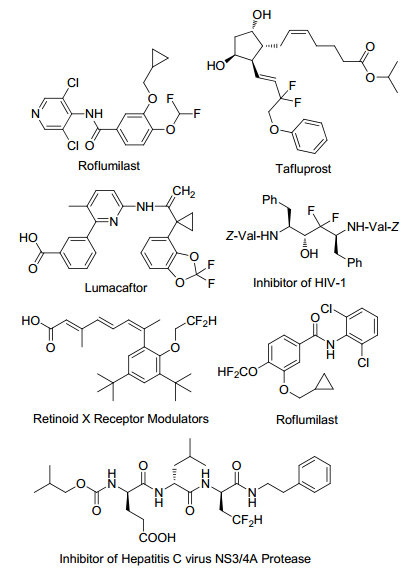
Figure 1.
Biologically active molecules
Progress of Difluoromethyl Heteroaryl Sulfones as Difluoroalkylation Reagents
Xuefen Tao , Rong Sheng , Kun Bao , Yuxin Wang , Yinxiu Jin

The selective introduction of a fluorine atom or a fluorine-containing group into a specific portion of an organic molecule can significantly change the original physical and chemical properties and biological activities of the parent molecule, due to the unique characteristics of fluorine, such as small atomic radius, maximum electronegativity, low polarizability, and strong C—F bond.[1, 2]
Among all the fluorine-containing groups, difluoromethyl (or difluoromethylene) is the most special one. It is generally believed that difluoromethyl group is similar in volume and polarity with hydroxymethyl. It can functionalize as a bioisostere of an oxygen or a carbonyl group. And it can also act as a lipophilic hydrogen bond donor better than hydroxymethyl. Therefore, difluoromethyl groups are often introduced into many biologically active molecules, such as anti-infectives, protease inhibitors, herbicides and so on (Figure 1).[3~6]
Researching on difluoromethyl-containing organic compounds has become one of the most popular areas in organic chemistry. Many efforts have been focused on the development of efficient difluoromethylation reagents. Over the past decade, a variety of difluoromethyl heteroaryl sulfones have been developed and widely used by introducing difluoromethyl-containing groups into organic compounds through various types of reactions, due to their ease of preparation, good functional group tolerance and universal applicability to a wide range of carbonyl compounds. They not only facilitate the generation of the specific reactive species for fluoroalkylation, but also provide opportunities to achieve synthetic diversity.[7~10]
Although several reviews on the fluoroalkylating agents have been appeared, [11~13] however, to the best of our knowledge, there is no review focus specifically on the applications of difluoromethyl heteroaryl sulfones. In this paper, we aim to provide a full review on the synthetic applications of these difluoromethyl heteroaryl sulfones in recent ten years.
Julia-Kocienski olefination reaction is a typical cascade process that involves three sequential steps: (1) the nucleophilic addition of a sulfonyl carbanion to a carbonyl group, (2) the ipso-substitution of the addition adduct at the heteroaryl ring (also known as the Smiles rearrangement), and (3) the fragmentation of the resulting sulfinate salt to give an alkene.
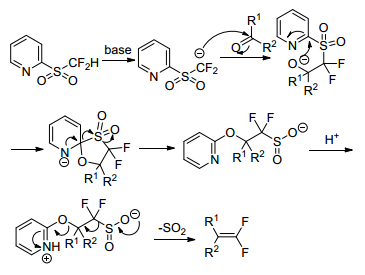
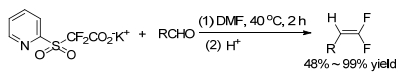
In 2010, Hu group[14] reported the first Julia-Kocienski olefination reaction for the synthesis of 1, 1-difluoro-alkenes. Alkyl ketones, aromatic and aliphatic aldehydes were treated with heteroaryl sulfones in the presence of base, such as t-BuOK, then followed by the treatment with acid to result in gem-difluoroolefins (Scheme 1).
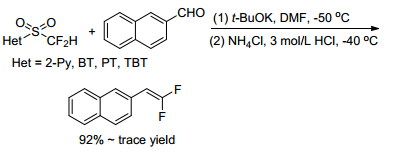
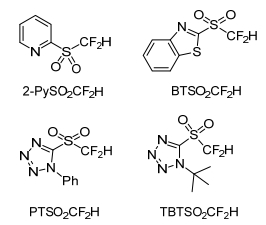
They demonstrated that difluoromethyl 2-pyridyl sulfone (2-PySO2CF2H) showed higher reactivity in the difluoroolefination reaction with 2-naphthaldehyde than other heteroaryl sulfones, such as difluoromethyl 1, 3-benzothiazol-2-yl sulfone (BTSO2CF2H), difluoromethyl 1-phenyl-1H-tetrazol-5-yl sulfone (PTSO2-CF2H), and difluoromethyl 1-tert-butyl-1H-tetrazol-5-yl sulfone (TBT-SO2CF2H) sulfone (Scheme 2). The yield of the reaction with BTSO2CF2H is 42%, while both PTSO2CF2H and TBTSO2CF2H gave almost no product.
The olefination reaction involved the formation of a relatively stable difluorinated sulfinate salt (Scheme 3).
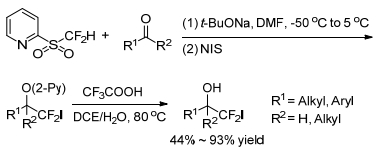
In 2012, Hu group[15] developed a formal nucleophilic iodo-, or bromo-difluoro-methylation for carbonyl compounds to obtain iodo-or bromo-difluoromethylated compounds via halogenation of the Julia-Kocienski intermediates and subsequent deprotection (Schemes 3, 4). The optimal reaction conditions for bromodifluoromethylation are using N, N-dimthylformamide (DMF) as solvent, sodium tert-butoxide (t-BuONa) as base to enhance the stability of the in-situ generated sulfinate intermediate, N-iodosuccin-imide (NIS) as the iodinating reagent to inhibite the formation of the undesired difluoroalkene, and the reactant molar ratio of 2-PySO2CF2H/carbonyl compound/t-BuO-Na/NIS (n:n:n:n=1:1.2:1.8:4). The reaction tolerates various substituents on the aryl aldehyde, aliphatic α, β-unsaturated aldehydes and ketones (Scheme 4).
By changing the base to LiHMDS, using phenyltrimethylammonium tribromide (PhMe3NBr3) as the halogenating reagent, and adjusting the reactant ratio of PY sulfone/ carbonyl compound/LiHMDS/PhMe3NBr3 (n:n:n:n=1:1.2:1.4:4), the bromodifluoromethylation of carbonyl compounds proceeded smoothly to afford the desired CF2Br-containing products mostly in good yields (Scheme 5).
The proposed reaction mechanism of this nucleophilic halogen-difluoromethylation is shown as Scheme 6.
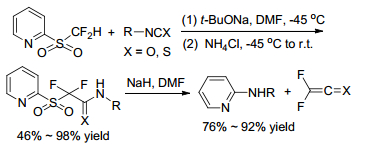
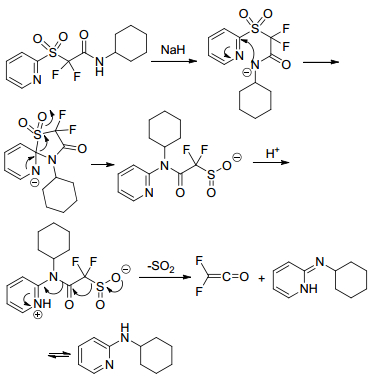
Sheng group[16] reported the first nucleophilic difluoroalkylation of isocyanates with difluoromethyl 2-pyridyl sulfone to get 2, 2-difluoro-2-pyridinylsulfonylacetamides and declared that the substrate scope of this reaction was including phenyl isocyanates, phenyl thioisocyanates and aliphatic isocyanates. The addition products converted to difluoroacetamide sulfinate and iododifluoroacetamide easily in the presence of sodium hydride (NaH) (Scheme 7). A proposed mechanism showed that the reaction experienced an intramolecular Julia-Kocienski reaction (Scheme 8).
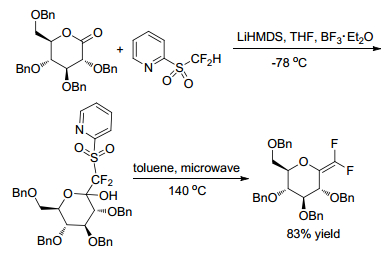
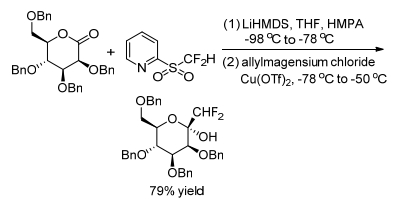
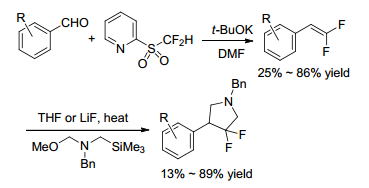
Gueyrard and Shen group[17, 18] reported the gem-difluoroolefination of benzylated sugar-derived lactones by a modified Julia olefination with difluoromethyl-2-pyridyl sulfone separately (Schemes 9, 10). The reaction was shown to proceed efficiently in both pyranose (D-gluco, D-galacto, D-manno, 2-deoxy-D-gluco) and furanose (D-arabino) sugar series. Meanwhile, Shen group[18] carried out a one-pot reduction with Grignard reagent of sugar lactones for the preparation of fluorinated 2-ketoses (Scheme 11).
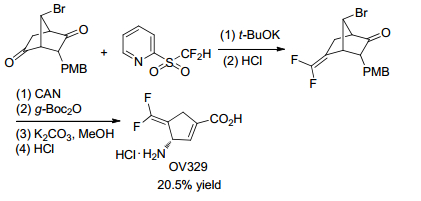
Later in 2018, Silverman and his coworker[19] optimized the synthesis of (S)-3-amino-4-(difluoromethylenyl)-cyclopent-1-en-1-carboxylic acid (OV329), a potent inactivator of GABA aminotransferase (GABA-AT) via Julia-Kocienski reaction (Scheme 12). Using Hu's reagent in the key step to furnish 1, 1'-difluoroalkene followed by methanolysis and subsequent elimination, the number of synthetic steps was reduced from 14 to 9, and the overall yield doubled.
Xiao group[20] reported an optimized Julia-Kocienski reaction which could be performed under mild conditions without additional base (Scheme 13). In this protocol the 2-PySO2CF2- was generated through the decarboxylation of potassium 2-pyridinyl sulfonyldifluoroacetate.
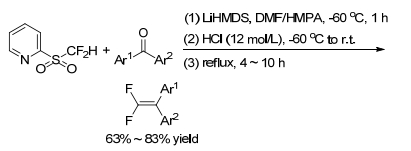
As for normal diaryl ketones, it is difficult to achieve efficient gem-difluoroolefination by typical Julia-Kocienski reaction under basic conditions. Hu group[21] further improved the classical Julia-Kocienski reaction and reported the acid-promoted Smiles rearrangement applying in Julia-Kocienski olefination reactions. Using LiHMDS as base and HMPA/DMF (V/V=1/10) as solvent, the gem-difluoroolefination of diaryl ketones is improved. Quenching the alcoholate intermediate with concentrated HCl at low temperature, such as -60 ℃, followed by acid-promoted elimination of the carbinol intermediate, 2, 2-diaryl-1, 1-difluoroalkenes were obtained in good yields (Scheme 14). By this strategy, both symmetric and asymmetric diaryl ketones, even some heteroaryl ketones, smoothly gave gem-difluoroalkenes in moderate-to-good yields.
The advantages of this protocol are one-pot operation, good functional-group tolerance, and high efficiency. But there still remain some limitations. As for nitrogen-containing ketones, such as phenyl 2-pyridyl ketone, the Smiles rearrangement failed to take place.
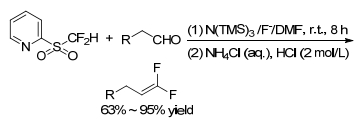
Hu group also studied the application of Julia-Kocienski reaction in the aliphatic aldehydes to achieve gem-difluoroolefination depending on amide base generated in situ (Scheme 15). This method could not only avoid the enolization of aliphatic aldehydes, but also be chemoselective of multi-carbonyl compounds. It demonstrated that aldehyde preferentially underwent gem-difluoroolefination than ketone, and ketone preferentially than ester.
McAlpine group[22] showed that vinyl difluorides could participate in 1, 3-dipolar cycloaddition with a simple azomethine ylide to generate gem-difluoropyrrolidines. They tried to synthesize 2, 2-difluorostyrenes with benzaldehydes and Hu's reagent, then found out that benzaldehydes with electron deficient groups gave higher yields in [3+2] cycloaddition reactions (Scheme 16).
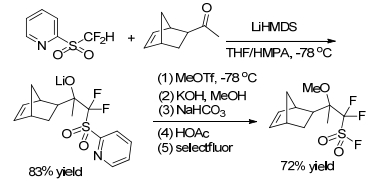
Difluoromethyl heteroaryl sulfones can be used to synthesize fluoropolymer. In 2011, Hu group[23] successfully synthesized the difluoroalkylsulfonyl chloride monomer for proton exchange membrane by nucleophilic addition (Scheme 17).
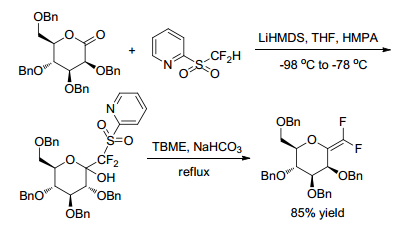
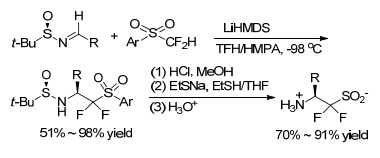
Taking 2-PySO2CF2H as a masked difluoro (sulfinato)-methylating agent, Prakash group[24] synthesized novel α, α-difluoro-β-amino sulfinics and sulfonic acids via nucleophilic difluoro(sulfinato)methylation of N-tert-butane-sulfinyl imine, using 2-PySO2CF2H as a masked difluoro(sulfinato)-methylating agent (Scheme 18). The so-obtained sulfinics and sulfonic acids are potentially useful blocks to construct difluorinated peptidosulfonamides. The reaction was carried out by using excess LiHMDS as the base, HMPA as an additive in THF at a low temperature (such as -98 ℃) to obtain target product in a high yield (95%) with a good diastereoselectivity (d.r. > 99:1).
By this protocol, the chiral optically active difluoro-β-amino sulfinic, sulfonic acids and complex fluorinated peptidosulfonamides were synthesized feasibly. The difluorinated β-amino sulfinates are expected to be suitable for constructing difluorinated peptidosulfonamides. The difluorinated β-amino sulfonic acids represent a novel class of functionalized fluoroalkyl sulfonic acids.
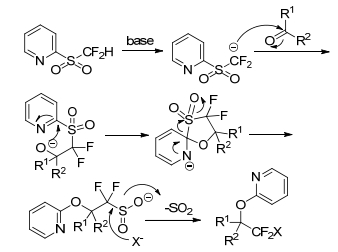
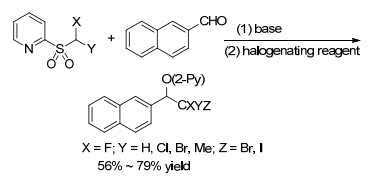
The key role that the traditional olefination changes to alkylation is the halogenation of in situ-generated sulfinate intermediates from Julia-Kocienski reaction. In addition, this method can also be extended to the introduction of other fluorinated groups, such as CFClBr, CFClI, CFBr2, and CFMeI by using various 2-pyridyl sulfonates (Scheme 19).[15]
However, this protocol is limited by its difficulty in removing the pyridioxyl group on the substrates bearing electron withdrawing substituents. Moreover, this method is senstitive to steric hindrance. Thus, it is not compatible with the aliphatic system and diaryl ketones.
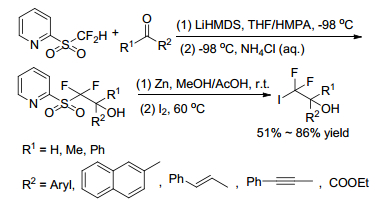
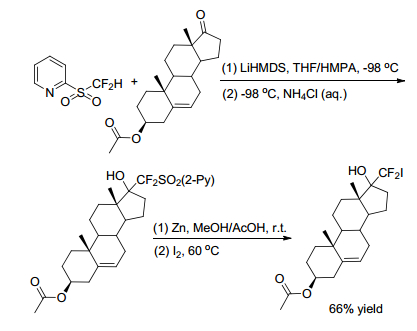
Later in 2016, Hu group[25] revealed a new approach for incorporation of CF2I group into organic molecules to achieve nucleophilic iododifluoromethylation of carbonyl compounds with 2-PySO2CF2H. In this method, the nucleophilic addition of 2-PySO2CF2H to carbonyl compounds was almost the same as previously reported, except that the reaction temperature was lowered to -98 ℃. The so-addition compounds went on depyridination smoothly by using a large excess of Zn in DMSO/AcOH system to give sulfinate in situ, followed by treatment of I2 at 60 ℃ to afford the corresponding iodinated products in high yield (Scheme 20). In this strategy, the Smiles rearrangement was avoided, and the substrate scope was rather broad. Benzaldehyde, 2-naphthaldehyde, aromatic aldehydes containing electron-donating and electron-withdrawing substituents, aliphatic and aromatic ketones were all well tolerated. Even some complex molecules such as steroidal ketones can result in excellent yields (Schemes 20, 21).
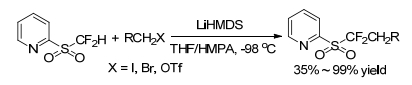
In 2011, Prakash and George group[14] declared the first nucleophilic substitution of difluoromethyl heteroaryl sulfones with alkyl halogens. The reaction went in the presence of a base, such as lithium hexamethyldisilazide (LiHMDS), with tetrahydrofuran (THF) as solvent and hexamethylphosphoramide (HMPA) as an additive, the temperature lowered to -98 ℃, and finally afforded α, α-difluoroalkyl heteroaryl sulfones (Scheme 22). They also decalard that sulfones such as BTSO2CF2H, PTSO2-CF2H, and TBTSO2CF2H were not as significantly reactive as 2-PySO2CF2H (Hu's reagent). The substrate scope of this protocol was primary alkyl halides and primary alcohol triflates. The secondary alkyl iodide couldn't give anticipated product. It indicated that the reaction was very sensitive to steric hindrance.
By treating with a less basic nucleophile, such as sodium ethanethiolate (EtSNa) or sodium difluoroethylsulfinate (DFES-Na), the α, α-difluoroalkyl sulfonates definitely underwent an intermolecular and dearylation to result in alkyl 2, 2-difluorosulfinate salts. Subsequently, oxidizing the so-obtained difluorosulfinate salts would give some efficient difluoroethylation agents (Scheme 23), which were difficult to prepare via direct nucleophilic difluoro (sulfonate)-methylation. This methodology was applicable for modification of functionalized molecules, such as carbohydrates, heterocycles and so on.[27, 28]
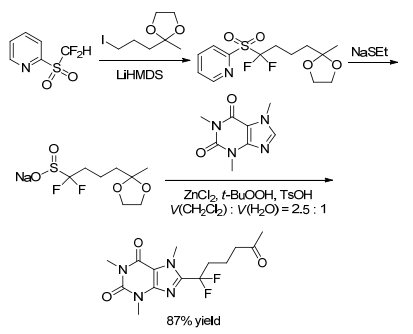
Shabat group[29] developed new difluoroalkyl ketal sulfinate salt reagent which was suitable for introducing ketone functional group into heteroarene bioactive compounds via one-pot reaction (Scheme 24). This protocol provided a general approach to obtain taggable ketone analogs directly from bioactive heteroarene compounds with limited options for conjugation.
Radical mediated difunctionalization of alkenes provides a powerful tactic for olefin utilization. Through this route, both CF2H and another functional group (H, C, N, O, Cl and Br) can be introduced into organic frameworks.[30~34]
In recent years, visible-light photoredox catalysis has emerged as a powerful synthetic method for bond activation and construction processes that are usually difficult to achieve with conventional methods.[35~38]
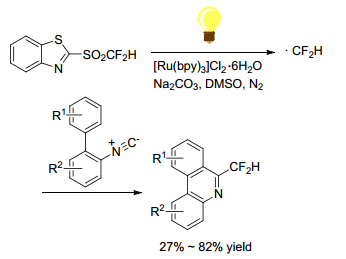
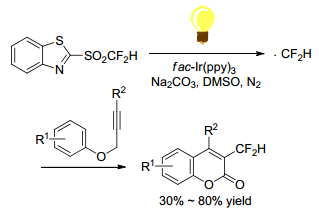
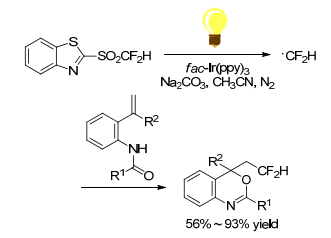
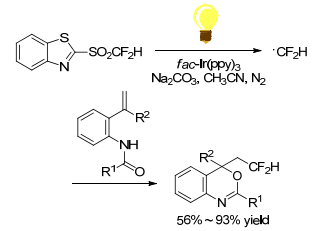
Hu and Ni group[39] reported the use of mono-, di-, and tri-fluorinated heteroaryl sulfones as a new class of radical fluoroalkylation reagents under visible-light photoredox catalysis (Scheme 25). With various isocyanides as radical acceptors, the radical fluoroalkylation progressed smoothly to afford fluoroalkylatedphenanthridine derivatives. This protocol opened a new door for the synthetic application of fluorinated sulfones as fluoroalkyl radical precursors. Using difluoromethyl sulfones as CF2H radical precursors, Fu group[40] realized an intramolecular difunctionalization of aryl alkynoates via radical initiating addition/cyclization (Scheme 26). This method provides a simple and efficient synthesis of 3-difluoromethylsubstituted coumarins in moderate to good yields. But the scope of alkynes was limited to alkynyl arenes. Later in 2016, Fu group[41] reported an efficient method for the synthesis of CF2H-containing benzoxazines and oxazolines through radical oxydifluoromethylation reaction of various olefinic amides with difluoromethyl sulfones. It was carried out in the presence of [fac-Ir(Ⅲ)(ppy)3] excited by 5 W blue LED for several hours (Scheme 27). Zou and his coworker[42] declared a similar radical cyclization of N-methacryloyl benzamides by using difluoromethyl sulfone to give difluoromethylated coumarins or isoquinolinediones (Scheme 28). Among the difluoromethyl sulfones, BTSO2CF2H was the best CF2H-transfer reagents.
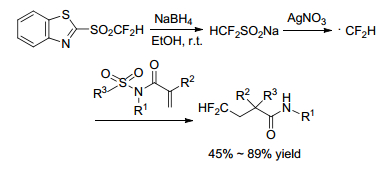
Hu group[43] developed a method for the straightforward introduction of CF2H and CH2F groups via a radical process (Scheme 29). The reaction went on in silver-catalyzed cascade fluoroalkylation/aryl migration/SO2 extrusion of conjugated N-arylsulfonylated amides. By treating ethanol solution of BTSO2CF2H with slight excess of NaBH4, followed by precipitation of the resulting salt in hexanes and washing with ethanol/hexanes, afforded fluorinated sulfinate salts. The later was excited by silver and gave out CF2H• radical, which reacted with conjugated N-arylsul-fonylated amides to produce α-aryl-β-fluoroalkyl amides. This reaction had a wide range of scope for substrates.
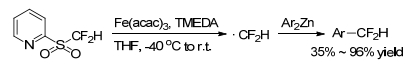
The direct introduction of difluoromethyl group onto arenes was often suffered from harsh reaction conditions, and poor functional group compatibility. Transition-metal-mediated difluoromethylation reactions were demonstrated as a viable approach for direct difluoromethylation. Recently, Hu group[44] tested this new strategy of iron-catalyzed aromatic difluoromethylation by using 2-PySO2CF2H as a precursor to generate difluoromethyl radical (Scheme 30).
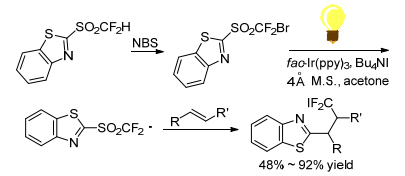
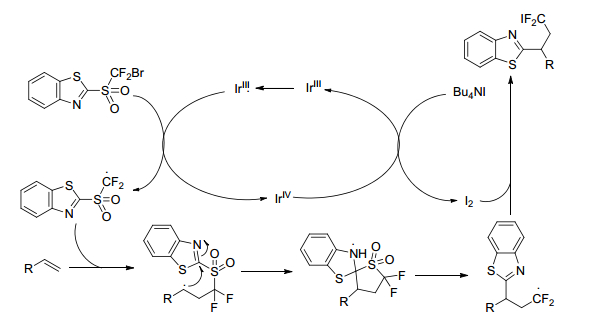
Zhu group[45] described a strategy for radical difunctionalization of alkenes. The heteroaryl and difluoromethyl incorporated into alkenes simultaneously by this process. After turning difluoromethyl heteroaryl sulfones into bromide difluoromethyl ones, the C—Br bond could readily promote by visible-light photoredox catalysis to form the difluoromethyl sulfonyl carbon radical species. The iododifluoromethylheteroarylation of alkenes promoted in the presence of tetrabutylammonium iodide (TBAI) and 4 molecular sieves (Scheme 31). The reaction demonstrated a highly broad substrate scope and functional group tolerance. The versatile adducts afforded by this protocol can be conveniently converted into a variety of valuable fluorine-containing compounds. The proposed reaction mechanism of radical difunctionalization is as shown in Scheme 32.
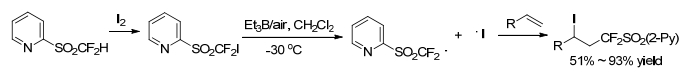
In 2014, Hu group[46] reported an Et3B/air-initiated radical (2-pyridylsulfonyl) difluoromethylation of terminal alkenes with iododifluoromethyl 2-pyridyl sulfone (2-Py-SO2CF2I). This synthetic methodology could also be extended to synthesize of (2-pyridylsulfonyl) difluoromethylated alkanes and alkenes (Scheme 33).
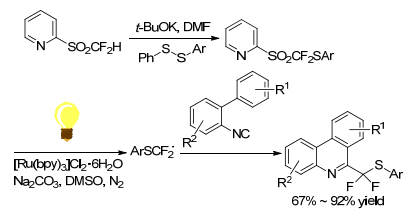
Using 2-PySO2CF2SAr as ArSCF2 radical precursor, Sheng group[47] first reported an efficient radical arylthiodifluoromethylation of various isocyanides to form phenanthridines and isoquinolines in good to excellent yields (Scheme 34). The optimal conditions for this protocol were taking [Ru(bpy)3Cl2]•6H2O (2 mol%) as photocatalyst, DMSO as solvent, Na2CO3 (3.0 equiv.) as base and under 6 W blue LED irradiation. It was proposed that 2-PySO2-CF2SAr was reduced by [Ru]+ through a SET process to release highly reactive radical species ArSCF2•.
This strategy gave a method for introducing an arylthiodifluoromethyl moiety into nitrogen containing heterocycles. The substrate scope of this radical reaction was wide. The ArSCF2 group could be introduced into both electron-rich and electron-deficient isocyanides under mild conditions. It demonstrated excellent functional group compatibility except the methyl-thiodifluoroalkyl sulfone.
Difluoromethyl heteroaryl sulfones are the newly developed nucleophilic difluoromethylation reagents. Among them, 2-PySO2CF2H is a bench-stable, crystalline solid that is now commercially available. It was widely used in nucleophilic reaction. 2-BTSO2CF2H is also a worthwhile novel reagent in addition. It opens up a straightforward access to difluoromethyl compounds. The substrate scope of these reagents has extended to carbonyl, alkyl halide, alkyne, imine derivatives, isocyanates and so on. There still are some unexplored applications worth studying. Finally, we hope that this review can provide new thoughts for the synthesis of difluoroalkyl compounds.
Kanda, E. Bull. Chem. Soc. Jpn. 1937, 12, 469. doi: 10.1246/bcsj.12.469
Premchandran, R. H.; Ogletree, M. L.; Fried, J. J. Org. Chem. 1993, 58, 5724. doi: 10.1021/jo00073a035
Huchet, Q. A.; Kuhn, B.; Wagner, B.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Fluorine Chem. 2013, 152, 119. doi: 10.1016/j.jfluchem.2013.02.023
廖斌, 廖清江, 药学进展, 2012, 36, 138. doi: 10.3969/j.issn.1001-5094.2012.03.012Liao, B.; Liao, Q. J. Prog. Pharm. Sci. 2012, 36, 138(in Chinese). doi: 10.3969/j.issn.1001-5094.2012.03.012
谭初兵, 时丽丽, 王士伟, 现代药物与临床, 2013, 28, 415.Tan, C. B.; Shi, L. L.; Wang, S. W. Drugs Clin. 2013, 28, 415(in Chinese).
陈玲, 赵天笑, 邹栩, 中国新药杂志, 2015, 24, 361.Chen, L.; Zhao, T. X.; Zou, X. Chin. J. New Drug 2015, 24, 361(in Chinese).
张建忠, 上海医药, 2015, 36, 79.Zhang, J. Z. Shanghai Med. Pharm. J. 2015, 36, 79(in Chinese).
Xiao, Y. L.; Zhang, B.; Feng, Z.; Zhang, X. G. Org. Lett. 2014, 16, 4822. doi: 10.1021/ol502121m
Zhang, X. X.; Cao, S. Tetrahedron Lett. 2017, 58, 375. doi: 10.1016/j.tetlet.2016.12.054
Ni, C. F.; Hu, M. Y.; Hu, J. B. Chem. Rev. 2015, 115, 765. doi: 10.1021/cr5002386
Zhou, Q. Q.; Zou, Y. Q.; Lu, L. Q.; Xiao, W. J. Angew. Chem., Int. Ed. 2019, 58, 1586. doi: 10.1002/anie.201803102
Grushin, V. V. A. Chem. Res. 2010, 43, 160. doi: 10.1021/ar9001763
Hine, J.; Porter, J. J. J. Am. Chem. Soc. 1960, 82, 6178. doi: 10.1021/ja01508a050
Zhao, Y. C.; Huang, W. Z.; Zhu, L. G.; Hu, J. B. Org. Lett. 2010, 12, 1444. doi: 10.1021/ol100090r
Zhao, Y. C.; Gao, B.; Hu, J. B. J. Am. Chem. Soc. 2012, 134, 5790. doi: 10.1021/ja301601b
Li, S.; Peng, P.; Wei, J.; Hu, Y. Z.; Hu, J. B.; Sheng, R. Adv. Synth. Catal. 2015, 357, 3429. doi: 10.1002/adsc.201500150
Habib, S.; Gueyrard, D. Eur. J. Org. Chem. 2015, 871.
Liu, X.; Yin, Q.; Yin, J.; Chen, G. H.; Wang, X.; You, Q. D.; Chen, Y. L.; Xiong, B.; Shen, J. K. Eur. J. Org. Chem. 2014, 6150.
Moschitto, M. J.; Silverman, R. B. Org. Lett. 2018, 20, 4589. doi: 10.1021/acs.orglett.8b01872
Wang, X. P.; Lin, J. H.; Xiao, J. C.; Zheng, X. Eur. J. Org. Chem. 2014, 928.
Gao, B.; Zhao, Y. C.; Hu, M. Y.; Ni, C. F.; Hu, J. B. Chem. Eur. J. 2014, 20, 1. doi: 10.1002/chem.201390210
McAlpine, I.; Tran-Dubé, M.; Wang, F.; Scales; S.; Matthews, J.; Collins, M. R.; Nair, S. K.; Nguyen, M.; Bian, J. W.; Alsina L. M.; Sun, J.; Zhong, J. Y.; Warmus, J. S.; O'Neill, B. T. J. Org. Chem. 2015, 80, 7266. doi: 10.1021/acs.joc.5b00853
赵延川, 张丽君, 许国峰, 郑吉, 胡金波, 中国科学•化学, 2011, 41, 1833.Zhao, Y. C.; Zhang, L. J.; Xu, G. F.; Zheng, J.; Hu, J. B. Sci. China Chem. 2011, 41, 1833(in Chinese).
Prakash, G. K. S.; Ni, C. F.; Wang, F.; Zhang, Z.; Haiges, R.; Olah, G. A. Angew. Chem., Int. Ed. 2013, 52, 10835. doi: 10.1002/anie.201304395
Miao, W. J.; Ni, C. F.; Zhao, Y. C.; Hu, J. B. Org. Lett. 2016, 18, 2766. doi: 10.1021/acs.orglett.6b01258
Prakash, G. K. S.; Ni, C. F.; Wang, F.; Hu, J. B.; George A. O. Angew. Chem., Int. Ed. 2011, 50, 2559. doi: 10.1002/anie.201007594
Zhou, Q.; Gui, J.; Pan, C. M.; Albone, E.; Cheng, X.; Suh, E. M.; Grasso, L.; Ishihara, Y.; Baran, P. S. J. Am. Chem. Soc. 2013, 135, 12994. doi: 10.1021/ja407739y
Zhou, Q.; Ruffoni, A.; Gianatassio, R.; Fujiwara, Y.; Sella, E.; Shabat, D.; Baran, P. S. Angew. Chem. 2013, 125, 4041. doi: 10.1002/ange.201300763
Gnaim, S.; Scomparin, A.; Li, X. L.; Baran, P. S.; Rader, C.; Ronit, S. F.; Shabat, D. Bioconjugate Chem. 2016, 27, 1965. doi: 10.1021/acs.bioconjchem.6b00382
Pintauer, T.; Matyjaszewski, K. Chem. Soc. Rev. 2008, 37, 1087. doi: 10.1039/b714578k
Eckenhoff, W. T.; Pintauer, T. Catal. Rev. 2010, 52, 1. doi: 10.1080/01614940903238759
Cao, M. Y.; Ren, X.; Lu, Z. Tetrahedron Lett. 2015, 56, 3732. doi: 10.1016/j.tetlet.2015.04.091
Chen, J. R.; Yu, X. Y.; Xiao, W. J. Synthesis 2015, 47, 604.
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527. doi: 10.1038/nchem.687
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102. doi: 10.1039/B913880N
Xuan, J.; Xiao, W. J. Angew. Chem., Int. Ed. 2012, 51, 6828. doi: 10.1002/anie.201200223
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322 doi: 10.1021/cr300503r
Koike, T.; Akita, M. Inorg. Chem. Front. 2014, 1, 561.
Rong, J.; Deng, L.; Tan, P.; Ni, C. F.; Gu, Y. C.; Hu, J. B. Angew. Chem., Int. Ed. 2016, 55, 2743. doi: 10.1002/anie.201510533
Zhu, M.; Fu, W. J.; Wang, Z. Q.; Xu, C.; Ji, B. M. Org. Biomol. Chem. 2017, 15, 9057. doi: 10.1039/C7OB02366A
Fu, W. J.; Han, X.; Zhu, M.; Xu C.; Wang, Z. Q.; Ji, B. M.; Hao, X. Q.; Song, M. P. Chem. Commun. 2016, 52, 13413. doi: 10.1039/C6CC07771D
Zou, G. L.; Wang, X. L. Org. Biomol. Chem. 2017, 15, 8748. doi: 10.1039/C7OB02226C
He, Z. B.; Tan, P.; Ni, C. F.; Hu, J. B. Org. Lett. 2015, 17, 1838. doi: 10.1021/acs.orglett.5b00308
Miao, W. J.; Zhao, Y. C.; Ni, C. F.; Gao, B.; Zhang, W.; Hu, J. B. J. Am. Chem. Soc. 2018, 140, 880. doi: 10.1021/jacs.7b11976
Yu, J. J.; Wu, Z.; Zhu, C. Angew. Chem., Int. Ed. 2018, 57, 1. doi: 10.1002/anie.201712460
Miao, W. J.; Ni, C. F.; Zhao, Y. C.; Hu, J. B. J. Fluorine Chem. 2014, 167, 231. doi: 10.1016/j.jfluchem.2014.05.012
Wei, J.; Gu, D. Y.; Wang, S. D.; Hu, J. B.; Dong, X. W.; Sheng, R. Org. Chem. Front. 2018, 5, 2568. doi: 10.1039/C8QO00644J

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
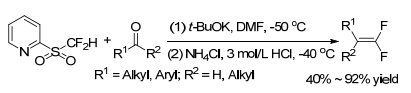

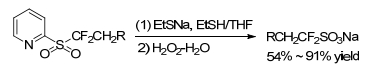

 下载:
下载:


































