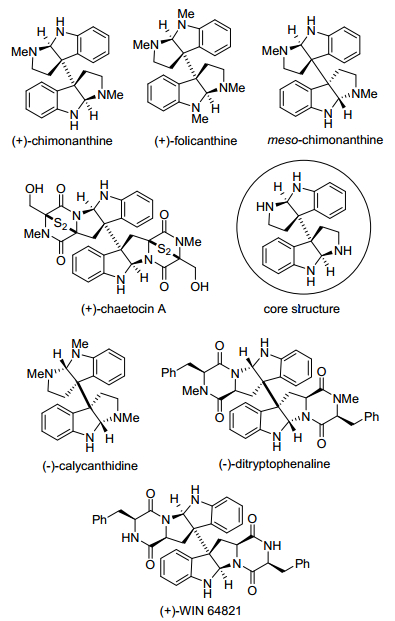
图 1.
具有代表性的二聚环色胺生物碱
Figure 1.
Representative dimeric cyclotryptamine alkaloids

二聚环色胺生物碱是吲哚生物碱家族中一类重要成员, 其生源途径源于色胺和色氨酸衍生物的氧化二聚.二聚环色胺生物碱由六氢吡咯吲哚(hexahydro-pyrrolo[2, 3-b]indole, HPI)结构单元通过易断裂的C3a—C3a' σ键方式连接而成. HPI类型天然产物组成了一类结构多样、生物学合成途径独特的生物碱家族化合物, 在植物、微生物和真菌资源中广为分布, 并拥有广泛的生物活性(Figure 1)[1].通过研究科学家发现二聚环色胺生物碱(+)-WIN 64821和(-)-ditryptophenaline是人类NK1受体上P物质的竞争性拮抗剂[2], (+)-chaetocin A具有抗菌活性、抑制细胞生长、抑制组蛋白赖氨酸特异性甲基转移酶的作用[3].
二聚环色胺生物碱结构中拥有相邻的C-3a(sp3)—C-3a'(sp3)季碳中心, 立体选择性合成这类生物碱中相邻的高位阻全碳季碳中心是化学合成的巨大挑战.由于其结构独特和优良的生物活性, 该类生物碱受到了合成化学工作者的广泛关注, 近年来一系列新颖的合成路线或策略被发展出来.关于二聚环色胺生物碱的合成研究, 国外已有了一些综述, 如2007年Overman等[4]从立体选择性构建季碳中心的角度, 综述了二聚和多聚环色胺生物碱的合成研究, 2008年Movassaghi等[5]从六氢吡咯吲哚生物碱生源合成假说启发的合成策略进行了综述.最近十多年来, 由于二聚环色胺生物碱全合成研究涌现出了许多新颖的合成策略和路线, 结合我们课题组的工作, 本文对2007年以来二聚环色胺生物碱的全合成研究进行简要的综述和总结.
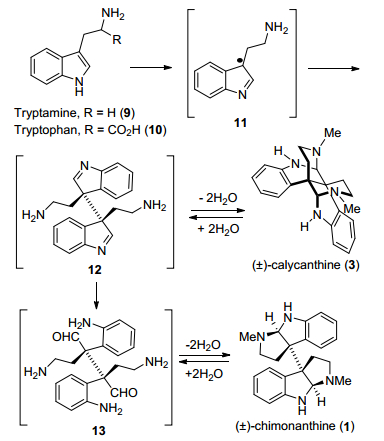
早在20世纪50年代, Woodward[6]和Robinson[7]等提出了二聚环色胺生物碱的生源合成途径假说, 即通过色胺和色氨酸衍生物的氧化二聚得到, 后来该假说得到Kirby等[8]同位素标记的实验验证.一系列结构研究表明, chimonanthine和calycanthine是来源于它们相应缩醛胺重排得到的异构体, 生物碱1和3只是假想缩醛胺中间体13重排得到异构体中的两个.通过后来实验中其它重排结构中间体化合物的分离[9], 表明中间体12来自化合物11苄位自由基氧化二聚, 而色胺是来自色氨酸的脱羧反应(Scheme 1).
2007年以来, 关于二聚环色胺生物碱的合成出现了许多新颖的合成路线, 本文按构建季碳中心的关键策略进行分类总结, 大致可以分为以下四类:还原型自由基二聚反应; 氧化型自由基二聚反应; 不对称催化; 其他类型.下面我们将从这四个类型对其合成路线进行综述总结.
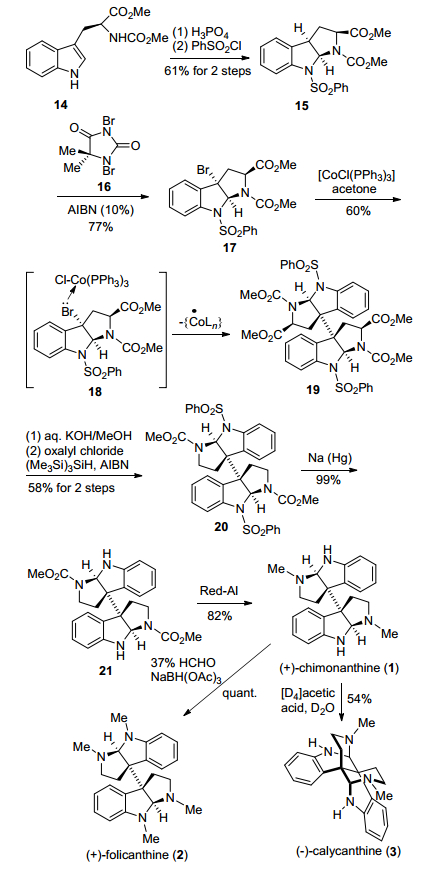
2007年, Movassaghi小组[10]发展了一种[CoCl-(PPh3)3]介导的还原型自由基二聚反应, 构建了二聚环色胺生物碱相邻手性季碳中心, 进而完成了二聚环色胺生物碱(+)-chimonanthine、(+)-folicanthine和(—)-caly-canthine的全合成.该方法报道后, 一系列基于[CoCl-(PPh3)3]介导的还原型自由基二聚反应被陆续报道.该路线以Na-甲氧基羰基-L-色氨酸甲酯14为起始原料, 在磷酸作用下, 吲哚环发生环化, 接着用苯磺酰基保护得到三环吡咯吲哚15.然后在溴代试剂16和自由基引发剂AIBN作用下, 15发生吡咯吲哚环C-3位溴代, 得到溴代化合物17, 再通过[CoCl(PPh3)3]介导的还原型自由基二聚反应, 以60%的产率得到关键二聚中间体19, 成功地实现了二聚环色胺生物碱核心骨架的构建.随后19经过甲酯基团水解、脱羧基、脱苯磺酰基、Red-Al还原氨基甲酸甲酯四步反应, 完成了天然产物(+)-chimonanthine合成; (+)-chimonanthine分别经过还原氨化甲基化或在氘代醋酸和氘水加热的条件下重排, 完成了另外两个天然产物(+)-folicanthine和(-)-calycan-thine的合成(Scheme 2).
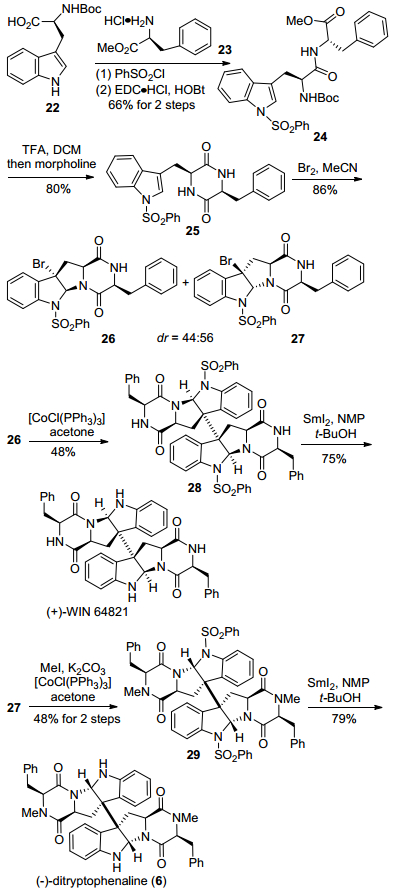
2008年, Movassaghi小组[11]采用他们报道的[CoCl-(PPh3)3]介导的还原型自由基二聚反应为关键步骤, 完成了天然产物(+)-WIN 64821和(—)-ditryptophenaline的全合成(Scheme 3).该路线从Na-叔丁氧羰基-L-色氨酸22出发, 经过苯磺酰基保护, 与L-苯丙氨酸甲酯盐酸盐23缩合, 得到酰胺化合物24, 再经过脱Boc基保护、氨解环化, 得到单一非对映异构体25.然后在Br2条件下, 25发生溴代环化, 得到C-3位溴代的内型产物26和外型产物27.接着26经过[CoCl(PPh3)3]介导的还原型自由基二聚反应, 得到关键二聚环色胺中间体28, 接着在SmI2条件下, 脱除苯磺酰基保护, 简洁高效地完成了(+)-WIN 64821的合成.化合物27经过氮上甲基化、[CoCl(PPh3)3]介导的还原型自由基二聚反应, 得到关键二聚中间体29, 再经过SmI2脱除苯磺酰基保护, 得到了天然产物(—)-ditryptophenaline.
2008年, de Lera小组[12]紧接着报道了天然产物(+)-WIN 64821和(+)-WIN 64745的合成, 采用类似Movassaghi小组[10, 11]的关键合成策略(Scheme 4).该合成路线以色氨酸衍生物30为原料, 在NBS溴代试剂和PPTS的作用下, 30发生溴代环化反应, 以85%的产率得到了单一非对映异构体31, 再经过[CoCl(PPh3)3]介导的还原型自由基二聚反应, 得到关键二聚环色胺中间体33.由于化合物33结构中C2/C2'的构型与目标天然产物构型刚好相反, 然后在LiHMDS/MeOH条件下, 翻转酯羰基α位碳原子构型, 以几乎定量的产率得到构型翻转产物34, 再经过三甲基碘硅烷(TMSI)脱Boc基保护, 得到四胺化合物35.随后在缩合剂O-(7-氮苯并三氮唑)-N, N, N, N-四甲基脲六氟磷酸酯(HATU)条件下, 化合物35与N-苄氧羰基-L-苯丙氨酸36缩合得到酰胺化合物37, 然后经过脱Cbz保护、氨解环化, 完成了(+)-WIN 64821的合成.天然产物(+)-WIN 64745则是经过化合物35在一锅法条件下, 先与N-苄氧羰基-L-亮氨酸38缩合、再与N-苄氧羰基-L-苯丙氨酸36缩合, 最后经过脱苄氧羰基保护、氨解环化得到.

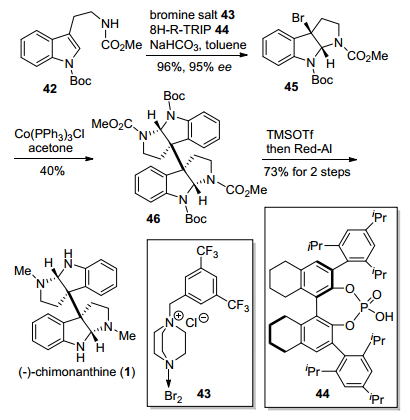
2013年, 马大为小组[13]发展了一种手性磷酸阴离子相转移催化剂, 利用该催化剂催化色胺衍生物吲哚环不对称溴代环化反应, 进而完成了天然产物(—)-chi-monanthine合成(Scheme 5).该合成方法以色胺衍生物42为原料, 然后在溴代试剂43和手性磷酸44的催化下, 实现了对色胺衍生物42的不对称溴代环化, 以96%的产率和95% ee值得到了C-3位溴代的三环吡咯吲哚关键中间体45, 然后采用Movassaghi小组[10]发展的[CoCl-(PPh3)3]介导还原型自由基二聚反应, 以40%的产率得到关键二聚环色胺中间体46, 最后经过脱Boc基保护、Red-Al还原氨基甲酸甲酯基团, 得到二聚环色胺生物碱(—)-chimonanthine.
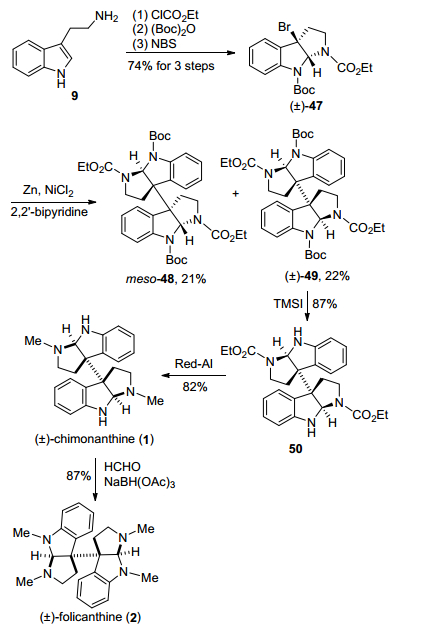
2013年, 彭羽小组[14]采用金属Ni/Zn催化的还原型自由基偶联二聚反应为关键步骤, 完成了二聚环色胺生物碱chimonanthine和(±)-folicanthine的合成(Scheme 6).该方法从商业可得原料色胺9出发, 经过氨基甲酸甲酯化、Boc基保护、NBS溴代环化, 得到消旋溴代环化产物47, 再采用Ni/Zn催化还原型自由基偶联反应, 得到了内消旋体二聚产物48和外消旋体二聚产物49.化合物49再经过TMSI脱Boc基保护、Red-Al还原氨基甲酸乙酯基团, 获得了二聚环色胺生物碱(±)-chimonanthine, 接着1经过氨基还原氨化甲基化, 得到天然产物(±)-folicanthine.
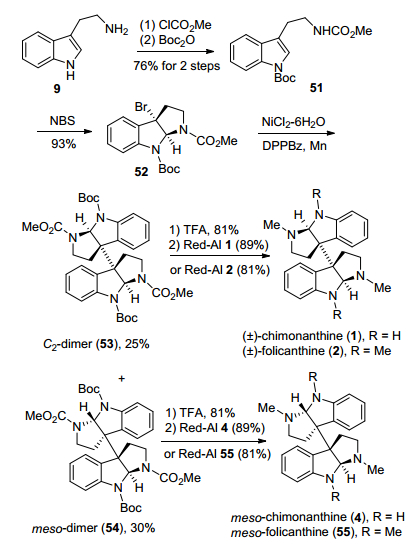
2014年, Oguri小组[15]采用与彭羽小组[14]类似的Ni/Mn催化的还原型自由基二聚反应为关键步, 以色胺9为起始原料, 经过氨基甲酸甲酯化、Boc基保护得到化合物51, 然后在NBS条件下溴代, 得到溴代环化产物52, 再采用Ni/Mn催化的还原型自由基二聚反应, 分别以25%、30%的产率得到了外消旋二聚体53和内消旋二聚体54, 然后分别经过脱Boc基保护、Red-Al还原氨基甲酸甲酯或直接用Red-Al还原氨基甲酸甲酯, 完成了(±)-chimonanthine、(±)-folicanthine、meso-chimo-nanthine和meso-folicanthine四个天然产物的合成(Scheme 7).
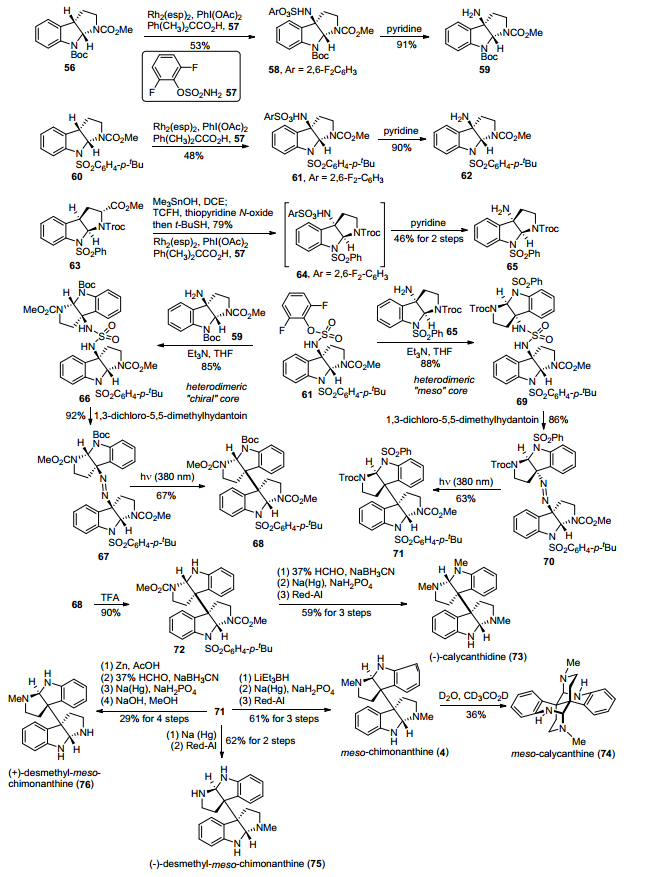
2014年, Movassaghi小组[16]发展了一种基于偶氮烯-定向断裂碎片化异源二聚反应, 完成了五个异源二聚环色胺生物碱的合成(Scheme 8).首先, 化合物56在Rh2(esp)2催化下, 与2, 6-二氟苯磺酸氨57发生C—H氨基化反应得到氨基磺酸酯58, 然后在吡啶条件下水解磺酸酯, 得到C-3位为氨基取代的吡咯吲哚化合物59.然后采用类似的合成方法, 完成了另外两个C-3位为氨基取代的吡咯吲哚化合物62和65的制备.在三乙胺条件下, 化合物61分别与59和65发生分子间磺酰胺化反应, 得到异源二聚体磺酰胺化合物66和69, 随后经过1, 3-二氯-5, 5-二甲基氧化, 脱去二氧化硫, 得到偶氮烯化合物67和70, 再在光照条件下, 化合物中偶氮烯键发生断裂, 得到关键异源二聚中间体68和71.随后68经过脱Boc基保护、氨基还原氨化甲基化、脱对叔丁基苯磺酰基保护、Red-Al还原氨基甲酸甲酯, 得到了异源二聚天然产物(—)-calycanthidine.异源二聚中间体71则经过LiEt3BH还原Troc基团为甲基、脱对叔丁基苯磺酰基保护、Red-Al还原氨基甲酸甲酯, 得到内消旋chimonanthine, 接着在D2O/CD3CO2D加热条件下发生重排, 完成了内消旋calycanthine的合成; 异源二聚天然产物(—)-desmethyl-meso-chimonanthine的合成则是通过化合物71脱出对叔丁基苯磺酰基保护、Red-Al还原氨基甲酸甲酯基团得到; (+)-desmethyl-meso-chimonanthine的合成是经过化合物71在锌粉/醋酸还原条件下脱除Troc基团、氨基还原氨化甲基化、脱除对叔丁基苯磺酰基保护、水解氨基甲酸甲酯基团得到.
2012年, 梁永民小组[17]发展了一种碘介导色胺衍生物环化-氧化自由基二聚串联反应, 实现了二聚环色胺生物碱(±)-folicanthine的合成(Scheme 9).该路线从色胺衍生物77出发, 在I2/Cs2CO3条件下, 化合物77发生环化-氧化自由基二聚串联反应, 通过一步反应得到关键二聚中间体78, 然后在萘钠条件下, 脱除对甲苯磺酰基保护, 再经过氨基还原氨化甲基化, 简洁高效地实现了天然产物(±)-folicanthine合成.

2013年, Ishikawa小组[18]发展了一种基于生源途径的氧化自由基二聚反应, 并以此为关键步骤实现了天然产物(+)-WIN 64821和(—)-ditryptophenaline的合成.该方法以L-色氨酸乙酯(80)为原料, 在甲磺酸条件下伯胺先成盐, 然后五氧化二钒对吲哚环选择性单电子氧化, 得到C-3, C-7和N-1位置的自由基, 再发生二聚反应, 得到C2对称二聚产物81和82, 以及非对称二聚产物83和84.虽然该反应的选择性不是很好, 但通过一步反应能得到了四个二聚关键中间体, 这些中间体通过几步化学转化, 可用于合成相应的天然产物.化合物81在一锅法条件下, 先与N-叔丁氧羰基-L-苯丙氨酸缩合, 然后在真空加热条件下脱Boc保护基、氨解环化形成二酮哌嗪环, 得到天然产物(+)-WIN 64821. (—)-ditryp-tophenaline则是经过化合物82先与N-叔丁氧羰基-N-甲基-L-苯丙氨酸缩合, 再脱Boc基保护、氨解环化两步反应得到(Scheme 10).

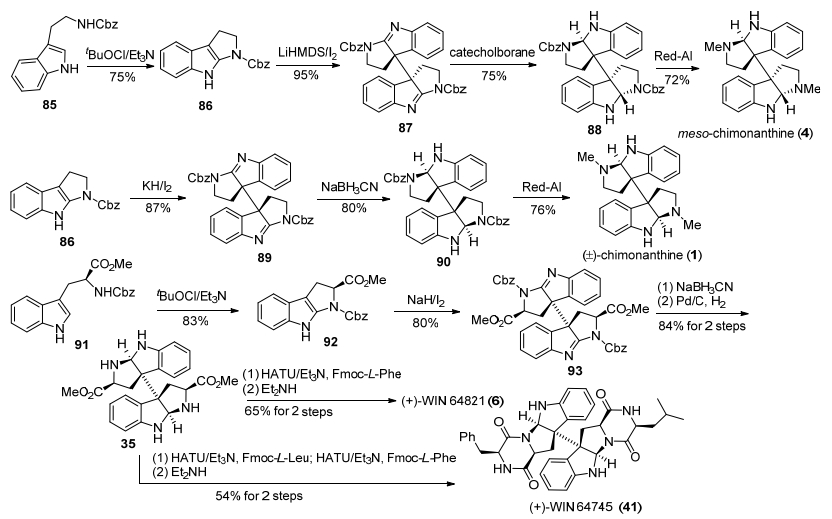
2014年, 李超忠小组[19]发展了一种基于Robinson[7]二聚环色胺生物碱的生源途径连续氧化-氧化-还原的策略.他们通过改变碱的类型, 得到不同类型二聚体, 进而完成天然产物meso-chimonanthine, (±)-chimonan-thine、(+)-WIN 64821和(+)-WIN 64745的合成.该路线以色胺衍生物85为原料, 首先, 在tBuOCl/Et3N条件下, 吲哚环发生氧化环化得到环化产物86, 随后在LiHMDS碱和碘氧化剂条件下, 氧化二聚得到了内消旋二聚中间体87, 然后经过亚胺还原、Red-Al还原氨基苄氧羰基, 完成了meso-chimonanthine的合成.化合物86在KH/I2条件下, 发生氧化二聚得到外消旋二聚关键中间体89, 再经过亚胺还原、Red-Al还原Cbz基团, 得到(±)-chimonanthine.天然产物(+)-WIN 64821和(+)-WIN 64745的合成, 则是以N-苄氧羰基-L-色氨酸甲酯91为原料, 在tBuOCl/Et3N条件下, 吲哚环发生氧化环化, 得到化合物92, 然后在NaH碱和碘氧化剂条件下, 92发生氧化自由基二聚反应, 得到关键二聚中间体93, 再经过亚胺还原、脱Cbz保护基, 得到了已知中间体35. 35经过与Fmoc-L-苯丙氨酸缩合, 接着在Et2NH条件下氨解环化, 得到了天然产物(+)-WIN 64821; (+)-WIN 64745的合成是经过35与Fmoc-L-亮氨酸、Fmoc-L-苯丙氨酸一锅法缩合, 再氨解环化得到的(Scheme 11).
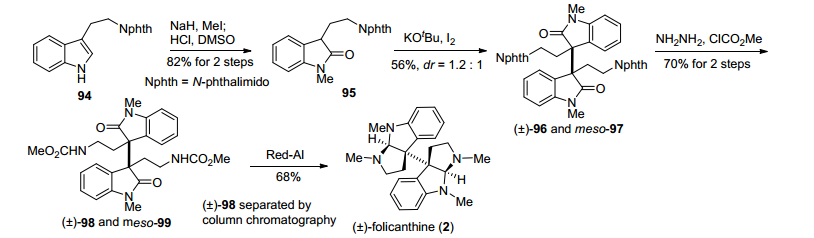
2015年, Bisai小组[20]在合成二聚环色胺生物碱(±)-folicanthine时, 报道了一种基于KOtBu/I2氧化色胺衍生物二聚反应的策略(Scheme 12).该策略以色胺衍生物94为原料, 通过吲哚环甲基化、吲哚环氧化, 得到氧化吲哚化合物95, 继而在KOtBu/I2条件下, 发生氧化自由基二聚反应, 以56%的收率得到外消旋二聚产物96和内消旋二聚产物97, 再经过肼解脱氮保护基邻苯二甲酰亚胺、氨基上甲酰甲酯基团、Red-Al还原氨化环化, 完成了(±)-folicanthine的合成.
2015年, 夏成峰小组[20a]报道了一种铜介导的色氨酸衍生物环化/氧化自由基二聚反应, 基于该关键反应进而完成了天然产物(+)-WIN 64821和(—)-ditrypto-phenaline的合成(Scheme 13).该合成方法以N-邻硝基苯磺酰基-L-色氨酸甲酯(100)为原料, 在CuCl2/DBU条件下, 色氨酸衍生物100发生吲哚环环化/氧化二聚串联反应, 得到以内型为主产物102和外型副产物101.内型产物102在苯硫酚条件下, 脱除邻硝基苯磺酰基, 随后在HATU条件下, 与N-苄氧羰基-L-苯丙氨酸36缩合得到酰胺化合物37, 再经过脱除苄氧羰基保护、氨解环化, 得到了(+)-WIN 64821.采用类似合成策略, 通过改变L-色氨酸甲酯保护基团, 可以实现内型和外型产物的比例控制.具体是以N-对硝基苯磺酰基-L-色氨酸甲酯(103)为原料, 在CuCl2/DBU条件下, 103发生吲哚环环化/氧化二聚反应, 得到以外型为主产物104, 再经过脱除对硝基苯磺酰基, 与N-苄氧羰基-N-甲基-L-苯丙氨酸(106)缩合, 得到酰胺107, 最后经过脱除苄氧羰基、氨解环化, 完成了(—)-ditryptophenaline的合成.

2015年, 夏成峰小组[21b]采用他们小组发展的铜介导色胺衍生物环化/氧化自由基二聚反应, 实现了其他二聚环色胺生物碱合成(Scheme 14).该方法以氯甲酸(+)-薄荷酯为手性辅基色胺衍生物108为原料, 在CuCl2/tBuOK条件下, 得到两种外消旋二聚体109和110, 且没有内消旋产物. 109再经过还原氨化甲基化、Red-Al还原酰胺得到(+)-folicanthine. 109在KOH加热条件下脱除手性辅基, 同时得到脱保护基产物112和重排产物113, 然后分别经过氨基甲酸甲酯化、Red-Al还原氨基甲酸甲酯, 完成了天然产物(+)-chimo-nanthine和(—)-calycanthine的合成.
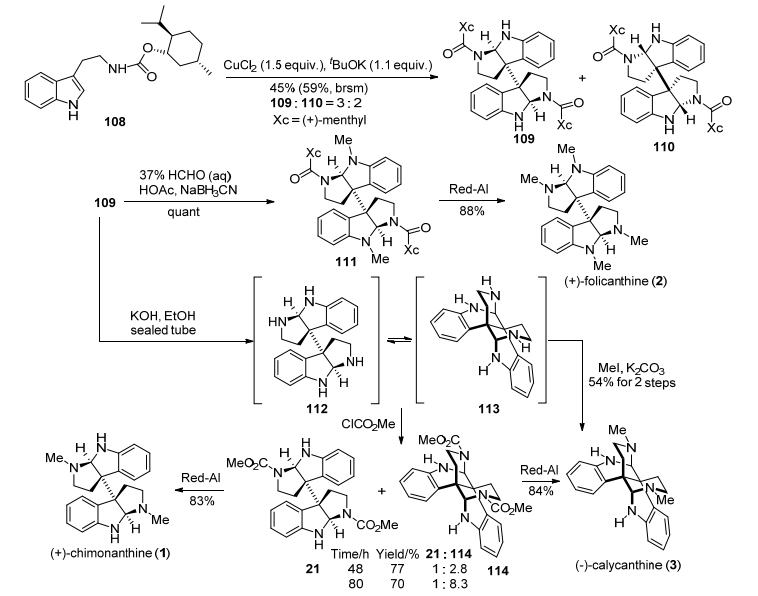
2015年, 我们小组[22a]发展了一种铜催化邻卤代苯胺类化合物芳基化串联氧化二聚反应, 基于该串联反应进而完成了二聚环色胺生物碱(+)-chimonanthine, (+)-folicanthine和(—)-calycanthine的合成(Scheme 15).该方法以2-溴苯胺(115)和1, 4-丁内酯(116)为原料, 通过氨解、苄基保护、醇氧化, 得到醛基化合物117, 再与R型手性叔丁基亚磺酰胺缩合得到亚胺化合物118, 接着用NaBH4还原、苄基保护, 得到铜催化前体化合物119.然后在CuI/LiN(SiMe3)2和tBuOOH一锅法条件下, 化合物119发生铜催化邻溴代苯胺类化合物芳基化串联氧化二聚反应, 得到了关键二聚中间体120和121, 尽管是非对映异构体, 该两个化合物未能够在柱层析上分离, 但经过稀盐酸脱叔丁基亚磺酰基保护、重结晶, 得到了99% ee值胺类化合物122, 再经过还原氨化甲基化、1-氯乙基氯甲酸酯脱苄基保护, 得到四胺化合物124, 随后经过还原氨化环化、脱苄基保护, 得到天然产物(+)-chimonanthine, 而(+)-folicanthine经过(+)-chimonanthine还原氨化甲基化得到, (+)-chimonanthine在AcOD/D2O加热条件下重排, 得到另外一个天然产物(—)-calycanthine.
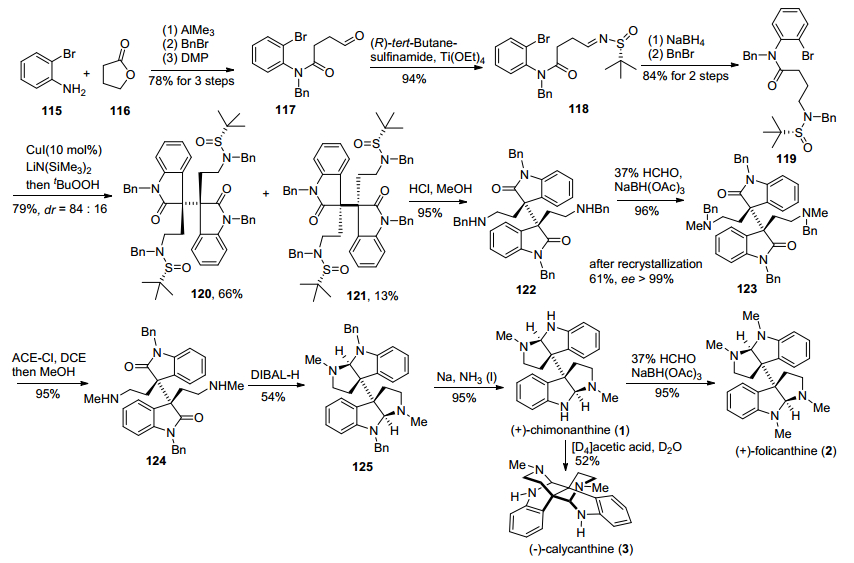
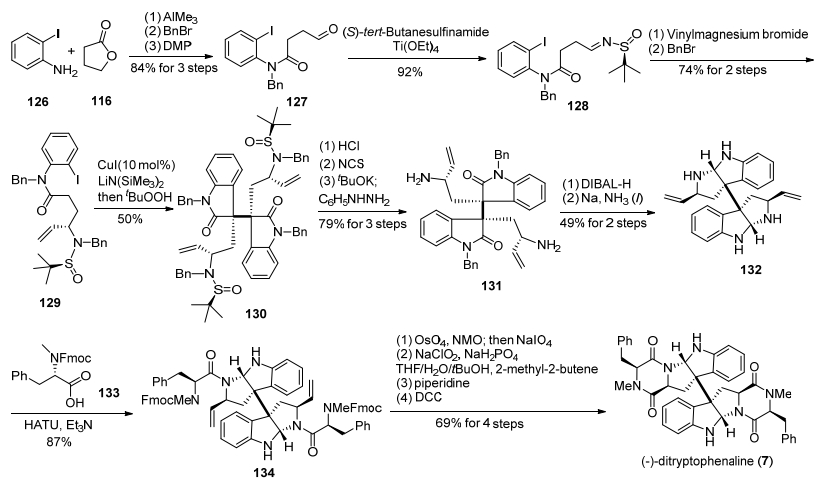
2015年, 我们小组[22b]仍然采用铜催化邻卤代苯胺类化合物芳基化串联氧化二聚反应为关键步骤, 实现了天然产物(—)-ditryptophenaline的全合成(Scheme 16).该合成路线从2-碘苯胺(126)和1, 4-丁内酯(116)出发, 通过氨解、苄基保护、醇氧化, 得到醛基化合物127, 再与S型手性叔丁基亚磺酰胺缩合得到亚胺化合物128, 经过与乙烯基溴化镁格氏试剂反应、苄基保护, 得到酰胺化合物129.然后在CuI/LiN(SiMe3)2和tBuOOH一锅法条件下, 129发生铜催化邻碘代苯胺类化合物芳基化串联氧化二聚反应, 以50%的收率得到了关键二聚中间体130, 紧接着经过脱叔丁基亚磺酰基、脱苄基保护, 得到伯胺化合物131.化合物131经过DIBAL-H还原氨化环化、脱苄基保护, 生成四胺化合物132.该中间体再在HATU条件下与FMOC-N-甲基-L-苯丙氨酸133缩合, 生成酰胺化合物134, 最后经过双键氧化切断、Pinnick氧化、脱Fmoc保护基、DCC酰胺缩合, 转化为天然产物(—)-ditryptophenaline.
2012年, Kanai小组[23]报道了利用手性锰盐/席夫碱复合物和Mg(OAc)2/苯甲酸复合体系, 催化氧化吲哚类化合物与硝基乙烯不对称双Michael加成反应, 构建二聚环色胺生物碱相邻手性季碳中心, 进而完成天然产物(+)-chimonanthine、(+)-folicanthine和(—)-calycanthine全合成的策略(Scheme 17).该路线以氧化吲哚135和靛红136为原料, 在醋酸和浓盐酸条件下发生Aldol反应, 经过Boc酐保护、双键氢化, 得到双氧化吲哚化合物137.化合物137在手性锰盐/席夫碱复合物催化下, 与硝基乙烯发生第一次不对称Michael加成反应, 引入了第一分子硝基乙烯, 构建了氧化吲哚化合物第一个手性季碳中心; 然后用硅胶去除手性金属锰盐/席夫碱复合物, 再加入Mg(OAc)2/苯甲酸催化, 串联发生第二次不对称Michael加成反应, 引入了第二分子硝基乙烯, 以69%的产率和95% ee值得到了关键中间体二聚氧化吲哚二硝基140.化合物140再经过硝基还原、氨基甲酸甲酯化, 得到酰胺化合物141, 随后在三乙基硼氢化锂条件下还原氨化环化、脱Boc保护, 得到已知中间体21, 然后参照Movassaghi小组[10]的方法, 完成了天然产物(+)-chimonanthine、(+)-folicanthine和(—)-calycanthine的合成.
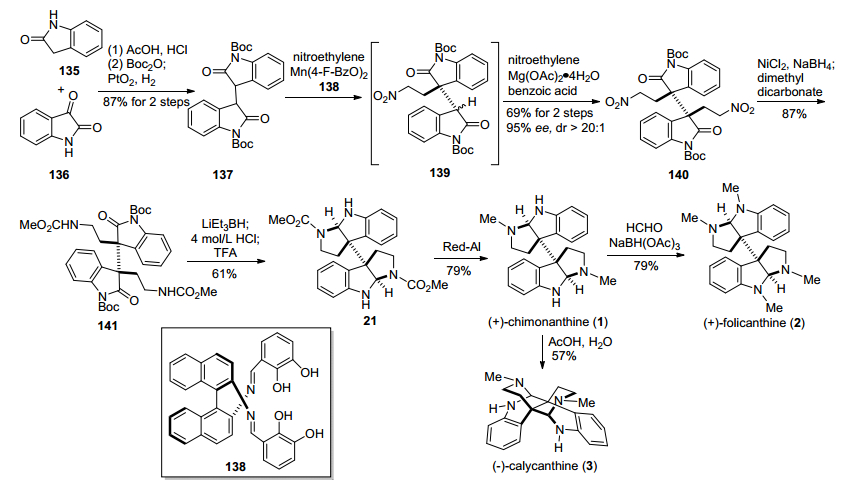
2013年, 龚流柱小组[24]报道了利用手性磷酸催化的3-羟基氧化吲哚类化合物与乙烯基氨基甲酸酯类化合物不对称亲核取代反应, 引入氧化吲哚C-3位手性季碳中心, 进而实现二聚环色胺生物碱(+)-folicanthine形式合成的方法(Scheme 18).该合成路线以3-羟基氧化吲哚类化合物142和乙烯基氨基甲酸酯类化合物143为原料, 在手性磷酸144催化下, 发生不对称亲核取代反应, 以82%的产率和90% ee值得到含手性季碳中心氧化吲哚类关键中间体145, 成功地引入了分子中C-3位的全碳季碳中心. 145再经过氮甲基化, 与盐酸羟氨盐酸盐反应, 生成酮肟化合物147和148, 接着在路易斯酸HgCl2条件下, 发生Beckmann重排, 得到酰胺化合物149.中间体149随后经过酰胺氮甲基化、吲哚环氧化, 生成氧化吲哚化合物150, 再在NaOH条件下, 与溴乙酸乙酯发生烷基化反应, 得到含相邻手性季碳化合物151, 接着在硝酸铈铵氧化条件下脱除PMP保护基, 与甲胺发生氨解反应, 得到已知酰胺中间体152, 最后参照Rodrigo等[25]的工作, 通过三步反应, 完成(+)-foli-canthine的合成.
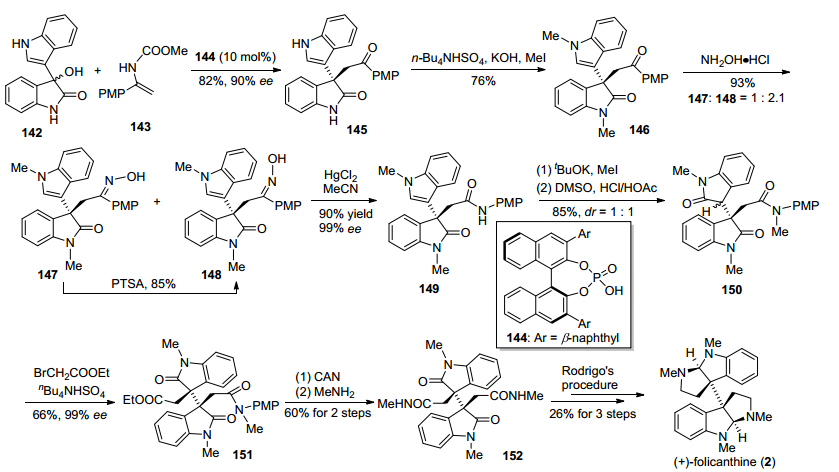
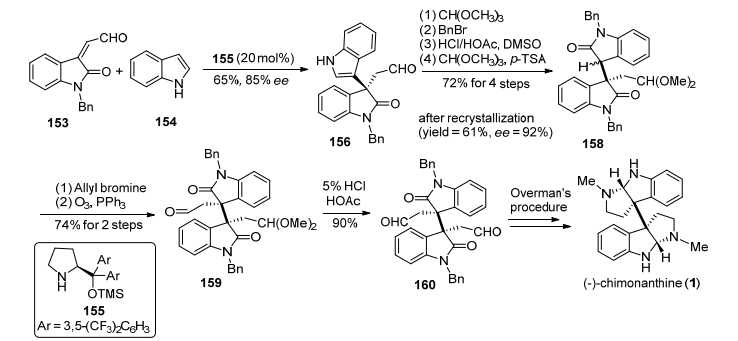
2013年, 张俊良小组[26]发展了一种有机小分子催化的吲哚与靛红衍生物不对称Michael加成反应, 构建了氧化吲哚C-3位关键的手性季碳中心, 进而完成了(—)-chimonanthine的形式合成.该路线利用靛红衍生物153和吲哚154在J rgensen-Hayashi有机小分子催化剂155作用下反应, 以65%的产率和85% ee值得到关键氧化吲哚中间体156, 然后通过重结晶使其ee值达到92%, 再经过醛基保护、苄基保护、吲哚环氧化、醛基保护, 得到氧化吲哚化合物158.在氧化吲哚化合物C-3位引入烯丙基, 再经过双键氧化切断、醛基脱保护, 得到了已知中间体160, 再参照Overman小组[27a]的合成方法, 可实现(—)-chimonanthine的合成(Scheme 19).
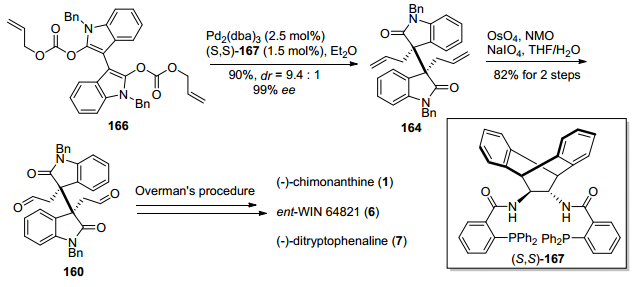
2013年, Trost小组[28]发展了一种基于钯催化的氧化吲哚类二碳酸二烯丙醇酯脱羧不对称烯丙基烷基化反应, 构筑了二聚氧化吲哚类化合物相邻手性季碳中心, 进而实现了(—)-chimonanthine, ent-WIN 64821和(+)-ditryptophenaline的形式合成(Scheme 20).该方法以氧化吲哚135和靛红136为原料, 经过Aldol反应、Boc酐保护、双键氢化三步反应, 得到了已知双氧化吲哚化合物137, 再与氯甲酸烯丙基酯反应, 得到二烯醇烯丙基碳酸酯化合物161, 然后在钯和手性氮/磷配体162条件下发生钯催化脱羧不对称烯丙基烷基化反应, 成功地构建了二聚环色胺生物碱中相邻手性季碳中心, 得到关键二聚氧化吲哚中间体163.接下来在OsO4/2, 6-lutidine条件下将中间体163的双键氧化切断, 得到已知化合物160, 参照Overman小组[27b]的方法, 实现ent-WIN 64821和(+)-ditryptophenaline的形式合成.二醛化合物160经过NaBH4还原, 得到已知中间体165, 参照Overman小组[25a]的方法, 也可以实现(—)-chimo-nanthine的形式合成.
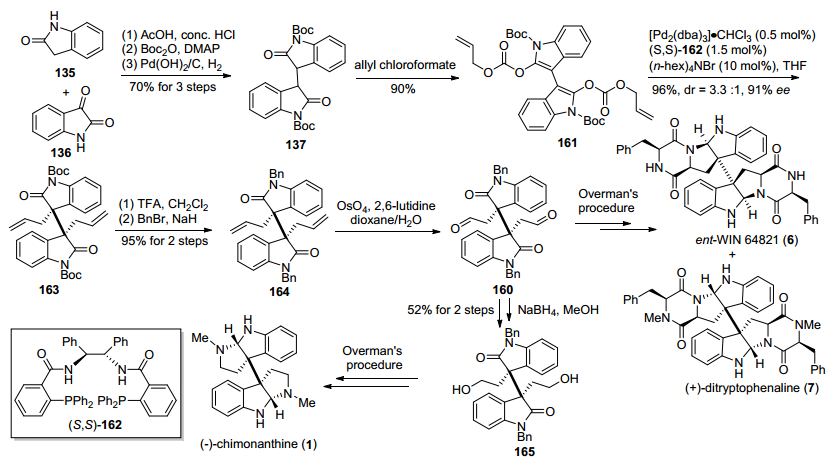
2014年, Bisai小组[29a]报道了与Trost小组[28]相类似的钯催化脱羧不对称烯丙基烷基化反应为关键的策略, 完成了二聚氧化吲哚类化合物相邻手性季碳的构筑.其产物通过后续化学转化得到已知中间体, 从而实现了(—)-chimonanthine、ent-WIN 64821和(+)-ditryptophen-aline的形式合成(Scheme 21).该方法基本上与Trost小组方法相类似, 与Trost小组不同的是, 钯催化剂由三(二亚苄基丙酮)二钯-氯仿加合物变为三(二亚苄基丙酮)二钯, 底物保护基由Boc变成了Bn, 采用了更大位阻的手性氮/磷配体167, 反应溶剂由四氢呋喃变成了乙醚, 反应体系及其他条件稍微有些不同, 但其dr值和ee值有了较大提升.
2018年, Bisai小组[29c]又发展了一种Cu(II)-tBu-PHOX催化3-羟基氧化吲哚类化合物与丙二酸苄基酯不对称烷基化反应, 构建了氧化吲哚类化合物C-3位手性季碳中心, 进而完成了(+)-folicanthine的形式合成(Scheme 22).首先, 化合物167和168在Cu(OTf)2催化剂和tBu-PHOX配体条件下, 167发生氧化吲哚C-3位脱羟基形成碳正离子, 然后受到丙二酸苄基酯168不对称亲核进攻, 以90%的产率和99% ee值得到关键化合物170.随后在LiCl加热条件下, 170发生β-酮酸苄酯Krapcho脱碳酸反应, 再经过吲哚环甲基化、吲哚环氧化, 得到双氧化吲哚化合物171.然后在NaOH条件下, 171发生氧化吲哚C-3位烷基化, 以82%的产率和大于(20 : 1)非对映选择性, 得到了含相邻手性季碳的化合物172, 再经过LiBH4还原, 得到已知二醇中间体165, 参照自己小组[29b]的方法, 即可实现(+)-folicanthine的形式合成.

2018年, Bisai小组[29d]还发展了一种基于硫脲催化3-对甲苯磺酰基氧化吲哚类化合物与丙二酸苄酯不对称亲核取代反应, 构建了氧化吲哚化合物手性季碳中心, 利用该策略进而实现了(—)-folicanthine, (—)-chimonanthine和(+)-calycanthine的形式合成.该方法以氧化吲哚类化合物174与丙二酸苄酯168在硫脲175催化作用下反应, 实现了丙二酸苄酯168对3-对甲苯磺酰基氧化吲哚类化合物174的不对称亲核取代, 分别以85%的产率、94% ee值和86%的产率、92% ee值, 得到了不同保护基的氧化吲哚类中间体176和177, 完成了氧化吲哚类化合物手性季碳中心的构筑(Scheme 23).化合物176在LiCl加热条件下发生β-酮酸苄酯Krapcho脱碳酸反应, 再经过甲基化, 生成化合物178. 178经过吲哚环氧化, 得到双吲哚化合物179, 然后179在NaOH条件下发生氧化吲哚环C-3位烷基化反应, 引入烷基侧链, 以82%产率和大于20:1的非对映选择性, 得到了含相邻手性季碳中心的中间体180.化合物180再经过LiBH4还原乙酯基团、Swern氧化, 得到已知中间体化合物181, 最后参照自己小组[29b]的方法, 实现了(+)-folicanthine的形式全合成.化合物177经过与化合物176类似的化学转化, 可实现(—)-chimonanthine和(+)-calycanthine的形式合成.
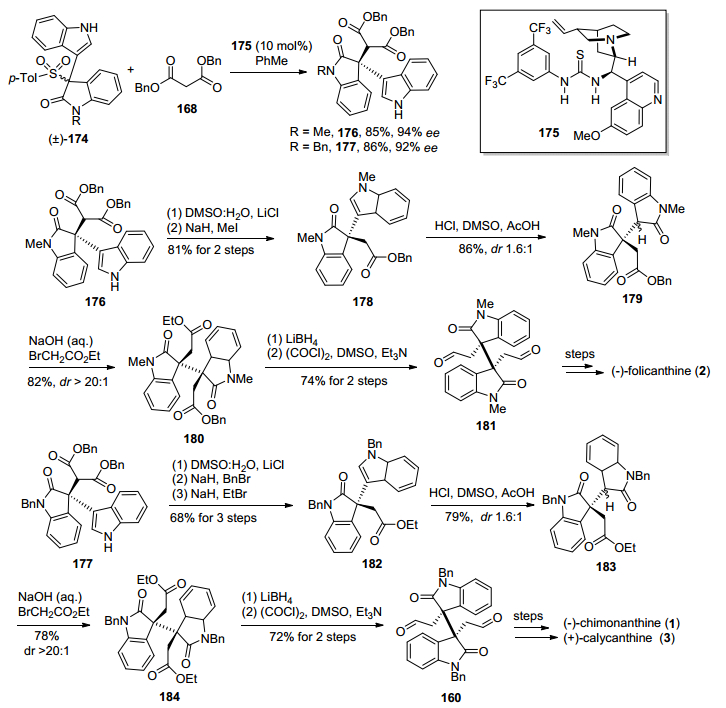
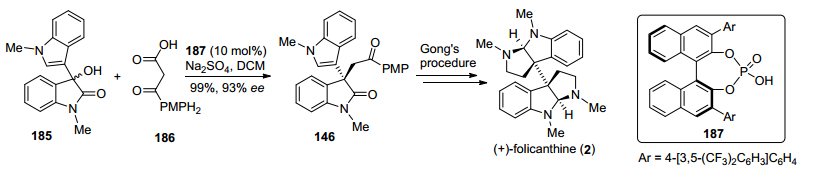
2015年, 马军安小组[30]发展了一种手性磷酸催化β-酮酸化合物与3-羟基氧化吲哚衍生物脱羧烷基化反应, 构筑了氧化吲哚化合物C-3位手性季碳中心, 进而实现了(+)-folicanthine的形式合成(Scheme 24).该方法以β-酮酸186和3-羟基氧化吲哚化合物185为原料, 在手性磷酸187催化下发生脱羧烷基化反应, 通过一步反应以99%的产率和93% ee值得到了已知关键中间体146, 然后参照龚流柱小组[24]和Rodrigo小组[25]的路线, 即可实现(+)-folicanthine的形式合成.
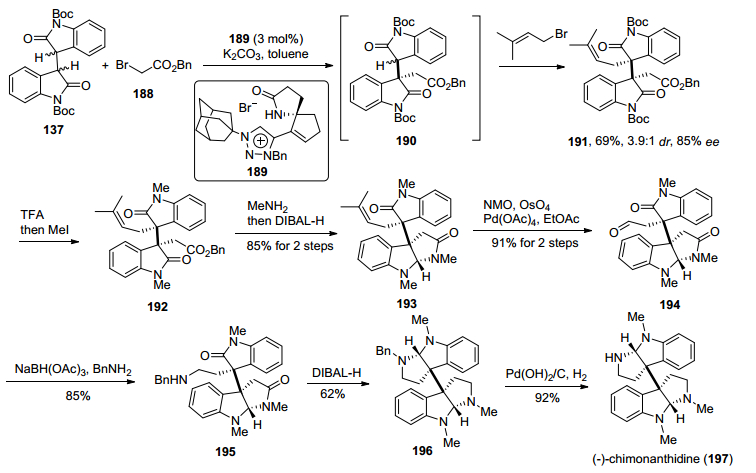
2018年, 涂永强小组[31]发展了一种新型手性螺环吡咯酰胺(SPA)衍生的三唑类有机阳离子, 以其催化二聚氧化吲哚类化合物与同种亲电试剂/不同种亲电试剂的不对称二烷基化反应, 从而实现了二聚氧化吲哚化合物相邻季碳中心对映选择性构建, 利用该方法实现了异源二聚环色胺生物碱(—)-chimonanthidine的首次不对称全合成(Scheme 25).该方法从已知二聚氧化吲哚类化合物137出发, 采用手性螺环吡咯酰胺(SPA)衍生的三唑类有机阳离子189催化二聚氧化吲哚类化合物137与烷基溴代物188和191的不对称二烷基化反应, 以69%产率、85% ee值得到了关键中间体化合物191, 然后经过TFA脱Boc基保护、氮甲基化, 得到了二聚氧化吲哚化合物192, 随后经过氨解、DIBAL-H还原氨化环化, 得到双氧化吲哚化合物193.他们将化合物193的双键氧化切断、继而醛基还原氨化、DIBAL-H还原氨化环化、脱苄基, 完成了异源二聚环色胺生物碱(—)-chimonanthidine的首次不对称全合成.
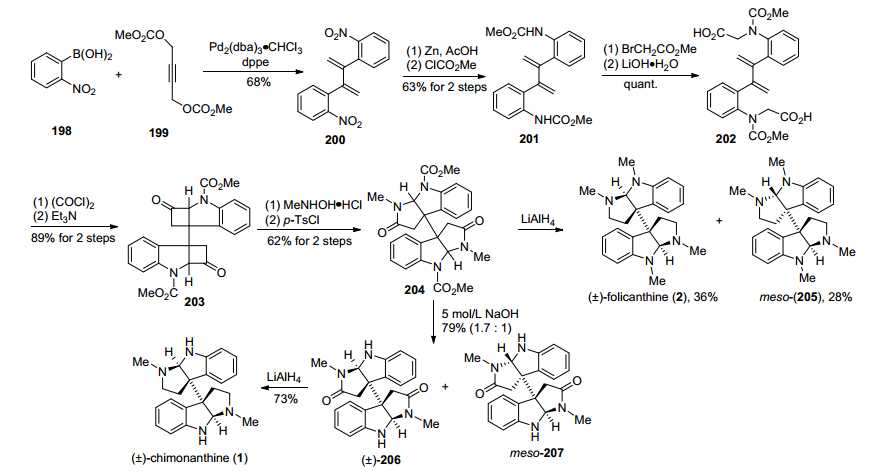
2013年, Shishido小组[32]报道了一种分子内氨甲酰基乙烯酮与烯烃[2+2]环加成反应作为关键步骤的合成策略(Scheme 26).该方法以2-硝基苯硼酸化合物198和1, 4-丁炔二醇碳酸甲酯化合物199为原料, 在钯催化下偶联得到1, 3-丁二烯化合物202, 接着在锌粉/醋酸条件下还原硝基为氨基、再经过氨基上甲酸甲酯基团, 得到酰胺化合物201, 然后经过氨基烷基化、酯水解, 得到羧酸化合物202.化合物202再经过羧酸酰氯化、三乙胺脱除HCl, 形成氨甲酰基乙烯酮, 与双键发生分子内[2+2]环加成反应, 得到关键二聚化合物203, 203与N-甲基羟胺盐酸盐反应, 得到酮肟化合物, 再与对甲苯磺酰氯反应, 得到的磺酸酯化合物经过扩环重排, 得到扩环酰胺化合物204.他们将化合物204的氨基甲酸甲酯经过LiAlH4还原, 完成了(±)-folicanthine全合成.将化合物204经过NaOH脱氨基甲酸甲酯、LiAlH4还原, 得到了(±)-chimonanthine.
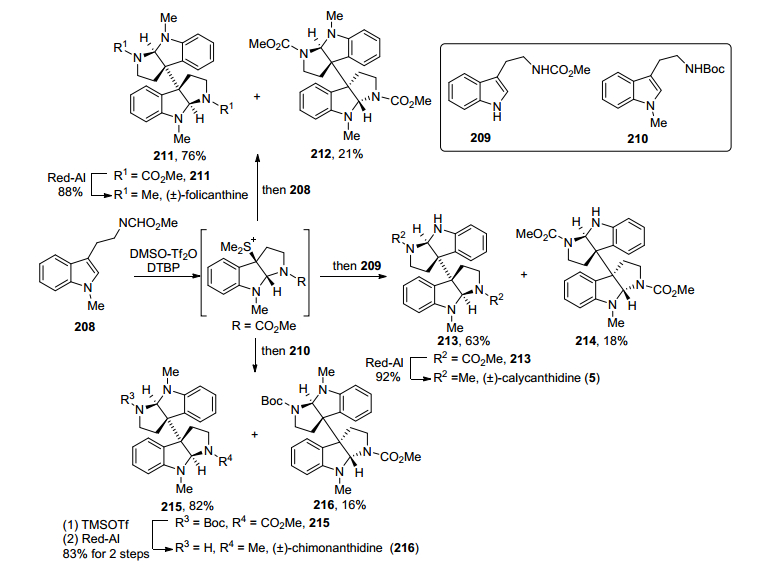
2014年Kawasaki小组[33]报道了一种基于硫鎓离子一锅法构建同源/异源二聚环色胺结构的方法, 通过该方法得到关键二聚中间体后, 经化学转化, 完成了同源二聚体(±)-folicanthine和异源二聚体(±)-calycan-thidine、(±)-chimonanthidine的全合成(Scheme 27).该合成路线以色胺类衍生物208为原料, 首先在DMSO和Tf2O体系下产生硫鎓离子, 然后发生色胺类衍生物吲哚环C-3位亲核进攻, 进而环化得到环化硫鎓离子中间体, 再分别加入色胺类衍生物208、209或210作为亲核试剂, 得到主要为外消旋的二聚体211、213或215和少量内消旋二聚体212、214或216.它们分别经过Red-Al直接还原氨基甲酸甲酯基团或先经过脱Boc保护基、再用Red-Al还原氨基甲酸甲酯基团, 完成了同源二聚体(±)-folicanthine或异源二聚体(±)-calycanthidine和(±)-chimonanthidine的合成.
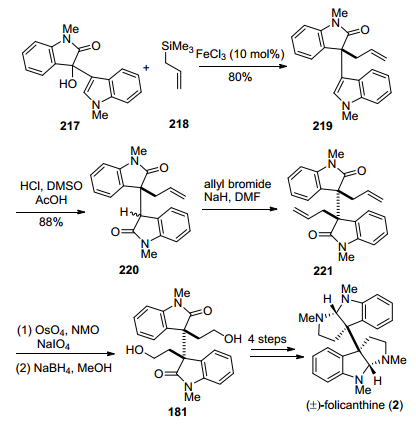
2017年, Bisai小组[29e]报道了一种FeCl3催化的3-羟基氧化吲哚类化合物与烯丙基三甲基硅烷的烯丙基烷基化反应(Scheme 28).他们将217和218在FeCl3催化下发生氧化吲哚C-3位烯丙基烷基化, 得到关键中间体219, 再经过吲哚环氧化、烯丙基烷基化, 得到二聚氧化吲哚中间体221, 经过双键的氧化切断, 得到已知中间体181, 再参照自己小组[27b]的方法, 实现了(±)-folicanthine的形式合成.
天然产物全合成是有机合成化学研究的重要内容之一, 对天然产物的结构鉴定、结构修饰、生源合成途径的验证以及新试剂、新反应的发展, 具有重要研究意义.本文对自2007年以来二聚环色胺生物碱的全合成进行了简要综述和总结, 由于二聚环色胺生物碱独特的相邻季碳中心, 如何立体选择性地构建该类分子中的季碳中心, 仍然是这类化合物合成中的难点.因此发展简洁高效地构建其骨架中相邻季碳中心的方法, 仍是有机合成化学家研究的热点内容, 今后的方向应该是利用简单的原料, 发展更高效、适用的催化体系或/和催化剂, 实现相邻全碳季碳中心的同步不对称合成.
For selected review, see:
(a) Crich, D.; Banerjee, A. Acc. Chem. Res. 2007, 40, 151.
(b) Ruiz-Sanchis, P.; Savina, S. A.; Albericio, F.; Álvarez, M. Chem. Eur. J. 2011, 17, 1388.
(c) Repka, L. M.; Reisman, S. E. J. Org. Chem. 2013, 78, 12314.
(d) Kim, J.; Movassaghi, M. Acc. Chem. Res. 2015, 48, 115.
(e) Liang, K. J.; Xia, C. F. Chin. J. Chem. 2017, 35, 255.
(a) Popp, J. L.; Musza, L. L.; Barrow, C. J.; Rudewicz, P. J.; Houck, D. R. J. Antibiot. 1994, 47, 411.
(b) Oleynek, J. J.; Sedlock, D. M.; Barrow, C. J.; Appell, K. C.; Casiano, F.; Haycock, D.; Ward, S. J.; Kaplita, P.; Gillum, A. M. J. Antibiot. 1994, 47, 399.
(c) Sedlock, D. M.; Barrow, C. J.; Brownell, J. E.; Hong, A.; Gillum, A. M.; Houck, P. R. J. Antibiot. 1994, 47, 391.
Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Nat. Chem. Biol. 2005, 1, 143. doi: 10.1038/nchembio721
Steven, A.; Overman, L. E. Angew. Chem. Int. Ed. 2007, 46, 5488. doi: 10.1002/anie.200700612
Schmidt, M. A.; Movassaghi, M. Synlett 2008, 313.
Woodward, R. B.; Yang, N. C.; Katz, T. J.; Clark, V. M.; Harley-Mason, J.; Ingleby, R. F. J.; Sheppard, N. Proc. Chem. Soc. 1960, 76.
Robinson, R.; Teuber, H. J. Chem. Ind. 1954, 783.
Kirby, G. W.; Shah, S. W.; Herbert, E. J. J. Chem. Soc. C 1969, 1916.
Steven, A.; Overman, L. E. Angew. Chem. Int. Ed. 2007, 46, 5488.
Movassaghi, M., Schmidt, M. A. Angew. Chem. Int. Ed. 2007, 46, 3725. doi: 10.1002/anie.200700705
Movassaghi, M.; Schmidt. M. A.; Ashenhurst, J. A. Angew. Chem. Int. Ed. 2008, 47, 1485. doi: 10.1002/anie.200704960
(a) Pérez-Balado, C.; de Lera, Á. R. Org. Lett. 2008, 10, 3701.
(b) Pérez-Balado, C.; Rodríguez-Graña, P.; de Lera, Á. R. Chem. Eur. J. 2009, 15, 9928.
Xie, W. Q.; Jiang, G. D.; Liu, H.; Hu, J. D.; Pan, X. X.; Zhang, H.; Wan, X. L.; Lai, Y. S.; Ma, D. W. Angew. Chem. Int. Ed. 2013, 52, 12924. doi: 10.1002/anie.201306774
Peng, Y.; Luo, L.; Yan, C. S.; Zhang, J. J.; Wang, Y. W. J. Org. Chem. 2013, 78, 10960. doi: 10.1021/jo401936v
Wada, M.; Murata, T.; Oikawa, H.; Oguri, H. Org. Biomol. Chem. 2014, 12, 298. doi: 10.1039/C3OB41918E
Movassaghi. M.; Lathrop. S. P. Chem. Sci.2014, 5, 333. doi: 10.1039/C3SC52451E
Li, Y. X.; Wang, H. X.; Ali, S.; Xia, X. F.; Liang, Y. M. Chem. Commun. 2012, 48, 2343 doi: 10.1039/c2cc16637b
Tadano, S.; Mukaeda, Y.; Ishikawa, H. Angew. Chem. Int. Ed. 2013, 52, 7990. doi: 10.1002/anie.201303143
Sun, D. Q.; Xing, C. Y.; Wang, X. Q.; Su, Z. Q.; Li, C. Z. Org. Chem. Front. 2014, 1, 956. doi: 10.1039/C4QO00165F
Ghosh, S.; Chaudhuri, S.; Bisai, A. Org. Lett. 2015, 17, 1373.
(a) Liang, K. J.; Deng, X.; Tong, X. G.; Li, D. S.; Ding, M.; Zhou, A. K.; Xia, C. F. Org. Lett. 2015, 17, 206.
(b) Ding, M.; Liang, K. J.; Pan, R.; Zhang, H. B.; Xia, C. F. J. Org. Chem. 2015, 80, 10309.
(a) Shen, X. F.; Zhou, Y. Y.; Xi, Y. K.; Zhao, J. F.; Zhang, H. B. Chem. Commun. 2015, 51, 14873.
(b) Shen, X. F.; Zhou, Y. Y.; Xi, Y. K.; Zhao, J. F.; Zhang, H. B. Nat. Prod. Bioprospect. 2016, 6, 117
Mitsunuma, H.; Shibasaki, M.; Kanai, M.; Matsunaga, S. Angew. Chem. Int. Ed. 2012, 51, 5217. doi: 10.1002/anie.201201132
Guo, C.; Song, J.; Huang, J.-Z.; Chen, P.-H.; Luo, S.-W.; Gong, L.-Z. Angew. Chem. Int. Ed. 2012, 51, 1046. doi: 10.1002/anie.201107079
Fang, C.-L.; Horne, S.; Taylor, N.; Rodrigo, R. J. Am. Chem. Soc. 1994, 116, 9480. doi: 10.1021/ja00100a010
Liu, R. R.; Zhang, J. L. Org. Lett. 2013, 15, 2266. doi: 10.1021/ol400845c
(a) Link, J. T.; Overman, L. E. J. Am. Chem. Soc.1996, 118, 8166.
(b) Paone, D. V.; Overman, L. E. J. Am. Chem. Soc.2001, 123, 9465.
Trost, B. M.; Osipov, M. Angew. Chem. Int. Ed. 2013, 52, 9176. doi: 10.1002/anie.201302805
(a) Ghosh, S.; Bhunia, S.; Kakde, B. N.; De, S.; Bisai, A. Chem. Commun. 2014, 50, 2434.
(b) Ghosh, S.; Chaudhuri, S.; Bisai, A. Chem. Eur. J. 2015, 21, 17479.
(c) Babu, K. N.; Kinthada, L. K.; Das, P. P.; Bisai, A. Chem. Commun. 2018, 54, 7963.
(d) Babu, K. N.; Roy, A.; Singh, M.; Bisai, A. Org. Lett. 2018, 20, 6327.
(e) Kinthada, L. K.; Medisetty, S. R.; Parida, A.; Babu, K. N.; Bisai, A. J. Org. Chem. 2017, 82, 8548.
Tang, X. D.; Li, S.; Guo, R.; Nie, J.; Ma, J. A. Org. Lett. 2015, 17, 1389. doi: 10.1021/acs.orglett.5b00159
Chen, S.-K.; Ma, W.-Q.; Yan, Z.-B.; Zhang, F.-M.; Wang, S.-H.; Tu, Y.-Q.; Zhang, X.-M.; Tian, J.-M. J. Am. Chem. Soc.2018, 140, 10099. doi: 10.1021/jacs.8b05386
Araki, T.; Manabe, Y.; Fujioka, K.; Yokoe, H.; Kanematsu, M.; Yoshida, M.; Shishido, K. Tetrahedron Lett. 2013, 54, 1012. doi: 10.1016/j.tetlet.2012.12.057
Tayu, M.; Higuchi, K.; Ishizaki, T.; Kawasaki, T. Org. Lett. 2014, 16, 3613. doi: 10.1021/ol5012373

图式1 二聚环色胺生物碱的生源途径假说
Scheme 1 Hypothesis for the biosynthesis of cyclotryptamine alkaloids

图式2 Movassaghi小组基于还原自由基二聚的合成路线
Scheme 2 Movassaghi's synthetic route based on reductive dimerization

图式3 Movassaghi小组的(—)-ditryptophenaline合成路线
Scheme 3 Movassaghi's synthetic route toward (—)-ditrypto-phenaline

图式4 de Lera小组的(+)-WIN 64745合成路线
Scheme 4 de Lera's synthetic route toward (+)-WIN 64745

图式8 Movassaghi小组的异源二聚生物碱合成路线
Scheme 8 Movassaghi's synthetic route toward heterodimeric alkaloids

图式9 梁永民小组的(±)-folicanthine合成路线
Scheme 9 Liang's synthetic route toward (±)-folicanthine

图式10 Ishikawa小组基于氧化二聚的合成路线
Scheme 10 Ishikawa's synthetic route based on oxidative dimerization

图式11 李超忠小组碘介导的氧化二聚合成路线
Scheme 11 Li's synthetic route based on iodine mediated dimerization

图式12 Bisai小组的(±)-folicanthine合成路线
Scheme 12 Bisai's synthetic route toward (±)-folicanthine

图式13 夏成峰小组的(—)-ditryptophenaline合成路线
Scheme 13 Xia's synthetic route toward (—)-ditryptophenaline

图式14 夏成峰小组的(+)-chimonanthine合成路线
Scheme 14 Xia's synthetic route toward (+)-chimonanthine

图式15 张洪彬小组的(+)-chimonanthine合成路线
Scheme 15 Zhang's synthetic route toward (+)-chimonanthine

图式16 张洪彬小组的(—)-ditryptophenaline合成路线
Scheme 16 Zhang's synthetic route toward (—)-ditryptophenaline

图式17 Kanai小组的催化不对称迈克尔加成合成路线
Scheme 17 Kanai's synthetic route based on asymmetric Michael additions

图式18 龚流柱小组的(+)-folicanthine合成路线
Scheme 18 Gong's synthetic route toward (+)-folicanthine

图式20 Trost小组的(+)-ditryptophenaline及(—)-chimonanthine形式合成路线
Scheme 20 Trost's formal synthesis of (+)-ditryptophenaline and (—)-chimonanthine

图式22 Bisai小组的(+)-folicanthine形式合成路线
Scheme 22 Bisai's formal synthesis of (+)-folicanthine

图式23 Bisai小组的(—)-chimonanthine和(+)-calycanthine形式合成路线
Scheme 23 Bisai's formal synthesis of (—)-chimonanthine and (+)-calycanthine

图式24 马军安小组(+)-folicanthine的形式合成路线
Scheme 24 Ma's formal synthetic route toward (+)-folicanthine

图式26 Shishido小组的(±)-folicanthine及(±)-chimonanthine合成路线
Scheme 26 Shishido's synthesis of (±)-folicanthine and (±)-chimonanthine

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:







