Key Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072
b.
Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072
c.
Department of Chemistry, Southern University of Science and Technology, Shenzhen Grubbs Institute, Shenzhen 518055
d.
Sauvage Center for Molecular Sciences, Wuhan University, Wuhan 430072
Received Date:
22 March 2019 Revised Date:
23 April 2019 Available Online:
25 June 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21871212) and the Natural Science Foundation of Hubei Province (No. 2018CFB430)
Abstract:
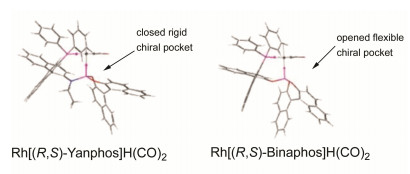
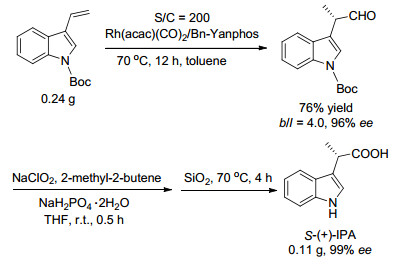
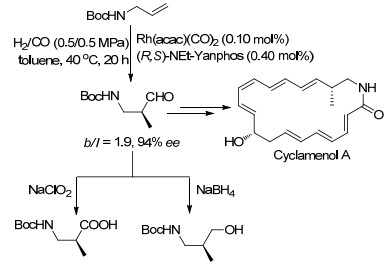
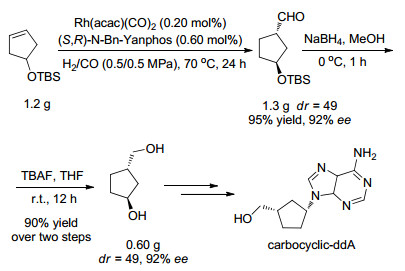
Asymmetric hydroformylation is one of the most important reactions for preparation of chiral aldehydes from alkenes. Recently, significant progress has been made in this field and a series of new ligands have been developed. Asymmetric hydroformylation of several important alkenes has been achieved, offering efficient and concise methods for the synthesis of chiral aldehydes. In this review, the achievements of asymmetric hydroformylation of typical alkenes and the development of ligands for asymmetric hydroformylation are summarized.
(a) Sakai, N.; Mano, S.; Nozaki, K.; Takaya, H. J. Am. Chem. Soc. 1993, 115, 7033. (b) Nozaki, K.; Nanno, T.; Takaya, H. J. Organomet. Chem. 1997, 527, 103. (c) Lambers-Verstappen, M. M. H.; de Vries, J. G. Adv. Synth. Catal. 2003, 345, 478. (d) Tanaka, R.; Nakano, K.; Nozaki, K. J. Org. Chem. 2007, 72, 8671.
(a) Noonan, G. M.; Fuentes, J. A.; Cobley, C. J.; Clarke, M. L. Angew. Chem., Int. Ed. 2012, 51, 2477. (b) Noonan, G. M.; Cobley, C. J.; Mahoney, T.; Clarke, M. L. Chem. Commun. 2014, 50, 1475.
[9]
(a) Babin, J. E.; Whiteker, G. T. US 5360938, 1993 [Chem. Abstr. 1995, 122, 186609]. (b) Buisman, G. J. H.; Vos, E. J.; Kamer, P. C. J.; Van Leeuwen, P. W. N. M. J. Chem. Soc., Dalton Trans. 1995, 409.
[10]
(a) Cobley, C. J.; Klosin, J.; Qin, C.; Whiteker, G. T. Org. Lett. 2004, 6, 3277. (b) Cobley, C. J.; Gardner, K.; Klosin, J.; Praquin, C.; Hill, C.; Whiteker, G. T.; Zanotti-Gerosa, A. J. Org. Chem. 2004, 69, 4031.
[11]
(a) Clark, T. P.; Landis, C. R.; Freed, S. L.; Klosin, J.; Abboud, K. A. J. Am. Chem. Soc. 2005, 127, 5040. (b) Watkins, A. L.; Landis, C. R. J. Am. Chem. Soc. 2010, 132, 10306. (c) Adint, T. T.; Landis, C. R. J. Am. Chem. Soc. 2014, 136, 7943.
[12]
Axtell, A. T.; Cobley, C. J.; Klosin, J.; Whiteker, G. T.; ZanottiGerosa, A.; Abboud, K. A. Angew. Chem., Int. Ed. 2005, 44, 4. doi: 10.1002/anie.200462726
Hua, Z.; Vassar, V. C.; Choi, H.; Ojima, I. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5411. doi: 10.1073/pnas.0307101101
[15]
(a) Kuil, M.; Goudriaan, P. E.; van Leeuwen, P. W. N. M.; Reek, J. N. H. Chem. Commun. 2006, 4679. (b) Bellini, R.; Reek, J. N. H. Chem.-Eur. J. 2012, 18, 13510.
[16]
Breit, B.; Breuninger, D. J. Am. Chem. Soc. 2004, 126, 10244. doi: 10.1021/ja0467364
[17]
Worthy, A. D.; Joe, C. L.; Lightburn, T. E.; Tan, K. L. J. Am. Chem. Soc. 2010, 132, 14757. doi: 10.1021/ja107433h
[18]
(a) Zhao, B.; Peng, X.; Wang, Z.; Xia, C.; Ding, K. Chem.-Eur. J. 2008, 14, 7847. (b) Peng, X.; Wang, Z.; Xia, C.; Ding, K. Tetrahedron Lett. 2008, 49, 4862.
Thomas, P. J.; Axtell, A. T.; Klosin, J.; Peng, W.; Rand, C. L.; Clark, T. P.; Landis, C. R.; Abboud, K. A. Org. Lett. 2007, 9, 2665. doi: 10.1021/ol070900l
[21]
Tan, R. C.; Zheng, X.; Qu, B.; Sarer, C. A.; Fandrick, K. R.; Senanayake, C. H.; Zhang, X. M. Org. Lett. 2016, 18, 3346. doi: 10.1021/acs.orglett.6b01452
Dingwall, P.; Fuentes, J. A.; Crawford, L.; Slawin, A. M. Z.; Bühl, M.; Clarke, M. L. J. Am. Chem. Soc. 2017, 139, 15921. doi: 10.1021/jacs.7b09164
[27]
Iu, L.; Fuentes, J. A.; Janka, M. E.; Fontenot, K. J.; Clarke, M. L. Angew. Chem., Int. Ed. 2019, 58, 2120. doi: 10.1002/anie.v58.7
[28]
Horiuchi, T.; Ohta, T.; Shirakawa. E.; Nozaki, K.; Takaya, H. J. Org. Chem. 1997, 62, 4285. doi: 10.1021/jo9624051
[29]
(a) Diéguez, M.; Pamies, O.; Claver, C. Chem. Commun. 2005, 1221. (b) Gual, A.; Godard, C.; Castillón, S.; Claver, C. Adv. Synth. Catal. 2010, 352, 463.
[30]
(a) Chikkali, S. H.; Bellini, R.; Berthon-Gelloz, G.; van der Vlugt, J. I.; de Bruin, B.; Reek, J. N. H. Chem. Commun. 2010, 46, 1244. (b) Chikkali, S. H.; Bellini, R.; de Bruin, B.; van der Vlugt, J. I.; Reek, J. N. H. J. Am. Chem. Soc. 2012, 134, 6607.
[31]
Rovira, L.; Vaquero, M.; Vidal-Ferran, A. J. Org. Chem. 2015, 80, 10397. doi: 10.1021/acs.joc.5b01805
[32]
Watkins, A. L.; Hashiguchi, B. G.; Landis, C. R. Org. Lett. 2008, 10, 4553. doi: 10.1021/ol801723a
[33]
Fuentes, J. A.; Pittaway, R.; Clarke, M. L. Chem.-Eur. J. 2015, 21, 10645. doi: 10.1002/chem.v21.30
[34]
Sherill, W. M.; Rubin, M. J. Am. Chem. Soc. 2008, 130, 13804. doi: 10.1021/ja805059f
(a) McDonald, R. I.; Wong, G. W.; Neupane, R. P.; Stahl, S. S.; Landis, C. R. J. Am. Chem. Soc. 2010, 132, 14027. (b) Abrams, M. L.; Foarta, F.; Landis, C. R. J. Am. Chem. Soc. 2014, 136, 14583.
[38]
Clemens, A. J. L.; Burke, S. D. J. Org. Chem. 2012, 77, 2983. doi: 10.1021/jo300025t
[39]
(a) Worthy, A. M.; Joe, C. L.; Lightburn, T. E.; Tan, K. L. J. Am. Chem. Soc. 2010, 132, 14757. (b) Joe, C. L.; Blaisdell, T. P.; Geoghan, A. F.; Tan, K. L. J. Am. Chem. Soc. 2014, 136, 8556.
[40]
Gadzikwa, T.; Bellini, R.; Dekker, H. L.; Reek, J. N. H. J. Am. Chem. Soc. 2012, 134, 2860. doi: 10.1021/ja211455j


 下载:
下载:



































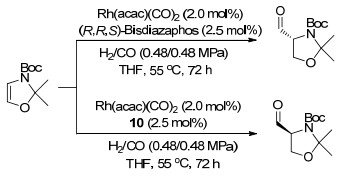







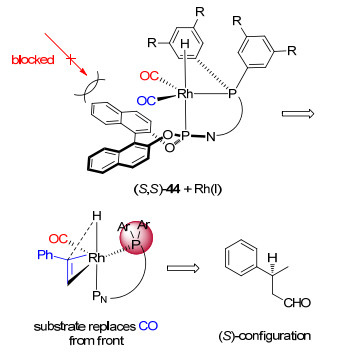



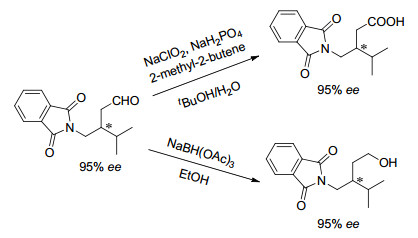






 下载:
下载:








