯酚(1)和炔丙基溴(2)为原料, 以丙酮为溶剂在碳酸钾作用下回流4 h, 以93%的收率得到炔丙基醚3.炔丙基醚3随后在m-CPBA作用下以93%的收率得到环氧化合物4, 然后环氧化合物4在Cp2TiCl促进下以52%的收率得到八元环醚化合物5和少量环氧化合物开环产物6.该研究工作的亮点是以Cp2TiCl作为自由基来源, 通过环氧化合物开环-环化反应构建八元环醚类化合物(Scheme 1).
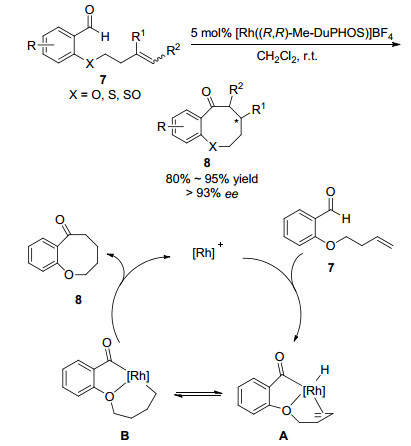
2009年Dong课题组[10]以(R,R)-Me-DuPHOS作为手性配体, 在手性金属铑催化剂催化下, 以二氯甲烷为溶剂室温下实现邻甲酰基苯基丁烯醚7发生分子内烯烃-氢酰基化反应, 以高收率和高对映选择性得到苯并八元环醚化合物8.该方法通过对分子7中杂原子的改变, 可以合成八元环硫醚和八元亚砜化合物.
反应机理研究表明, 邻甲酰基苯基丁烯醚7在金属铑催化下首先发生C—H键活化生成金属中间体A, 然后A对端基烯烃发生插入反应得到中间体B, 最后通过还原-消除反应并伴随着催化剂的离去得到最终产物8.利用过渡金属催化实现大环化合物对映选择性合成的报道较少, 该工作首次报道了通过手性铑催化剂催化分子内氢酰基化反应, 对映选择性合成八元环酮醚化合物8 (93% ee).该方法反应条件温和, 符合原子经济性原则, 为不对称合成八元环醚化合物提供了新的合成策略(Scheme 2).
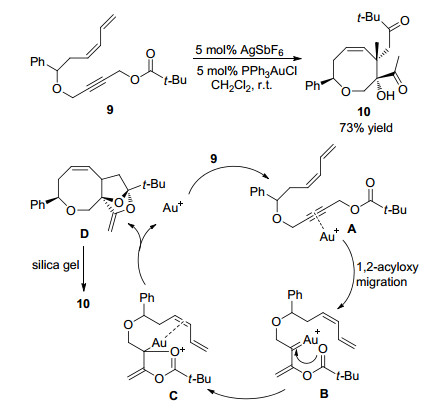
2014年, 厍学功课题组[11]报道了在5 mol%金和5 mol%六氟锑酸银(AgSbF6)存在条件下, 以烯炔脂9为反应底物, 室温下反应2 h, 最终以73%收率得到多取代八元环醚化合物10.反应机理研究表明, 首先金催化剂活化烯炔脂9中的碳-碳叁键形成金属中间体A, 然后发生1,2-酰基迁移生成金卡宾中间体B.伴随着羰基氧的亲核进攻生成1,3-偶极中间体C, 然后通过分子内[3+2]环加成串联反应得到缩酮中间体D, 同时释放金催化剂.中间体D在硅胶上发生水解反应, 得到最终产物10.该反应的特点是多环结构中间体D不稳定, 接触硅胶后发生开环反应得到八元环醚化合物.由于反应底物烯炔脂9分子内存在大体积的叔丁基和苯基取代基, 利用空间位阻效应可以有效控制该反应的非对映选择性(Scheme 3).
2017年, Lloyd-Jones课题组[12]报道了在金催化下利用分子内交叉偶联反应, 高效构建八元环醚类化合物的研究成果.芳基-三甲基硅烷11在4 mol% thtAuBr3催化下以CHCl3/MeOH (V:V=50:1, 0.1 mol/L)为溶剂, PhI(OAc)2和樟脑磺酸(CSA)为添加剂, 在室温下反应1 h, 即以75%的收率得到含有八元环醚骨架的化合物12.实验结果显示, 反应底物对产物结构的影响非常明显, 通过调节连接两个芳基的醚键结构, 可以得到五元-九元环不同结构的醚类化合物, 为合成联苯并环醚化合物特别是八元环醚化合物提供了新的合成策略(Scheme 4).
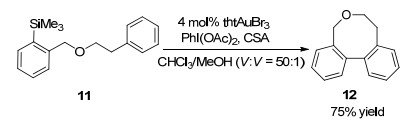
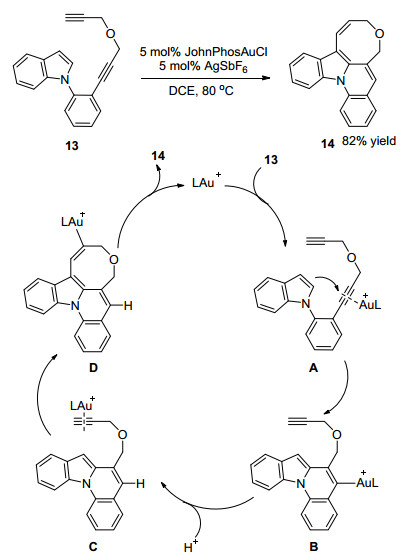
2018年施敏课题组[13]报道了以包含双炔的吲哚衍生物13为底物, 在5 mol% JohnPhosAu(Ⅰ)Cl和5 mol% AgSbF6为催化剂条件下, 发生分子内氢氨化反应构建八元环醚并吲哚骨架化合物的合成方法.该反应的机理是金正离子首先活化双炔衍生物13得到中间体A, 然后吲哚C-2位发生分子内的亲核加成反应得到中间体B.金催化剂继续活化分子内的端炔得到中间体C, 然后经历吲哚C-3的第二次亲核加成反应得到中间体D, 最终脱除金催化剂得到预期产物14.研究发现化合物14具备独特的荧光发射能力, 该化合物几乎不发射254~400 nm的荧光, 但是在400~580 nm可见光范围内可以发射绿色荧光, 表明该类化合物在有机发光二极管研究领域具有应用前景(Scheme 5).
一些八元环醚类化合物可以通过小环的环扩张重排反应进行合成, 利用环扩张反应制备八元环醚化合物是一类典型的分子内重排反应.反应在加热或者路易斯酸催化条件下, 小环分子首先生成一个稳定的过渡态或活性中间体, 然后发生[3, 3]和[1, 1]单键迁移重排反应生成更稳定的八元环醚类化合物.
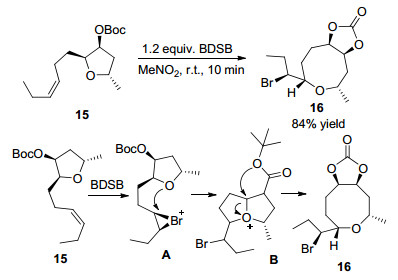
2011年Snyder课题组[14]报道了四氢呋喃衍生物15, 在1.2 equiv.的Et2SBr·SbBrCl5 (BDSB)催化下, 以MeNO2为溶剂在室温条件下反应10 min, 以84%的收率得到八元环醚类化合物16.反应机理研究表明, 四氢呋喃衍生物15首先在BDSB的作用下生成溴正离子中间体A, 然后氧原子进攻溴正离子A得到中间体B, 最终在酯羰基的进攻下再次发生开环反应得到产物16.该方法反应时间短, 具有高区域选择性和立体选择性, 反应合成了一系列含八元环醚骨架天然产物Laurencia的系列衍生物, 为天然产物Laurencia的全合成以及中尺寸环状化合物的立体选择性合成提供了有益借鉴(Scheme 6).
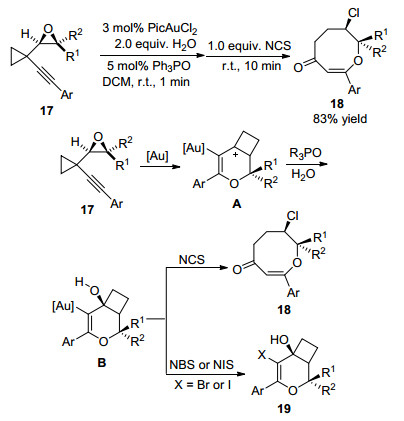
2011年, 刘瑞雄课题组[15]报道了利用开环反应合成八元环醚化合物的新方法.反应以苯乙炔基-氧杂环丙烷双取代环丙烷17为反应底物, 在金催化下以1 equiv. N-氯代丁二酰亚胺(NCS)和2 equiv.水为添加剂, 二氯甲烷为溶剂在室温下反应10 min, 以83%的收率得到含有八元环醚结构的化合物18.该反应的机理是苯乙炔基-氧杂环丙烷双取代环丙烷17在金催化剂和水的共同作用下生成烯基金中间体A, 进一步在三烷基膦酸酯(R3PO)和水的作用下生成含羟基的二氢吡喃并环丁烷中间体B.由于R3PO和体系中氢质子的存在, 使得中间体B羟基部分的氧氢键发生电子转移并促使断裂C—C键, 同时进攻NCS成功捕获Cl+离子, 伴随着环丁烷开环从而得到八元环醚18.作者提出阳离子中间体A水解过程中产生的氢质子有助于NCS解离出Cl+离子, 进而生成目标化合物18.当将NCS换成NBS或NIS时, 由于Br+和I+离子对金有着更强的结合作用, 直接发生取代反应生成化合物19 (Scheme 7).
烷基苯基醚在高温下是很稳定的化合物, 但是Claisen发现烯丙基-芳基醚在高温(200 ℃)下可以重排为邻-烯丙基苯酚, 后人称该反应为Claisen重排反应.邻-烯丙基苯酚还可以再进一步发生Claisen重排反应, 得到对-烯丙基苯酚. Claisen重排反应的机理是σ[3, 3]重排, 中间产物烯酮不稳定(无芳香性), 会互变异构为有芳香性的酚.这个反应的特点是高度的区域选择性, 反应的驱动力是生成热力学稳定的取代程度最大的“烯烃”[16]. Claisen重排反应起初是在芳香化合物中发现并得到应用, 后来发现该反应可以应用到非芳香化合物, 在醚类化合物中如果存在烯丙氧基与碳-碳双键相连的结构, 就有可能发生Claisen重排反应, 这种拓展使得Claisen重排反应在有机合成领域得到广泛应用(Scheme 8).
1993年Boeckman课题组[17]首次运用Claisen重排反应制备八元环醚类化合物, 反应以1,1-二羟甲基-2-烯丙基环丁烷20为反应底物, 在Dess-Martin氧化剂存在条件下以二氯甲烷作为溶剂室温条件下发生氧化反应, 通过Claisen重排反应以88%的收率得到八元环醚化合物21 (Scheme 9).该反应巧妙应用Claisen重排反应制备八元环醚类化合物, 通过调节反应底物的分子结构可以高效合成不同结构的七元和八元环醚化合物.

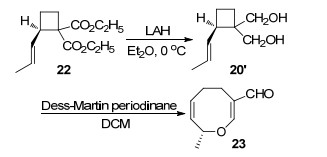
1997年, Boeckman课题组[18]在前期利用Claisen重排反应成功合成八元环醚类化合物的基础上, 再次利用Claisen重排反应合成出光学纯的手性八元环醚类化合物.作者以光学纯手性1,1-二羧酸乙酯-2-丙烯基环丁烷(22)为反应底物, 乙醚为溶剂, 0 ℃条件下利用四氢铝锂(LAH)还原22得到手性1,1-二羟甲基-2-烯丙基环丁烷(20'), 化合物20’进一步被3 equiv. Dess-Martin试剂氧化, 在二氯甲烷溶剂中发生Claisen重排反应得到具有光学活性手性八元环醚类化合物23 (Scheme 10).
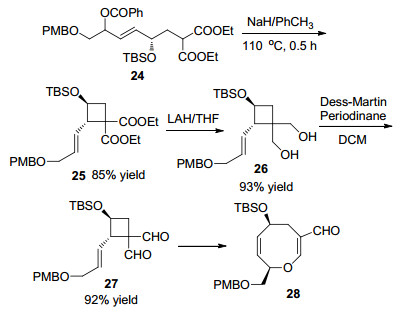
2002年, Boeckman课题组[19]利用不对称合成策略合成天然产物(+)-Laurenyne, 产物中关键的八元环醚结构通过立体选择性SN2'环化反应和Claisen重排反应进行构筑.反应以2, 6-二甲基苯基碳酸脂24为底物, 碱性条件下以甲苯为溶剂110 ℃反应0.5 h, 以85%的收率得到单一构型的多取代烯丙基环丁烷25. 1,1-双酯基取代环丁烷25在四氢铝锂(LAH)作用下发生还原反应, 以93%的收率得到1,1-双醇环丁烷化合物26, 进一步在Dess-Martin试剂的氧化下以92%的收率得到1,1-双醛取代环丁烷27.在45 ℃条件下化合物27发生Claisen重排反应得到热力学更加稳定的八元环醚化合物28 (Scheme 11).
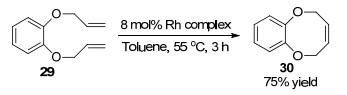
从非环前体出发合成八元环醚化合物, 由于存在较强的环张力和跨环排斥的原因往往造成关环困难[20].当底物存在构象约束不易形成八元环醚化合物时, 利用烯烃换位反应实现关环目标是一个很好的选择. 1995年, Grubbs课题组[21]报道了以邻苯二烯丙基醚29为底物, 发生烯烃换位反应合成苯并八元环醚类化合物的新方法.该反应以8 mol%金属铑卡宾为催化剂, 以甲苯为溶剂在55 ℃条件下反应3 h, 以75%的收率得到含苯并八元环醚结构的化合物30.以往文献报道的烯烃换位反应只能合成五到七元环状化合物, Grubbs通过对非环状反应底物的改造成功实现八元环醚的高效合成(Scheme 12).
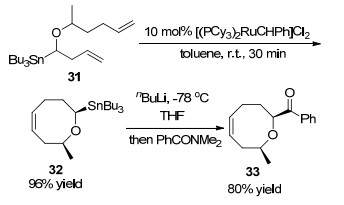
1997年, Linderman课题组[22]报道了开链三丁基锡取代双烯化合物31发生烯烃换位反应合成八元环醚化合物的研究成果.反应以三丁基锡取代双烯化合物31为反应底物, 在10 mol%金属铑卡宾催化下, 以甲苯为溶剂, 室温条件下反应12 h, 以96%的收率得到三丁基锡取代八元环醚化合物32.为了探讨三丁基锡取代八元环醚化合物的应用范围, 在-78 ℃条件下以四氢呋喃(THF)为溶剂与正丁基锂发生转金属反应, 然后与N,N-二甲基苯甲酰胺反应以80%收率得到苯甲酰基取代八元环醚化合物33 (Scheme 13).
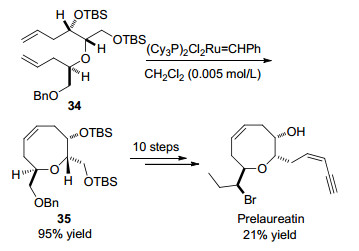
2000年, Crimmins课题组[23]在研究天然产物(+)-Prelaureatin的手性不对称全合成过程中, 利用烯烃换位反应成功构建出目标分子中关键的八元环醚骨架. TBS基团保护的双烯化合物34在Grubbs金属铑催化剂作用下, 选择二氯甲烷为溶剂在室温条件下反应2 h, 以95%的收率得到八元环醚化合物35, 进一步经过10步化学转化以21%的收率得到天然产物(+)-Prelaureatin (Scheme 14).
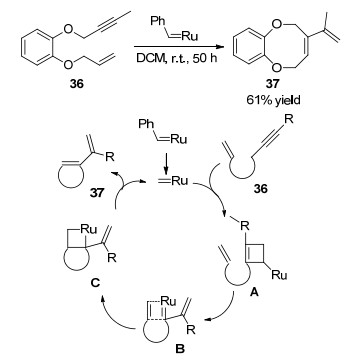
2000年, Mori课题组[24]报道了一种新颖的利用烯烃换位反应合成八元环醚的方法.邻苯二酚衍生的烯炔化合物36在10 mol%金属铑催化剂作用下, 在二氯甲烷溶剂中室温条件下反应过夜, 以61%的收率得到含环外双键的八元环双醚类化合物37.研究反应的机理表明烯炔化合物36在金属铑卡宾催化下, 与炔烃发生[2+2]环加成反应得到环状铑中间体A, 中间体A快速转变成烯基卡宾中间体B, 然后发生分子内[2+2]环加成反应得到环铑中间体C.中间体C通过两次化学键断裂得到双烯八元环双醚化合物37和铑卡宾化合物.该反应提供了一种高效合成八元环双醚化合物的新方法, 因为产物中含有双醚结构, 拓展了八元环双醚的骨架种类(Scheme 15).
2006年, Martin课题组[25]报道了通过烯烃换位反应高立体选择性合成八元环醚化合物的新方法.该反应以炔丙醇38和烯丙基醇39为反应底物, 通过加入Co2(CO)6形成烯丙基醚中间体40, 进一步在Grubbs催化剂催化下以二氯甲烷为溶剂35 ℃反应12 h得到Co2(CO)6环醚中间体41, 最终脱掉Co2(CO)6以83%的收率和高非对映选择性(d.r.>17:1)得到双烷基取代的八元环醚类化合物42 (Scheme 16).该方法操作简单, 合成步骤少, 每一步反应收率高立体选择性好, 是立体选择性合成八元环醚化合物的有效方法.
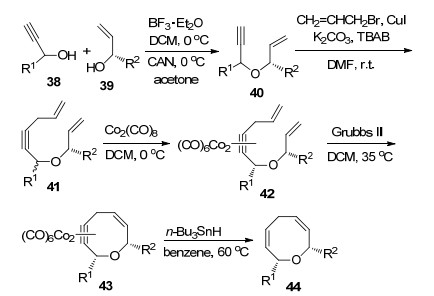
2006年, Martin课题组[25]报道了通过烯烃复分解关环反应高立体选择性合成八元环醚化合物的新方法.该反应以炔丙醇38和烯丙基醇39为反应底物首先形成烯丙基醚中间体40, 进一步在碘化亚铜催化下发生炔烃烷基化反应得到双烯炔中间体41, 41与Co2(CO)8反应得到含Co2(CO)6的中间体42, 42在Grubbs催化剂催化下以二氯甲烷为溶剂35 ℃反应12 h得到Co2(CO)6环醚中间体43, 最终脱掉Co2(CO)6以83%的收率和高非对映选择性(d.r.>17:1)得到双烷基取代八元环醚化合物44 (Scheme 16).该方法操作简单合成步骤少, 每一步反应收率高立体选择性好, 是立体选择性合成八元环醚化合物的有效方法.
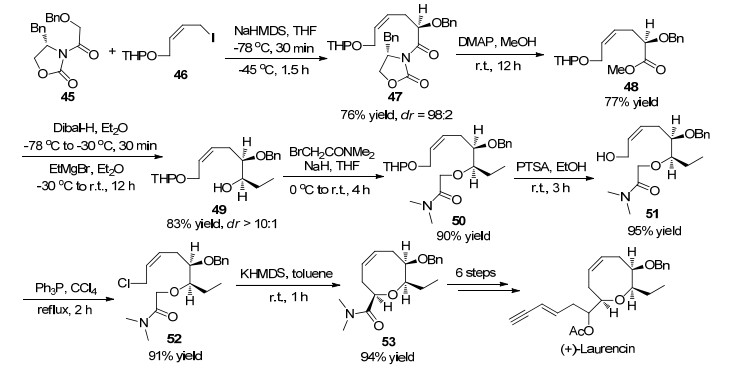
2005年, Kim课题组[26]以噁唑烷酮45为原料首先在NaHMDS存在条件下以四氢呋喃为溶剂-78 ℃反应30 min, 然后加入THP保护的1-碘-2-丁烯醇46, 在-45 ℃条件下继续反应1.5 h, 以76%的收率和98:2的dr值得到化合物47.接着在4-二甲氨基吡啶(DMAP)作用下以甲醇为溶剂室温反应12 h, 以77%收率得到羧酸甲酯化合物48.羧酸甲酯化合物48在乙醚溶剂中-78 ℃条件下加入还原剂DIBAL-H, -30 ℃反应30 min, 然后加入乙基格氏试剂, 以83%的收率和大于10:1的dr值得到化合物49.
化合物49在NaH催化下以四氢呋喃为溶剂在0 ℃到室温条件下反应4 h, 以90%的收率得到氧烷基化产物50.化合物50在对甲苯磺酸(PTSA)条件下以乙醇为溶剂室温条件下反应3 h, 以95%的收率得到脱除THP保护基的醇51.化合物51在三苯基膦催化下以四氯化碳为溶剂, 加热回流2 h以91%的收率得到氯化产物52.最后一步反应是合成八元环醚的关键步骤, 在六甲基二硅基胺基钾(KHMDS)催化下以甲苯为溶剂, 在室温条件下反应1 h, 以高达94%的收率得到产物53.化合物53进一步经过六步化学转化得到天然产物(+)-Laurencin.该方法合成步骤较长, 但是每一步反应操作简单, 收率高, 立体选择性好, 是不对称合成天然产物(+)-Laurencin的有效方法(Scheme 17).
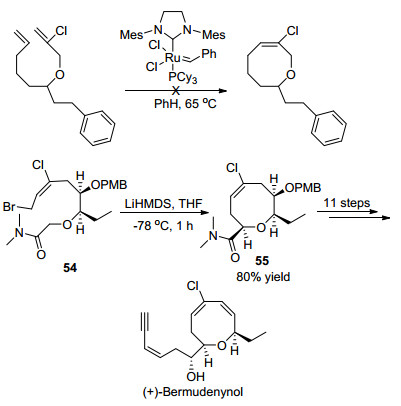
2014年, Kim课题组[27]报道了不对称全合成天然产物(+)-Bermudenynol的新方法. (+)-Bermudenynol中八元环醚骨架中包括氯代烯烃结构, 由于烯烃上有氯原子取代, 按照之前的烯烃换位反应构建八元环醚骨架策略, 不能够实现(+)-Bermudenynol的合成.为了解决上述问题, Kim课题组采用分子内酰胺烯醇烷基化策略, 利用溴取代的内酰胺54为反应底物, 以LiHMDS为催化剂, 选择四氢呋喃为溶剂在-78 ℃条件下反应1 h, 以80%的收率得到含烯基氯结构的八元环醚化合物55, 进一步经过多步化学转化, 成功合成出(+)-Bermudenynol化合物(Scheme 18).
近年来利用有机小分子催化分子间串联环化反应合成八元环醚化合物得到发展.有机小分子催化剂作为继酶催化和金属催化之后的第三类催化剂, 在有机合成研究领域得到广泛应用.有机小分子催化剂和传统的有机金属络合物催化剂相比, 具有反应条件温和、环境友好、易于回收利用等优点, 符合绿色化学的要求.目前, 通过有机小分子催化串联环加成反应构建八元环醚类化合物只有两篇报道, 不对称催化合成八元环醚尚未有报道.
2015年, 黄有课题组[28]报道了炔酮类化合物56和α-氰基-α,β-不饱和酮化合物57发生串联环化反应合成八元环醚的新方法, 在等物质的量的DABCO促进下, 以乙二醇和氯仿为混合溶剂, 在60 ℃条件下发生[4+4]串联环加成反应, 能够以78%的收率得到八元环醚类化合物58 (Scheme 19).该方法反应条件温和, 底物适用范围广, 原子经济性好, 反应原料易得, 为方便合成八元环醚类化合物提供了有效手段.
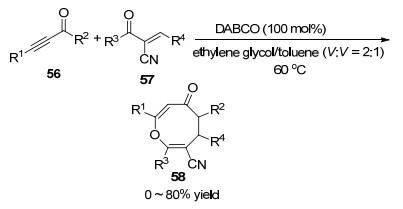
2018年, 苗志伟课题组[29]报道了Lewis碱DBU促进下苯基甲基取代炔酮与吡唑酮衍生的α,β-不饱和活泼烯烃发生[4+4]环加成反应合成八元环醚的反应, 苯基甲基取代的炔酮类化合物59与吡唑酮衍生的α,β-不饱和活泼烯烃60在150 mol% DBU促进下, 以二氯甲烷为溶剂在室温下反应5 min, 以99%的收率得到含有吡唑骨架结构的八元环醚类化合物61.
反应机理是苯基甲基取代炔酮59在Lewis碱DBU作用下失去质子得到烯醇中间体A, 中间体A对吡唑酮衍生物60的碳-碳双键发生亲核进攻得到中间体B.中间体B捕获质子后变成关键的中间体C, 接着发生DBU对炔发生第二次迈克尔加成反应得到两性中间体D.中间体D经过烯醇互变得到中间体E, 最终伴随分子内的氧负离子对碳-碳双键的亲核加成脱掉DBU得到八元环醚并吡唑衍生物61 (Scheme 20).该方法具有条件温和、反应时间短、原料廉价易得的特点, 可以很高的收率得到吡唑并八元环醚类化合物, 为合成八元环醚类化合物提供了一种新的选择.
八元环醚骨架结构广泛存在于天然产物分子和药物分子中, 目前已经发现的该类化合物合成方法包括, 过渡金属催化法、环扩张法、Claisen重排法、烯烃换位反应法以及有机小分子催化串联环化反应法等.比较上述五种合成八元环醚化合物的方法各有优缺点, 具体表现为: (1)过渡金属催化法的优点是反应高效, 可以快速构建八元环醚骨架结构.缺点是反应所使用的过渡金属催化剂价格昂贵, 一般不能回收循环使用. (2)环扩张法的优点是反应条件温和, 操作步骤简单.缺点是底物合成往往需要多步反应才能够实现. (3) Claisen重排法的优点是反应的区域选择性好, 收率高.缺点是底物适用范围窄. (4)烯烃换位反应法的优点是催化剂体系完善, 反应能够实现八元环醚化合物的不对称合成.缺点是反应底物结构单一, 需要多步反应进行合成. (5)有机小分子催化串联环化反应法的优点是催化剂价格便宜, 来源广泛.缺点是适用于该方法合成八元环醚化合物的反应体系比较单一, 底物范围有待进一步拓展.未来该研究领域将主要围绕以下两方面开展工作: (1)开发新的合成策略, 实现八元环醚化合物的高效合成; (2)发展新型手性催化剂或者手性诱导试剂, 实现八元环醚化合物的不对称合成.随着有机合成技术的不断发展, 含有八元环醚骨架结构的有机化合物合成一定会迎来新的更大发展.
Zhou, Z. F.; Menna, M.; Cai, Y. S.; Guo, Y. W. Chem. Rev. 2014, 115, 1543. https://www.ncbi.nlm.nih.gov/pubmed/25525670
(a) Gonzaĺez, A. G.; Martín, J. D.; Martín, V. S.; Norte, M.; Peŕez, R.; Ruano, J. Z.; Drexler, S. A.; Clardy, J. Tetrahedron 1982, 38, 1009.
(b) Noite, M.; Gonzalez, A. G.; Cataldo, F.; Rodríguez, M. L.; Brito, I. Tetrahedron 1991, 47, 9411.
(c) Kim, H.; Choi, W. J.; Jung, J.; Kim, S.; Kim, D. J. Am. Chem. Soc. 2003, 125, 10238.
(a) Irie, T.; Suzuki, M.; Masamune, T. Tetrahedron Lett. 1965, 6, 1091.
(b) Irie, T.; Suzuki, M.; Masamune, T. Tetrahedron 1968, 24, 4193.
(c) Crimmins, M. T.; Choy, A. L. J. Am. Chem. Soc. 1999, 121, 5653.
(c) Irie, T.; Suzuki, M.; Masamune, T. Tetrahedron Lett. 1965, 6, 109.
Fukuzawa, A.; Takasugi, Y.; Murai, A. Tetrahedron Lett. 1991, 32, 5597. doi: 10.1016/0040-4039(91)80093-L
Suzuki, M.; Takahashi, Y.; Matsuo, Y.; Masuda, M. Phytochemistry 1996, 41, 1101. doi: 10.1016/0031-9422(95)00726-1
(a) Singh, S. B.; Zink, D. L.; Quamina, D. S.; Pelaez, F.; Teran, A.; Felock, P.; Hazuda, D. J. Tetrahedron Lett. 2002, 43, 2351.
(b) Ramana, C. V.; Reddy, C. N.; Gonnade, R. G. Chem. Commun. 2008, 3151.
(c) Tadross, P. M.; Bugga, P.; Stoltz, B. M. Org. Biomol. Chem. 2011, 9, 5354.
(d) Foot, J. S.; Giblin, G. M. P.; Taylor, R. J. K. Org. Lett. 2003, 5, 4441.
(a) Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G.; Fronczek, F. R. Tetrahedron Lett. 1993, 34, 1999.
(b) Macías, F. A.; Molinillo, J. M. G.; Varela, R. M.; Torres, A.; Fronczek, F. R. J. Org. Chem. 1994, 59, 8261.
(c) Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. J. Nat. Prod. 1999, 62, 1636.
(a) Burton, J. W.; Clark, J. S.; Derrer, S.; Stork, T. C.; Bendall, J. G.; Holmes, A. B. J. Am. Chem. Soc. 1997, 119, 7483.
(b) Tsushima, K.; Murai, A. Tetrahedron Lett. 1992, 33, 4345.
(c) Bratz, M.; Bullock, W. H.; Overman, L. E.; Takemoto, T. J. Am. Chem. Soc. 1995, 117, 5958.
(d) Mujica, M. T.; Afonso, M. M.; Galindo, A.; Palenzuela, J. A. Synlett 1996, 983.
(e) Krüger, J.; Hoffmann, R. W. J. Am. Chem. Soc. 1997, 119, 7499.
(f) Mujica, M. T.; Afonso, M. M.; Galindo, A.; Palenzuela, J. A. J. Org. Chem. 1998, 63, 9728.
Mandal, S. K.; Roy, S. C. Tetrahedron Lett. 2006, 47, 1599. doi: 10.1016/j.tetlet.2005.12.131
Coulter, M. M.; Dornan, P. K.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 6932. doi: 10.1021/ja901915u
Zhao, C. G.; Xie, X. G.; Duan, S. S.; Li, H. L.; Fang, R.; She, X. G. Angew. Chem., Int. Ed. 2014, 53, 10789. doi: 10.1002/anie.201406486
Corrie, T. J. A.; Ball, L. T.; Russell, C. A.; Lloyd-Jones, G. C. J. Am. Chem. Soc. 2017, 139, 245. doi: 10.1021/jacs.6b10018
Liu, R. X.; Wang, Q.; Wei, Y.; Shi, M. Chem. Commun. 2018, 54, 1225. doi: 10.1039/C7CC09250D
Snyder, S. A.; Treitler, D. S.; Brucks, A. P.; Sattler, W. J. Am. Chem. Soc. 2011, 133, 15898. doi: 10.1021/ja2069449
Liao, H. H.; Liu, R. S. Chem. Commun. 2011, 47, 1339. doi: 10.1039/C0CC03309J
Cao, T. X.; Kong, Y.; Luo, K.; Chen, L. F.; Zhu, S. F. Angew. Chem., Int. Ed. 2018, 57, 8707. https://www.ncbi.nlm.nih.gov/pubmed/29697177
Boeckman, R. K.; Shair, M. D.; Vargas, J. R.; Stolz. L. A. J. Org. Chem. 1993, 58, 1295. doi: 10.1021/jo00058a001
Boeckman, R. K.; Reeder, M. R. J. Org. Chem. 1997, 62, 6456. doi: 10.1021/jo9712254
Boeckman, R. K.; Zhang, J.; Reeder, M. R. Org. Lett. 2002, 4, 3891. doi: 10.1021/ol0267174
李正邦, 王锋鹏, 陈东林, 有机化学, 2000, 20, 282. doi: 10.3321/j.issn:0253-2786.2000.03.002Li, Z. B.; Wang, F. P.; Chen, D. L. Chin. J. Org. Chem. 2000, 20, 282(in Chinese). doi: 10.3321/j.issn:0253-2786.2000.03.002
Miller, S. J.; Kim, S. H.; Chen, Z. R.; Grubbs, R. H. J. Am. Chem. Soc. 1995, 117, 2108. doi: 10.1021/ja00112a031
Linderman, R. J.; Siedlecki, J.; ONeill, S. A.; Sun, H. J. Am. Chem. Soc. 1997, 119, 6919. doi: 10.1021/ja9711674
Crimmins, M. T.; Tabet, E. A. J. Am. Chem. Soc. 2000, 122, 5473. doi: 10.1021/ja0007197
Mori, M.; Kitamura, T.; Sakakibara, N.; Sato, Y. Org. Lett. 2000, 2, 543. doi: 10.1021/ol991398a
Ortega, N.; Martin, T.; Martin, V. S. Org. Lett. 2006, 8, 871. doi: 10.1021/ol052932j
Baek, S.; Jo, H.; Kim, H.; Kim, H.; Kim, S.; Kim, D. Org. Lett. 2005, 7, 75. doi: 10.1021/ol047877d
Kim, G.; Sohn, T.; Kim, D.; Paton. R. S. Angew. Chem., Int. Ed. 2014, 53, 272. doi: 10.1002/anie.201308077
Liang, L.; Li, E. Q.; Dong, X. L.; Huang, Y. Org. Lett. 2015, 17, 4914. doi: 10.1021/acs.orglett.5b02498
Cheng, C.; Zhang, J. Y.; Wang, X.; Miao, Z. W. J. Org. Chem. 2018, 83, 5450. doi: 10.1021/acs.joc.8b00352

图 1 具有八元环醚结构的天然产物分子和药物活性分子
Figure 1 Representative examples of eight-membered ethers possessing biological activities

图式 1 Cp2TiCl催化八元环醚化合物的合成
Scheme 1 Synthesis of eight-membered cyclic ethers catalyzed by Cp2TiCl

图式 2 金属铑催化分子内氢酰基化反应及反应机理
Scheme 2 Rh-catalyzed intramolecular olefin hydroacylation and reaction mechanism

图式 3 金催化1,2-酰基迁移及分子内环加成反应及反应机理
Scheme 3 Gold-catalyzed 1,2-acyloxy migration/intramolecular cycloaddtion and reaction mechanism

图式 4 金催化环加成反应合成联苯并环醚化合物
Scheme 4 Gold-catalyzed cycloaddtion to synthesize of biphenyl cycloether compound

图式 5 金催化双炔双氢芳基化反应及反应机理
Scheme 5 Gold-catalyzed twofold hydroarylation of diynes and reaction mechanism

图式 6 溴正离子引导的环扩张反应及反应机理
Scheme 6 Bromonium-induced ring expansion and reaction mechanism

图式 7 金催化1-环氧丙烷-1-乙炔基环丙烷开环反应及反应机理
Scheme 7 Gold-catalyzed ring expansion of 1-oxiranyl-1-alkynylcyclopropanes and reaction mechanism

图式 9 利用Claisen重排反应合成八元环醚化合物
Scheme 9 Synthesis of eight-membered cyclic ethers by Claisen rearrangement reaction

图式 10 利用Claisen重排反应不对称合成八元环醚化合物
Scheme 10 Asymmetric synthesis of eight-membered cyclic ethers by Claisen rearrangement reaction

图式 12 催化烯烃换位反应(RCM)合成八元环醚化合物
Scheme 12 Catalytic ring-closing metathesis synthesis of eightmembered rings

图式 13 开链三丁基锡取代双烯化合物发生烯烃换位反应
Scheme 13 Ring-closing metathesis of acyclic tributylstannyl-substituted dienes

图式 14 利用烯烃换位反应构建八元环醚骨架全合成(+)-Prelaureatin
Scheme 14 Construction of the oxocene core of (+)-prelaureatin by ring-closing metathesis

图式 15 通过烯烃换位反应合成苯并八元环醚化合物
Scheme 15 Synthesis of benzocyclooctene derivatives by ringclosing metathesis

图式 16 通过烯烃换位反应立体选择性合成八元环醚化合物
Scheme 16 Stereoselective synthesis of eight-membered cyclic ethers by ring-closing metathesis

图式 17 高立体选择性全合成天然产物(+)-Laurencin
Scheme 17 Stereoselective and efficient total synthesis of (+)-laurencin

图式 18 高立体选择性高效全合成天然产物(+)-Bermudenynol
Scheme 18 Stereoselective and efficient total synthesis of (+)-bermudenynol

图式 19 通过DABCO催化[4+4]环加成反应合成八元环醚化合物
Scheme 19 Synthesis of eight-membered cyclic ethers by DABCO-mediated [4+4] annulation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:


