图式 1.
Huisgen内盐的形成
Scheme 1.
Formation of Huisgen zwitterion

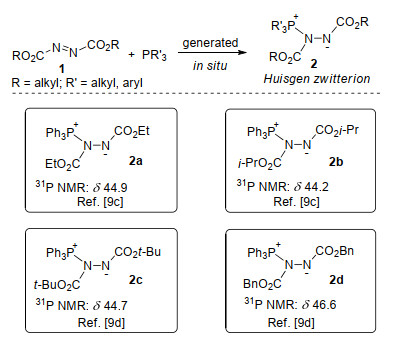
有机磷化合物作为有机合成试剂、配体以及催化剂, 在有机合成中具有重要用途[1].近二十年来, 叔膦促进的有机反应受到了人们的广泛关注, 大量高效、高选择性的有机合成方法得以报道.这一研究趋势主要与叔膦化合物独特而又丰富的化学性质有关.叔膦具有较强的亲核性, 如通过对缺电子多重键的亲核进攻, 易产生内盐形式的活性中间体; 同时, 叔膦又具有良好的离去能力.鉴于此, 叔膦已成为一类优良的催化剂.众多叔膦催化的反应[2]:包括Rauhut-Currier反应、Morita-Baylis- Hillman反应、活化烯或炔的迈克尔加成反应, 炔烃的异构化反应, 以及形式多样的环化反应等得以报道, 并在近年来取得重要进展.此外, 叔膦还具有强的亲氧性, 可在反应中以磷氧化物的形式发生消除, 完成化学计量磷的反应[3].叔膦促进的经典有机人名反应[4], 如Wittig烯化反应[5]、Mitsunobu反应[6]、Staudinger反应[7]和Appel反应[8]在有机合成中发挥着重要作用.在叔膦促进的反应中, 大都经叔膦对底物中缺电子多重键加成形成的内盐中间体进行, 常用的反应底物包括缺电子烯、炔、联烯酸酯, 以及Morita-Baylis-Hillman (MBH)衍生物, 而对其他类型的底物则研究较少[2, 3].因此, 继续拓展叔膦促进的反应底物类型, 将进一步促进叔膦化合物在有机合成中的应用.
Huisgen内盐是叔膦与偶氮二甲酸酯加成形成的两性离子中间体, 其准确结构在20世纪60年代由化学家Huisgen等[9]确定(Scheme 1).其中, 常用的叔膦试剂主要有三苯基膦(Ph3P)、三甲基膦(Me3P)、三正丁基膦(n-Bu3P)等, 由于烷基膦在空气中容易被氧化, 且价格相对较高, 因此最常用的叔膦是Ph3P.常见的偶氮试剂包括偶氮二甲酸二乙酯(DEAD, 1a)、偶氮二甲酸二异丙酯(DIAD, 1b)、偶氮二甲酸二叔丁酯(DTBAD, 1c)、偶氮二甲酸二苄酯(DBAD, 1d)、偶氮二甲酸二甲酯(DMAD, 1e), 考虑到价格等因素, 最常用的偶氮试剂是DEAD和DIAD. Huisgen内盐具有较强的亲核性, 能够被一系列亲电试剂原位捕获, 通常经过磷-氧四元杂环中间体, 以氮杂-Wittig的方式脱除氧磷实现碳-氮键的构筑, 高效合成结构多样的氮杂环化合物.此外, Huisgen内盐还是Mitsunobu反应[6]的关键中间体.由于Huisgen内盐在氮杂环化合物的合成上显示出独特的高效性, 受到了众多有机化学家的关注[3, 10~12].本文根据亲电试剂的类型不同, 对Huisgen内盐参与的环化反应研究进展进行综述, 为了让读者对这一主题有较全面的了解, 文中还简要介绍了早期的相关报道.
Huisgen内盐参与的环化反应最早可以追溯至20世纪60年代, Cookson等[13]首次报道了Ph3P促进下DEAD 1a与丁炔二甲酸二甲酯(3)的环化反应, 以中等收率生成了吡唑衍生物4a.作者认为该反应起始于Ph3P与1a形成的鏻盐中间体2a' (Scheme 2a). 1969年, Huisgen等[9b]研究发现, 在Ph3P促进下DMAD 1e与3反应同样可以得到相应的吡唑化合物4b; 此外, 进一步研究发现, 异氰酸苯酯或异硫氰酸苯酯5也能发生类似的环化反应, 生成三唑类化合物6 (Scheme 2b).根据实验结果, Huisgen等[11]将上述环化反应的关键内盐中间体的结构更正为2e, 即Huisgen内盐.
在Huisgen内盐发现初期, 其独特的化学反应性并没有引起人们的注意.直到1987年, Miller和Kolasa[14]通过Mitsunobu反应合成α-氨基酸的过程中, 意外发现除了预期的Mitsunobu反应产物之外, 还生成了一种作者认为是1, 2, 3-噁二唑的环状产物.例如, α-羟基丙酸甲酯(7)与N-苄氧羰基-O-苄基羟胺(8)在Ph3P/DIAD存在下发生Mitsunobu反应, 以中等收率生成预期产物9, 此外还以10%的收率得到环状化合物10 (Scheme 3).

对于上述实验结果, Lee等[15]通过系统研究Huisgen内盐与羰基化合物的反应模式, 将环化产物的结构更正为1, 3, 4-噁二唑化合物10', 并提出了可能的反应机理:反应物7在Mitsunobu试剂的作用下氧化为丙酮酸酯11, 经原位形成的Huisgen内盐进攻得到中间体12; 随后经过鏻盐转移、分子内关环等过程脱除三苯基氧磷, 形成环化产物10' (Scheme 4).
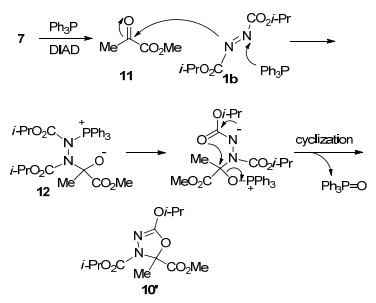
近年来, 随着叔膦促进的环化反应大量涌现[2, 16], Huisgen内盐重新受到化学家的重视. 2005年, Lee课题组[15]报道了Huisgen内盐与α-二羰基化合物的环化反应, 实现了1, 3, 4-噁二唑衍生物的合成(Scheme 5).研究发现, 芳基及烷基取代的α-酮酸酯11与Huisgen内盐反应, 生成多取代的1, 3, 4-噁二唑衍生物13; 烷基α-二酮14同样具有较好的反应活性, 当使用不对称的烷基α-二酮时, 环化反应发生在位阻较小的甲基酮一侧, 以中等收率得到1, 3, 4-噁二唑化合物15.值得一提的是, 环状α-二酮16也能发生反应, 区域选择性地生成螺环1, 3, 4-噁二唑衍生物17, 作者认为热力学平衡及分子内氢键控制了反应的立体化学.

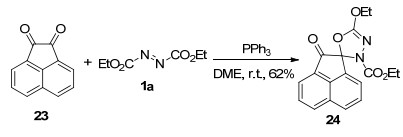
同年, Nair等[17]进一步将上述反应底物拓展至靛红衍生物18, 以中等至较高收率实现了螺环1, 3, 4-噁二唑衍生物19的合成.研究表明, 该类化合物具有抗痉挛和抗惊厥活性[18].在相同的反应条件下, 邻苯醌20与Huisgen内盐反应生成了一种结构新颖的苯并-1, 2, 3-噁二唑衍生物21.可能的反应机理如Scheme 6所示, 反应起始于Huisgen内盐亲核进攻邻苯醌20的羰基, 经分子内关环脱除三苯基氧磷, 得到类似19的螺环中间体22; 该中间体经过分子内开环重排/环化过程恢复苯环的芳香性, 得到产物21.研究表明, 环状二酮苊醌23也能与Huisgen内盐反应, 以中等收率生成螺噁二唑啉衍生物24 (Scheme 7).
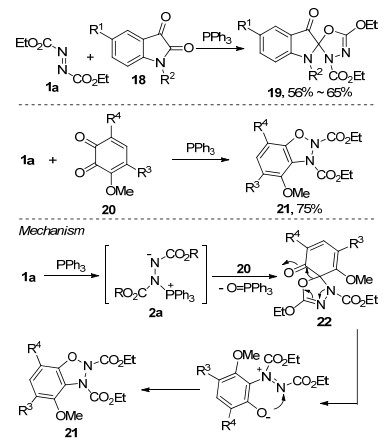
随后, 程津培等[19]报道了苯并呋喃二酮25与Huisgen内盐的环化反应, 以中等至较高收率合成了一系列含有苯并呋喃酮骨架的螺环衍生物26.该反应具有较好的底物普适性, 机理上同样经过了Husigen内盐亲核进攻-分子内环化-磷氧化合物消除的过程(Scheme 8).
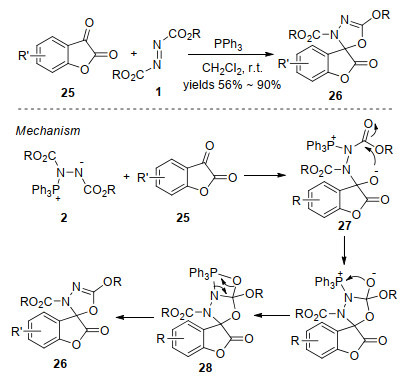
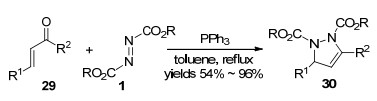
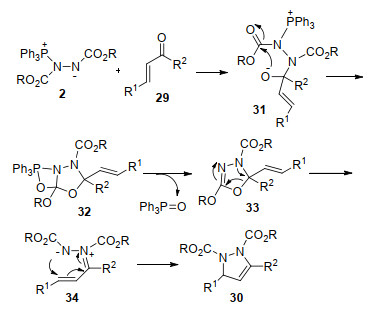
2007年, Nair等[20]研究发现, 原位形成的Huisgen内盐能与查尔酮29发生环化反应, 生成吡唑啉衍生物30 (Scheme 9).该反应具有较温和的反应条件、较好的底物普适性, 为吡唑啉衍生物的合成提供了一种高效的方法.值得一提的是, 官能化的吡唑啉衍生物作为一类重要的氮杂环化合物, 具有丰富的生物活性及广泛的用途[21].
对上述实验结果, 作者提出了可能的反应机理: Huisgen内盐对查尔酮29的羰基亲核进攻, 得到中间体31, 随后发生分子内关环生成四元杂环中间体32, 32脱除三苯基氧磷形成中间体33.该中间体经分子内开环得到α, β-烯基腙类两性离子中间体34, 34通过分子内1, 4-氮杂-Michael加成生成吡唑啉衍生物30 (Scheme 10).
环状α, β-不饱和酮也表现出较好的反应性, 以中等收率生成稠环吡唑啉衍生物(Scheme 11).
进一步的研究发现, 当芳基取代的1, 4-二烯-3-酮(35)作反应底物时, 在Ph3P/DIAD的存在下, 以54%的收率生成了吡唑并哒嗪衍生物36.该类化合物具有优良的抗菌、抗炎和止痛活性[22].机理上, 在Ph3P的作用下, 1, 4-二烯-3-酮(35)与DIAD首先形成乙烯基吡唑啉中间产物37, 该中间产物随后与第二分子DIAD发生分子间Diels-Alder反应, 得到产物36.当采用结构上不对称的1, 4-二烯-3-酮时, 反应的区域选择性取决于取代基R1及R2的电子效应.对于后续Diels-Alder反应, 利用外加亲双烯体代替DIAD时, 同样取得了成功.在相同的反应条件下, 底物38与1 equiv. DIAD及马来酰亚胺39反应时, 生成了三元稠环化合物40, 从而为上述反应机理提供了有力的证据(Scheme 12).
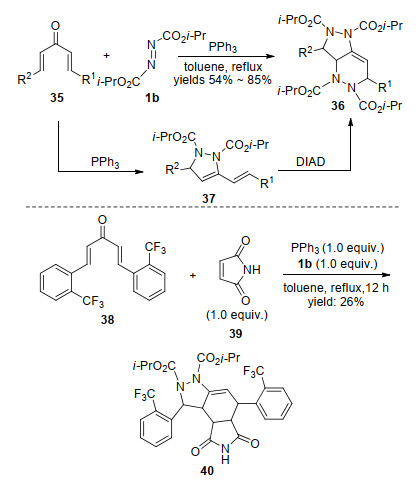
2011年, Kumar小组[23]采用色酮衍生的α, β-不饱和酮41 (R2≠H)作为反应底物, 实现了与Husigen内盐的环化反应, 生成了色酮取代的吡唑啉衍生物42.然而, 当使用3-色酮-α, β-不饱和醛时(R2=H), 反应还生成了少量色酮并二氮杂环庚二烯43, 这是首例利用Huisgen内盐合成七元氮杂环的报道.可能的反应机理如Scheme 13所示.首先原位形成的Huisgen内盐进攻底物中的羰基生成磷氧五元杂环, 随后脱除三苯基氧磷得到氧鎓盐中间体44.该中间体的氮负离子经1, 4-及1, 2-加成, 分别得到吡唑啉衍生物42及七元氮杂环化合物43.
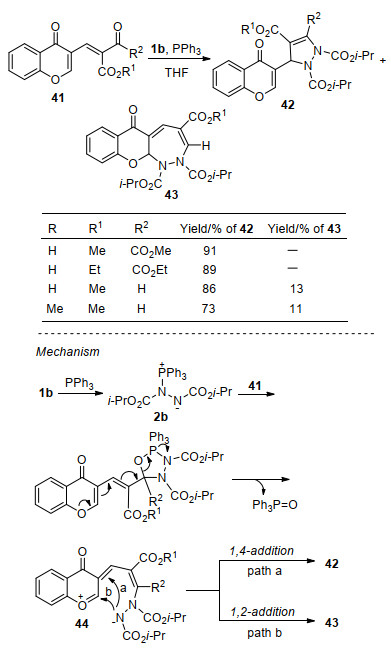
随后, 该课题组[24]报道了3-甲酰基色酮45与Huisgen内盐的串联环化反应, 合成了具有三个连续季碳中心的多环化合物47.研究发现, 在反应体系中加入叔胺添加剂4-二甲胺基吡啶(DMAP)可提高反应效率.有趣的是, 将等量的3-甲酰基色酮、Ph3P和DIAD反应时, 以26%的收率生成预期环化产物46, 同时还以27%的收率得到了多环化合物47, 并回收约40%的3-甲酰基色酮原料; 当Ph3P增加至2.5 equiv., DIAD增加至2.2 equiv.时, 产物47的收率提高了约两倍.机理上, 首先Huisgen内盐与反应物45中α, β-不饱和醛结构单元发生环化反应, 经过类似上述与查尔酮的反应过程[20], 生成吡唑啉中间产物46; 随后第二分子Huisgen内盐以SN2'的方式进攻46的碳碳双键单元, 得到两性离子中间体48; 该中间体经连续分子内亲核加成, 生成四元磷氧杂环49, 最后脱除三苯基氧磷得到产物47 (Scheme 14, a).为了拓展该串联环化反应的应用价值, 作者利用缺电子联烯酸酯50成功捕获了中间产物46, 由色酮45经一锅两次环化过程合成了稠环化合物51 (Scheme 14, b).上述反应进一步丰富了Huisgen内盐与α, β-不饱和羰基化合物的环化反应模式, 实现了复杂稠环氮杂环化合物的高效合成.
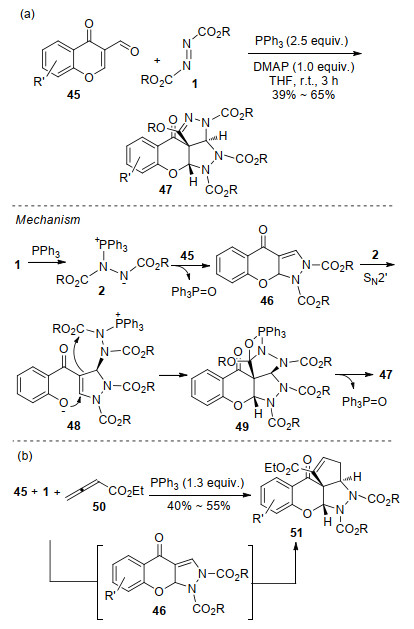
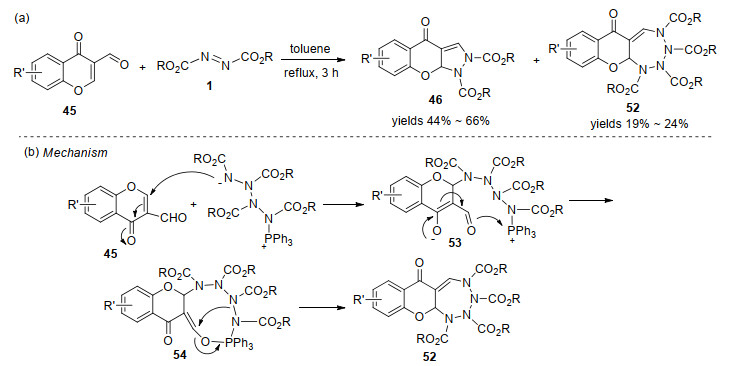
采用相同的反应底物, Terzidis等[25]实现了色酮并[2, 3-c]吡唑啉化合物的合成.有趣的是, 反应还以较低的收率分离到了四氮杂䓬衍生物52 (Scheme 15, a).研究认为, 先生成的Huisgen内盐与另一分子偶氮二甲酸酯反应形成两性离子中间体53, 该中间体对色酮亲核进攻得到的中间产物发生分子内关环, 得到大环中间体54, 最后消除三苯基氧磷生成四氮杂䓬衍生物52 (Scheme 15, b).
缺电子烯烃是指在C=C键上连有吸电子基团的化合物.受吸电子基团的影响, 这类化合物具有较高的反应活性, C=C键容易受亲核试剂进攻, 经过后续转化, 可以合成结构多样的碳环及杂环化合物.为了总结方便, 本节将含有缺电子C=C键结构单元的反应底物统一归纳为“缺电子烯烃”进行讨论.
作为一类特殊的“缺电子烯烃”, 缺电子联烯(如联烯酸酯55)指一端带有拉电子基团的累积双烯, 受拉电子基团的影响, 联烯酸酯的α, β-碳碳双键具有较高的反应活性.当有叔膦存在时, 叔膦容易进攻联烯酸酯的β位, 生成1, 3-偶极中间体56, 该中间体具有较高的反应活性, 能与不同反应底物完成多种类型的化学转化(Scheme 16, a)[26]. 然而, 当反应体系中同时存在联烯酸酯和偶氮二甲酸酯时, 叔膦优先与偶氮二甲酸酯原位生成Huisgen内盐, 而联烯酸酯则作为Michael受体参与反应.

2006年, Nair小组[27]首次报道了Huisgen内盐与联烯酸酯的环化反应.当底物为α-取代联烯酸酯57时, 反应以中等收率生成具有环外双键的吡唑啉衍生物58 (Scheme 16, b).机理上, 原位形成的Huisgen内盐2b进攻α-取代联烯酸酯57的β-位, 形成两性离子中间体59, 中间体59发生分子内亲核加成得到中间体60;最后脱除三苯基氧磷, 生成吡唑啉衍生物58 (Scheme 16, b).有趣的是, 当采用γ-取代的联烯酸酯61时, 化学选择性地生成了高度官能化的吡唑衍生物62, 反应过程中偶氮化合物的酯基发生了迁移(Scheme 17).作者提出了可能的反应机理:首先, Huisgen内盐亲核进攻γ-取代联烯酸酯61, 形成中间体63.与上述α-取代联烯酸酯的反应不同, 中间体63的碳负离子在对偶氮化合物的酯基进行亲核进攻时, 发生了分子内酯基的迁移, 得到的中间体64经氮杂-Wittig反应脱除三苯基氧磷, 形成吡唑啉中间体65.最后, 经双键迁移生成吡唑衍生物62.
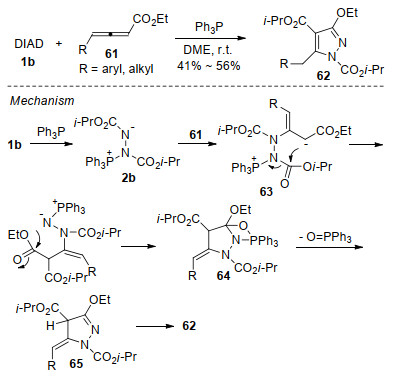
受Nair小组研究工作的启发, Swamy等[28]随后报道了α-联烯膦酸酯66、γ-联烯磷酸酯67与Huisgen内盐类似的环化反应, 分别合成了膦酰基取代的吡唑啉衍生物68和吡唑衍生物69 (Scheme 18), 进一步拓展了这类反应的应用范围.
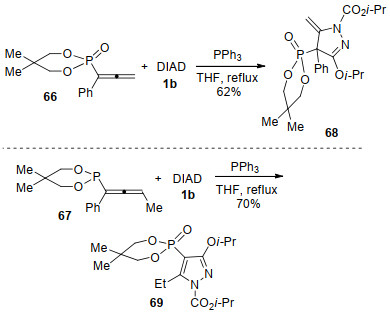
2014年, 麻生明等[29]探索了联烯甲酸70与Huisgen内盐的环化反应, 高效合成了吡唑-3-酮衍生物71.吡唑-3-酮是多种药物活性分子的核心骨架, 并具有丰富的药物活性, 如作为除草剂、杀虫剂、止痛剂等[30].对于反应机理, 作者做了如Scheme 19中的阐述.原位形成的Huisgen内盐首先夺取联烯甲酸70的质子, 形成离子对72-A, 经鏻盐转移生成离子对72-B; 随后72-B的氮负离子进攻羰基形成内盐中间体73, 73脱除三苯基氧磷后得到酰基肼中间体74; 该中间体发生分子内氮杂- Michael加成, 生成内盐中间体75; 最后经双键迁移、质子转移等过程生成产物71.
除了上述缺电子联烯, Huisgen内盐与普通缺电子烯烃同样能够发生环化反应, 但一般需要碳碳双键上连有两个或以上拉电子基团的高活性烯烃底物. 2012年, Yamazaki等[31]报道了Huisgen内盐与乙烯三羧酸酯76的环化反应, 以中等至较高收率合成了多取代吡唑啉衍生物.研究发现, 当反应物为双酯基取代的烯烃78时, 环化产物79的收率大幅降低.此外, 丙烯酸酯80需要在α-位有拉电子基团(如磷酰基, 三氟甲基, 氰基, 酯基等)时才能发生反应, 以中等收率生成吡唑啉化合物81 (Scheme 20).该反应仅对高活性的烯烃底物才能获得较好的结果, 反应的底物范围相对较窄.

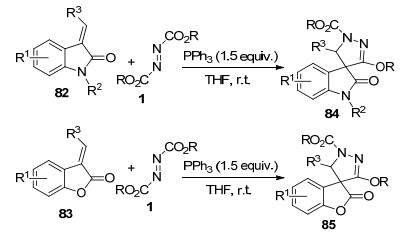
2015年, 贺峥杰课题组[32]报道了Huisgen内盐与3-亚甲基-2-吲哚酮82及3-亚甲基苯并呋喃酮83的环化反应, 分别以较高的收率和优秀的立体选择性合成了含有羟吲哚骨架及苯并呋喃酮骨架的螺环吡唑啉衍生物84和85 (Scheme 21).该反应具有条件温和、底物适用范围广、立体选择性好等优点, 为螺环吡唑啉衍生物提供了一种高效的合成方法.初步生物活性测试表明, 部分合成的螺环吡唑啉衍生物在50 μg•mL-1的浓度下对多种农作物真菌具有良好的抗菌活性.
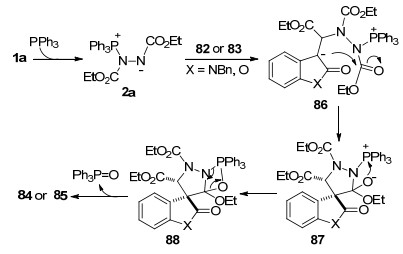
可能的反应机理是:首先, 原位形成的Huisgen内盐与3-亚甲基吲哚酮82或3-亚甲基苯并呋喃酮83发生共轭加成得到内盐中间体86, 随后, 中间体86的碳负离子进攻酯羰基形成中间体87, 经磷-氧键形成得到的中间体88脱除三苯基氧磷, 生成螺环吡唑啉产物84或85 (Scheme 22).
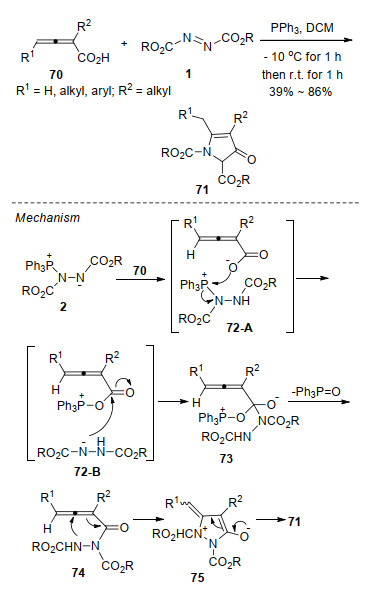
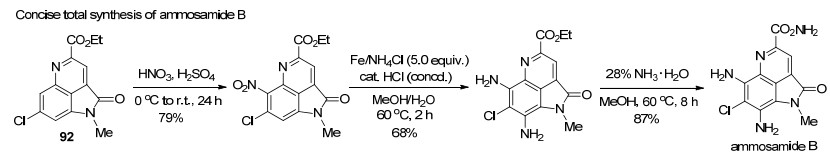
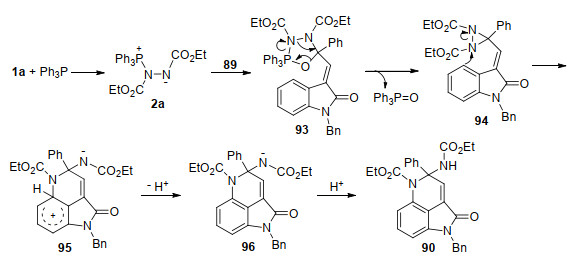
随后, 该课题组[33]报道了Huisgen内盐与3-酰亚甲基-2-吲哚酮89一种新颖的环化反应, 合成了官能化的吡咯并[4,3,2-de]二氢喹啉酮衍生物90.该反应不仅为构建吡咯并[4,3,2-de]喹啉核心骨架提供了高效便捷的新方法, 而且代表了Huisgen内盐一种新颖的反应模式, 反应中偶氮化合物的氮氮键发生了断裂, 并作为N1合成子参与氮杂环的构筑.在该反应基础上, 作者采取“两步一锅法”策略, 还实现了吡咯并[4,3,2-de]喹啉酮91的合成(Scheme 23).为进一步证明该反应的应用价值, 作者以上述方法为关键步骤, 从产物92出发, 经过硝化、还原和氨解三步转化, 以46.7%的总收率, 实现了海洋生物碱ammosamide B的全合成(Scheme 24).该合成路线简短、高效、取代基变化灵活, 为ammosamides天然生物碱及其类似物的生物活性与构效关系研究提供了合成方法学基础.
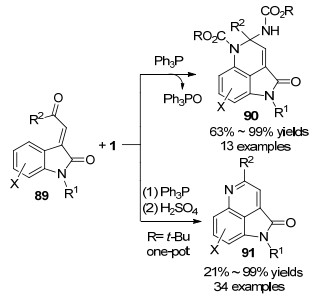
所提出的反应机制如Scheme 25所示:原位形成的Huisgen内盐亲核进攻89的酮羰基, 并经P—O键形成得到五元杂环中间体93, 93脱除三苯基氧磷生成二氮杂环丙烷中间体94.在分子内富电子苯环的进攻下, 具有高度环张力的二氮杂环丙烷94发生氮—氮键异裂, 生成三环中间体95, 95脱质子恢复苯环结构得到中间体96, 最后经质子化生成吡咯并[4,3,2-de]二氢喹啉酮产物90.
最近, 苗志伟等[34]报道了Huisgen内盐与2-芳叉基- 1, 3-茚满二酮97的环化反应, 立体选择性地合成了茚并吡唑啉衍生物98.研究发现, 茚满二酮底物中两个酮羰基的存在是反应化学选择性发生的关键.当采用2-苄叉基-1-茚满酮99为反应底物时, 在相同的反应条件下生成了与Nair等[20]所报道类似的吡唑啉衍生物100.不同于前述Nair[20]及贺峥杰等[32]的报道, 在该反应中原位生成的Huisgen内盐2先进攻底物中α, β-不饱和酮单元的C=C键, 经转化后得到的氮负离子中间体103再进攻分子中第二个酮羰基, 最后经三苯基氧磷的消除, 得到环化产物98 (Scheme 26).

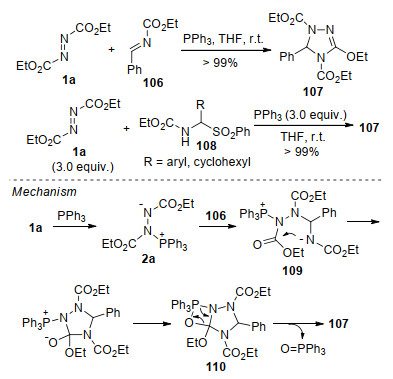
在化学结构及反应活性方面, 亚胺的C=N键与羰基化合物的C=O键具有较大的相似性, 亚胺化合物同样是叔膦介导的环化反应中常用的亲电试剂. 2011年, 施敏团队[35]报道了N-取代芳香醛亚胺106与Huisgen内盐的环化反应, 以较高的收率合成了1, 2, 4-三唑啉衍生物107.当使用亚胺前体108替代亚胺时, 在3 equiv. DEAD和PPh3的作用下, 环化反应同样可以顺利进行, 且反应底物范围能够拓展到烷基如环己基取代的α-磺酰基胺(R=cyclohexyl).作者认为, Huisgen内盐可以作为路易斯碱促进化合物108原位形成亚胺106.可能的反应机理如Scheme 27所示, 原位形成的Huisgen内盐2a亲核进攻亚胺106, 形成两性离子中间体109; 该中间体通过分子内环化形成中间体110, 最后以氮杂- Wittig的方式脱除三苯基氧磷, 生成环化产物107, 机理上类似于Huisgen内盐与羰基化合物的反应[17].
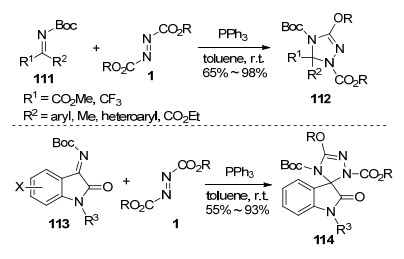
一般来说, 酮亚胺的C=N键由于具有较大的空间位阻而降低了反应活性, 因此要实现酮亚胺与Huisgen内盐的环化反应, 需要在酮亚胺C=N键上引入拉电子基团, 从而增强其亲电反应活性. 2013年, Kumar等[36]报道了酮亚胺111与Huisgen内盐的环化反应, 在温和条件下高效合成了多官能团的1, 2, 4-三唑啉衍生物112.拉电子基团R1 (如CO2R, CF3等)的存在提高了酮亚胺C=N键的亲电性, 使反应能够顺利进行.反应底物中R2为芳基、烷基、杂芳基或酯基时, 均表现出较高的反应活性.此外, 靛红衍生的酮亚胺113同样是优良的反应底物, 不同取代的靛红酮亚胺能以中等至优秀的收率生成相应羟吲哚螺三唑啉化合物114 (Scheme 28).
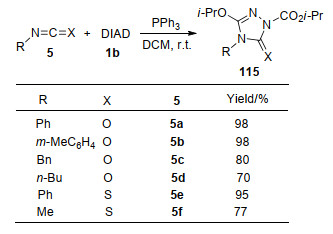
在早期研究中, Huisgen等[9]探索了Ph3P/DMAD与异氰酸苯酯或异硫氰酸苯酯的环化反应. 2005年, Alizadeh等[37]对该反应的底物范围进行了拓展, 实现了Huisgen内盐与芳基、烷基取代的异氰酸酯或异硫氰酸酯5的环化反应, 合成了1, 2, 4-三唑衍生物115 (Scheme 29).
2008年, 王彦广课题组[38]报道了Huisgen内盐与酰基氮杂环丙烷116的环化反应, 在温和的反应条件下以优秀的收率合成了2-氨基甲酸酯取代的吡唑啉衍生物117.该反应具有较宽的底物适用范围, 氮杂环丙烷的取代基R1、R2不论是富电子或缺电子基的芳基, 均表现出很好的反应性, 从而为氨基甲酸酯取代的吡唑啉衍生物提供了一种高效的合成途径(Scheme 30, a).有趣的是, 当底物中具有较大位阻的取代基时, 反应除生成相应的吡唑啉化合物外, 还以中等收率生成了烯基腙衍生物118 (Scheme 30, b).基于氘同位素标记结果, 提出的可能反应机理如下:原位形成的Huisgen内盐与反应底物116的酮羰基发生环化反应, 生成噁二唑中间体119; 由于氮杂环丙烷较大的环张力, 119经开环及一系列分子内串联反应, 生成中间体120.当取代基空间位阻较小时, 中间体120经分子内氮杂-Michael加成、质子迁移过程, 生成吡唑啉产物117; 而当取代基空间位阻较大时, 中间体120直接经分子内质子迁移转化为烯基腙衍生物118 (Scheme 30, c).
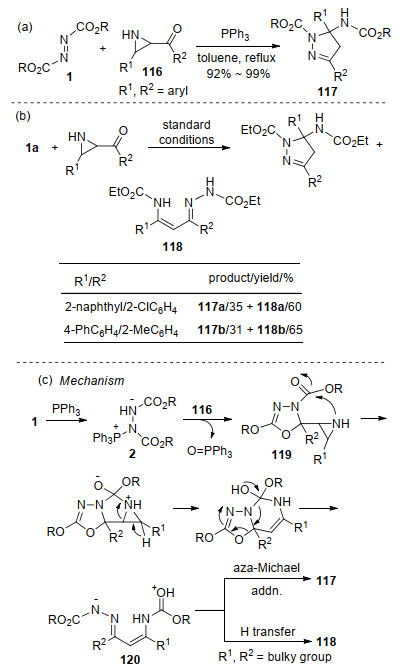
2010年, 该研究小组[39]还成功实现了Huisgen内盐与芳基醛腙化合物121的环化反应, 合成了1, 2, 4-三唑啉酮衍生物122.该反应具有较好的底物普适性, R1为富电子芳基、缺电子芳基、及杂环芳基时, 均能以较高至优秀的收率生成目标产物(Scheme 31, a).有趣的是, 当采用脂肪醛腙(R1=alkyl)时, 则与偶氮二甲酸酯1加成生成了链状的肼类衍生物123 (Scheme 31, b).进一步研究发现, 该加成反应中PPh3并不是必需的, 在加热的条件下即可直接进行(Scheme 31, c).得到的链状产物123在三氟乙酸(TFA)的促进下可以发生分子内关环, 生成与122类似的三唑啉酮124 (Scheme 31, d).作者认为, 链状产物是由偶氮化合物对脂肪醛腙发生氮杂- Michael加成形成.
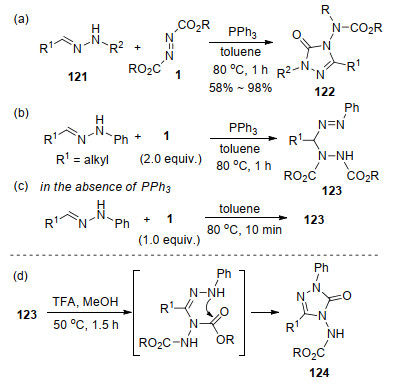
基于上述实验结果, 提出了如Scheme 32的反应机理, 首先, PPh3与DEAD 1a原位形成Huisgen内盐2a; 同时, 在分子内N—H质子迁移过程的诱导下, 醛腙121异构化为偶极化合物121', 该化合物被Huisgen内盐亲核进攻生成中间体125.该中间体经分子内环化、P—O键形成、三苯基氧磷消除过程, 得到偶极中间体126. 126经过分子内乙基迁移生成1, 2, 4-三唑啉化合物, 最后在DEAD参与下脱去质子生成产物122.随后, 唐明生及朱艳艳等[40]利用密度泛函理论(DFT)计算辅证了该反应机制.
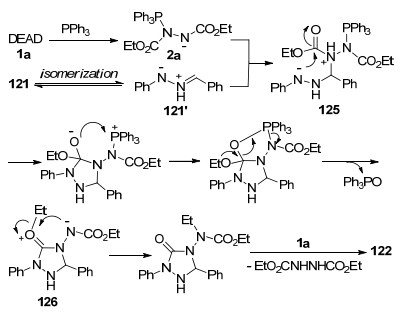
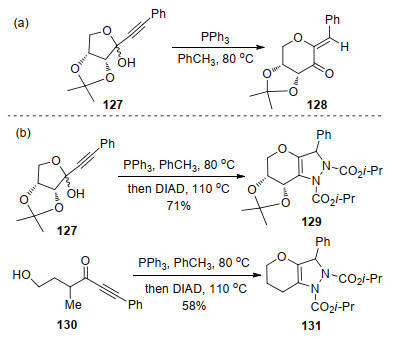
2012年, Peczuh等[41]研究发现, 在PPh3催化下, 环状芳基乙炔半缩酮127可经开环并重新环化, 得到含有环外双键的α, β-不饱和酮128 (Scheme 33, a).为进一步拓展该方法的应用价值, 并受到Huisgen内盐与查尔酮的环化反应[20]启发, 作者利用两步一锅法合成策略, 实现了吡喃并[3, 2-c]吡唑啉化合物的合成.如芳基乙炔半缩酮127或其异构体130与PPh3和DIAD反应, 分别以中等收率生成了吡喃并吡唑啉化合物129和131 (Scheme 33, b).
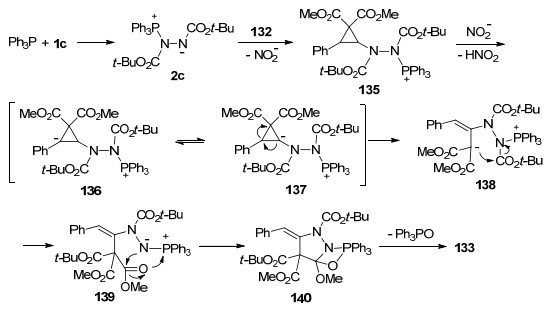
2016年, 贺峥杰等[42]报道了原位形成的Huisgen内盐与硝基环丙烷132的环化反应, 以较高的收率及优秀的立体选择性合成了高度官能化的3-烷氧基吡唑啉衍生物133, 产物经酸解/加热脱羧两步一锅法反应, 可以转化为相应的3-烷氧基-1H-吡唑化合物134.该反应具有良好的底物适用范围, 为3-烷氧基吡唑啉和1H-吡唑衍生物的高效合成提供了有效途径(Scheme 34).
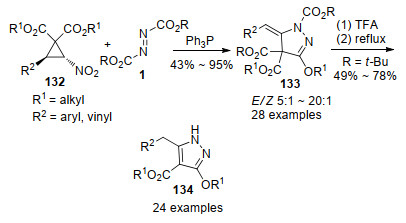
作者通过机理探究实验, 提出了Scheme 35所示的反应机制.反应起始于Ph3P与DTBAD 1c原位形成Huisgen内盐中间体2c, 2c亲核进攻硝基环丙烷132, 离去硝基负离子生成中间体135, 该中间体经硝基负离子脱质子, 得到可以相互转化的两性离子中间体136和137, 随后经分子内开环重排形成中间体138, 138中碳负离子进攻N-酯基, 并经分子内酯基迁移得到中间体139; 最后中间体139的氮负离子进攻位阻较小的甲酯, 以氮杂-Wittig的方式经中间体140脱除三苯基氧磷, 生成吡唑啉产物133.
综上所述, 尽管距Huisgen内盐的发现已经过去了半个多世纪, 其化学反应性早期也有少量报道, 然而直到近二十年, 随着叔膦促进下具有内盐结构的两性离子中间体参与的环化反应大量涌现, Huisgen内盐才重新受到有机化学家们的重视, 并由此发展了一系列基于Huisgen内盐的环化反应, 为氮杂环化合物的合成提供了高效的新途径.在这些反应中, 羰基化合物、缺电子烯烃及亚胺等亲电试剂是常用的反应底物.机理上一般起始于Husigen内盐对反应底物亲核加成, 经过连续转化后脱除磷氧化合物, 完成环化过程.因此, 通常需要化学计量的叔膦试剂参与, 导致反应具有较低的原子经济型及较差的绿色化学特征.鉴于此, 开发叔膦催化的基于Husigen内盐的环化反应, 将显著拓展Huisgen内盐在有机合成中的应用, 也是继续发展这一领域的重要方向.
Cadogan, J. I. G. Organophosphorus Reagents in Organic Synthesis, Academic Press, London, 1979.
(a) Ye, L. -W.; Zhou, J.; Tang, Y. Chem. Soc. Rev. 2008, 37, 1140.
(b) Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102.
(c) Marinetti, A.; Voituriez, A. Synlett 2010, 174.
(d) Wang, S.; Han, X.; Zhong, F.; Wang, Y.; Lu, Y. Synlett 2011, 2766.
(e) Zhao, Q.-Y.; Lian, Z.; Wei, Y.; Shi, M. Chem. Commun. 2012, 48, 1724.
(f) Fan, Y.-C.; Kwon, O. Chem. Commun. 2013, 49, 11588.
(g) Wang, Z.; Xu, X.; Kwon, O. Chem. Soc. Rev. 2014, 43, 2927.
(h) Xiao, Y.; Sun, Z.; Guo, H.; Kwon, O. Beilstein J. Org. Chem. 2014, 10, 2089.
(i) Xie, P.; Huang, Y. Org. Biomol. Chem. 2015, 13, 8578.
(j) Xiao, Y.; Guo, H.; Kwon, O. Aldrichim. Acta 2016, 49, 3.
(k) Wang, T.; Han, X.; Zhong, F.; Yao, W.; Lu, Y. Acc. Chem. Res. 2016, 49, 1369.
(l) Zhou, R.; He, Z. Eur. J. Org. Chem. 2016, 1937.
(a) Xu, S.; He, Z. RSC Adv. 2013, 3, 16885.
(b) Xu, S. L.; He, Z. J. Chin. J. Org. Chem. 2014, 34, 2438 (in Chinese).
(徐四龙, 贺峥杰, 有机化学, 2014, 34, 2438.)
(c) Karanam, P.; Reddy, G. M.; Koppolu, S. R.; Lin, W. Tetrahedron Lett. 2018, 59, 59.
(d) Liu, Y.; Sun, F.; He, Z. Tetrahedron Lett. 2018, 59, 4136.
Quin, L. D. A Guide to Organophosphorus Chemistry, Wiley, New York, 2000, p. 379.
Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863. doi: 10.1021/cr00094a007
Swamy, K. C. K.; Kumar, N. N. B.; Balaraman, E.; Kumar, K. Chem. Rev. 2009, 109, 2551. doi: 10.1021/cr800278z
Gololobov, Y. G.; Kasukhin, L. F. Tetrahedron 1992, 48, 1353. doi: 10.1016/S0040-4020(01)92229-X
Appel, R. Angew. Chem., Int. Ed. 1975, 14, 801. doi: 10.1002/anie.197508011
(a) Huisgen, R.; Blaschke, H.; Brunn, E. Tetrahedron Lett. 1966, 7, 405.
(b) Brunn, E.; Huisgen, R. Angew. Chem., Int. Ed. 1969, 8, 513.
(c) Itzstein, M. v.; Jenkins, I. D. Aust. J. Chem. 1983, 36, 557.
(d) Monge, D.; Jensen, K. L.; Marín, I.; Jø rgensen, K. A. Org. Lett. 2011, 13, 328.
Nair, V.; Menon, R. S.; Sreekanth, A. R.; Abhilash, N.; Biju, A. T. Acc. Chem. Res. 2006, 39, 520. doi: 10.1021/ar0502026
Nair, V.; Biju, A. T.; Mathew, S. C.; Babu, B. P. Chem. Asian J. 2008, 3, 810. doi: 10.1002/asia.200700341
Košmrlj, J.; Kočevar, M.; Polanc, S. Synlett 2009, 2217.
Cookson, R. C.; Locke, J. M. J. Chem. Soc. 1963, 6062.
Kolasa, T.; Miller, M. J. J. Org. Chem.1987, 52, 4978. doi: 10.1021/jo00231a026
Otte, R. D.; Sakata, T.; Guzei, I. A.; Lee, D. Org. Lett. 2005, 7, 495. doi: 10.1021/ol051956n
(a) Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535.
(b) Wei, Y.; Shi, M. Acc. Chem. Res. 2010, 43, 1005.
(c) Huang, Y.; Xie, P. Eur. J. Org. Chem. 2013, 6213.
(d) Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659.
(e) Li, W.; Zhang, J. Chem. Soc. Rev. 2016, 45, 1657.
Nair, V.; Biju, A. T.; Vinod, A. U.; Suresh, E. Org. Lett. 2005, 7, 5139. doi: 10.1021/ol051956n
Dogan, H. N.; Duran, A.; Rollas, S.; Sener, G.; Armutak, Y.; Keyer Uysal, M. Med. Sci. Res. 1998, 26, 755. https://www.sciencedirect.com/science/article/abs/pii/S138614251200203X
Wang, Z.-D.; Dong, N.; Wang, F.; Li, X.; Cheng, J.-P. Tetrahedron Lett. 2013, 54, 5473. doi: 10.1016/j.tetlet.2013.07.129
Nair, V.; Mathew, S. C.; Biju, A. T.; Suresh, E. Angew. Chem., Int. Ed. 2007, 46, 2070. doi: 10.1002/anie.200604025
(a) Elguero, J. In Comprehensive Heterocyclic Chemistry, Vol. 5, Eds.: Katritzky, A. R.; Rees, C. W.; Potts, K. T., Pergamon, Oxford, 1984, p. 167.
(b) Elguero, J. In Comprehensive Heterocyclic Chemistry Ⅱ, Vol. 3, Eds.: Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Pergamon, Oxford, 1996, p. 1.
(a) BraLa, M. F.; Cacho, M.; GarcMa, M. L.; Mayoral, E. P.; LNpez, B.; Pascual-Teresa, B.; Ramos, A.; Acero, N.; Llinares, F.; MuLozMingarro, D.; Lozach, O.; Meijer, L. J. Med. Chem. 2005, 48, 6843.
(b) Witherington, J.; Bordas, V.; Haigh, D.; Hickey, D. M. B.; Ife, R. J.; Rawlings, A. D.; Slingsby, B. P.; Smith, D. G.; Ward, R. W. Bioorg. Med. Chem. Lett. 2003, 13, 1581.
(c) Tewari, A. K.; Mishra, A. Bioorg. Med. Chem. 2001, 9, 715.
Liu, W.; Khedkar, V.; Baskar, B.; Schürmann, M.; Kumar, K. Angew. Chem., Int. Ed. 2011, 50, 6900. doi: 10.1002/anie.201102440
Baskar, B.; Wittstein, K.; Sankar, M. G.; Khedkar, V.; Schürmann, M.; Kumar, K. Org. Lett. 2012, 14, 5924. doi: 10.1021/ol3028412
Papafilippou, A.; Terzidis, M. A.; S.-S.Julia.; Tsoleridis, C. A. Tetrahedron Lett. 2011, 52, 1306. doi: 10.1016/j.tetlet.2011.01.063
(a) McCleverty, J. A.; Meyer, T. J. Comprehensive Coordination Chemistry Ⅱ, Elsevier Ltd, Boston, 2003.
(b) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(c) Bö rner, A. Phosphorus Ligands in Asymmetric Catalysis, Wiley-VCH, Weinheim, Germany, 2008.
(d) Zhou, Q.-L. Privileged Chiral Ligands and Catalysts, Wiley- VCH, Weinheim, Germany, 2011.
(e) Zhang, C.; Lu, X. J. Org. Chem. 1995, 60, 2906.
Nair, V.; Biju, A. T.; Mohanan, K.; Suresh, E. Org. Lett. 2006, 8, 2213. doi: 10.1021/ol0604623
Chakravarty, M.; Kumar, N. N. B; Sajna, K. V.; Swamy, K. C. K. Eur. J. Org. Chem. 2008, 4500. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21235279
Lü, R.; Cheng, X.; Zheng, X.; Ma, S. Chem. Commun. 2014, 50, 1537. doi: 10.1039/c3cc48215d
(a) Kazmo, Y.; Akira, M.; Norihiko, M.; Toshiio, M.; Kazutaka, A. Shigera, I. Pestic. Sci. 1999, 55, 161.
(b) Gursoy, A.; Demirayak, M. S.; Ç apan, G.; Erol, K.; Vural, K. Eur. J. Med. Chem. 2000, 35, 359.
(c) Badawey, E. A. M.; El-Ashmawey, I. M. Eur. J. Med. Chem. 1998, 33, 349.
Yamazaki, S.; Maenaka, Y.; Fujinami, K.; Mikata, Y. RSC Adv. 2012, 2, 8095. doi: 10.1039/c2ra21249h
Yang, C.; Li J.; Zhou, R.; Chen, X.; Gao, Y.; He, Z. Org. Biomol. Chem. 2015, 13, 4869. doi: 10.1039/C5OB00258C
Yang, C.; Chen, X.; Tang, T.; He, Z. Org. Lett. 2016, 18, 1486. doi: 10.1021/acs.orglett.6b00456
Li, Y.; Zhang, H.; Wei, R.; Miao, Z. Adv. Synth. Catal. 2017, 359, 4158. doi: 10.1002/adsc.201701013
Lian, Z.; Guan, X.-Y.; Shi, M. Tetrahedron 2011, 67, 2018. doi: 10.1016/j.tet.2011.01.072
Sankar, M. G.; Garcia-Castro, M.; Wang, Y.; Kumar, K. Asian J. Org. Chem. 2013, 2, 646. doi: 10.1002/ajoc.201300120
Alizadeh, A. Helv. Chim. Acta 2005, 88, 2777. doi: 10.1002/hlca.200590218
Cui, S.-L.; Wang, J.; Wang, Y.-G. Org. Lett. 2008, 10, 13. doi: 10.1021/ol7022888
Sun, Y.; Jiang, Z.; Hong, D.; Lu, P.; Wang, Y.; Lin, X. Tetrahedron 2010, 66, 2427. doi: 10.1016/j.tet.2010.01.087
Zhang, W.; Zhu, Y.; Wei, D.; Tang, M. J. Comput. Chem. 2012, 33, 715. doi: 10.1002/jcc.22906
Saha, J.; Lorenc, C.; Surana, B.; Peczuh, M. W. J. Org. Chem. 2012, 77, 3846. doi: 10.1021/jo3001854
Yang, C.; Liu, W.; He, Z.; He, Z. Org. Lett. 2016, 18, 4936. doi: 10.1021/acs.orglett.6b02415

图式 4 由Huisgen内盐合成1, 3, 4-噁二唑
Scheme 4 Synthesis of 1, 3, 4-oxadiazoles via Huisgen zwitterion

图式 5 Huisgen内盐与α-二羰基化合物的环化反应
Scheme 5 Annulation of Huisgen zwitterion with α-dicarbonyl compounds

图式 6 Huisgen内盐与靛红及邻苯醌的环化反应
Scheme 6 Annulations of Huisgen zwitterion with isatins and o-benzoquinones

图式 8 Huisgen内盐与苯并呋喃二酮的环化反应
Scheme 8 Annulation of Huisgen zwitterion with benzofuranediones

图式 12 基于Huisgen内盐的串联环化反应
Scheme 12 Cascade annulation reactions involving Huisgen zwitterion

图式 13 Huisgen内盐与色酮衍生物的环化反应
Scheme 13 Annulation of Huisgen zwitterion with chromone derivatives

图式 14 Huisgen内盐与3-甲酰基色酮的串联环化反应
Scheme 14 Cascade annulation of Huisgen zwitterion with 3-formylchromones

图式 15 Terzidis报道的Huisgen内盐与3-甲酰基色酮的环化反应
Scheme 15 Annulation reaction of Huisgen zwitterion with 3-formylchromones reported by Terzidis

图式 16 Huisgen内盐与α-取代联烯酸酯的环化反应
Scheme 16 Annulation of Huisgen zwitterion with α-substituted allenoates

图式 17 Huisgen内盐与γ-取代联烯酸酯的环化反应
Scheme 17 Annulation of Huisgen zwitterion with γ-substituted allenoates

图式 18 Huisgen内盐与联烯膦酸酯的环化反应
Scheme 18 Annulation of Huisgen zwitterion with allenylphos- phonates

图式 19 Huisgen内盐与联烯甲酸的环化反应
Scheme 19 Annulation of Huisgen zwitterion with allenoic acids

图式 20 Huisgen内盐与缺电子烯烃的环化反应
Scheme 20 Annulation of Huisgen zwitterion with electron- deficient alkenes

图式 23 Huisgen内盐与3-酰亚甲基-2-吲哚酮的环化反应
Scheme 23 Annulation of Huisgen zwitterion with 3-acylme- thylidene oxindoles

图式 26 Huisgen内盐与2-芳叉基-1, 3-茚满二酮的环化反应
Scheme 26 Annulation of Huisgen zwitterion with 2-arylideneindane-1, 3-diones

图式 31 Huisgen内盐与醛腙的反应
Scheme 31 Annulation of Huisgen zwitterion with aldehyde hydrazones

图式 32 Huisgen内盐与醛腙的环化反应机制
Scheme 32 Mechanism of the annulation reaction of Huisgen zwitterion with aldehyde hydrazone

图式 34 Huisgen内盐与硝基环丙烷的环化反应
Scheme 34 Annulation of Huisgen zwitterion with nitrocyclopropanes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:















