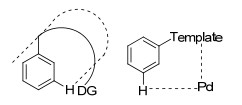
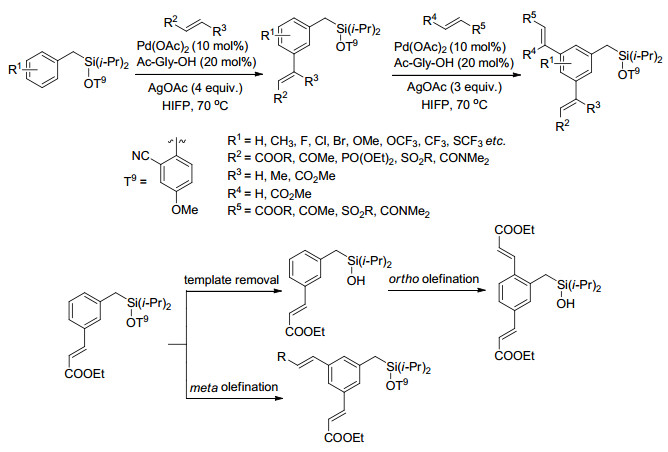
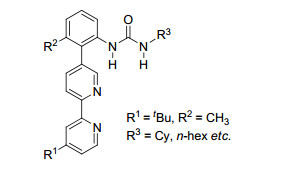
图 1.
U型模板及模板导向间位活化过渡态
Figure 1.
U template and transition state of meta-C—H activation

过渡金属催化的惰性碳氢键直接官能化是合成有机分子的重要方法之一, 被广泛应用于生物制药、农药、食品和有机材料等领域, 是非常活跃的研究领域.由于一个分子中往往存在多个反应活性相近的碳氢键, 因此位点选择性的进行碳氢活化是碳氢键活化面临的挑战之一.在过去的几年里, 间位碳氢键活化取得了一定的进展, 一是通过位阻和电性来调节[1~3], 但这仅局限于1, 3-二取代的底物, 大大限制了反应底物.二是通过导向基来诱导[4~7], 常用的策略包括模板导向、降冰片烯介导及配体次级效应等, 此种方法克服了底物中固有的电子效应和空间效应, 扩大了底物的适用范围, 具有一定的优势.本文将对近几年导向基团诱导及过渡金属催化的间位碳氢键活化进行详细综述, 从催化体系的特点、碳氢键活化的关键中间体中导向基团的构象及底物的构象对反应的区域选择性的影响进行了探讨.
自2012年Yu课题组[8]报道了含有氰基导向基的模板, 实现了导向基定位的间位烯烃化, 模板导向过渡金属催化的远程碳氢键活化取得了一定的进展, 主要以氰基和氮杂环为导向基, 以U型模板(图 1)实现了甲苯、酚、苄醇、苯基取代乙酸、3-苯丙酸、氢化喹啉和苯胺等衍生物中苯环间位的碳氢官能化.该策略通过模板构象、底物构象以及活化C—H与导向基团的相对位置的控制, 使催化剂接近间位C—H, 来实现间位的选择性活化(图 1), 导向基团距离间位C—H一般10~12个原子, 模板和底物的几何结构要使配位后的金属催化剂接近活化位置.
直线型的氰基模板可缓解间位官能团化过渡态的大环张力, 氰基为导向基的模板主要有氰化苄、羟基苯腈和氨基苯腈.
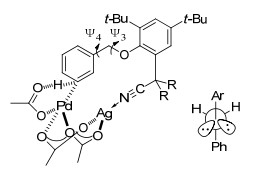
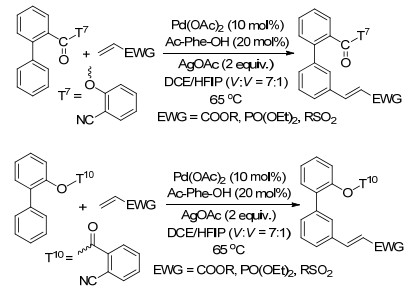
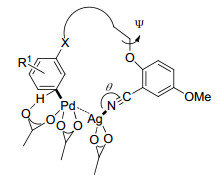
2012年, Yu课题组[8]在模板导向间位碳氢键活化领域做出了开创性工作, 首次以氰基为导向基实现了苄基醚的间位烯烃化(Eq. 1), 该反应以Pd(OPiv)2为催化剂、AgOPiv为氧化剂, 丙烯酸乙酯为烯烃化试剂, 以中等到优秀产率得到间位取代的产物, 对各种化学性质的取代基(吸电子和给电子)取代的单醚都有很好的区域选择性, 苯环上取代基的位置对反应的区域选择性影响不大, 但是对于苯环上无取代基或对位取代的底物会伴随一定比例两个间位同时被取代的二取代产物.对于烯烃化试剂, 乙烯基乙基酮、丙烯酸酯和乙烯基膦酸二乙酯都能与间甲基苄醚发生烯烃化反应, 甚至一些二取代烯烃和三取代的环状烯烃也能高活性、高选择性地生成间位烯烃化和烯丙基化产物.他们认为弱配位的直线型氰基可使催化剂接近被活化的碳氢键, 降低大环金属过渡态的环张力, 促进碳氢键活化.他们对苄基醚间位烯烃化反应进行了机理研究[9], 认为反应过程经过脱质子碳氢键活化、烯烃插入、β-氢消除和还原消除四个步骤, 其中碳氢键活化决定反应速率以及产物的区域选择性. PdAg(OAc)3是反应活性物种, 因为该活性物种活化邻、间和对位碳氢键得到的大环过渡态分别是14、15和16元环, 更稳定(图 2).银盐在反应中一是作为氧化剂, 二是与导向基配位形成大环过渡态.碳氢活化过渡态中C-N-Ag的角度以及二面角Ψ3对产物的区域选择性有很大的影响, Ψ3在底物中为174°, 在过渡态中C-N-Ag角度和二面角Ψ3越接近180° (Ph和Ar处于对位交叉式)越稳定(图 2), 在碳氢活化步骤中, 活化邻、间和对位碳氢键得到的大环过渡态中Ψ3分别为-166°, -173°和154°, 活化间位的过渡态更接近底物的分子构象, 底物不易变形, C-N-Ag的角度分别为160°, 156°和135°, 此外二面角Ψ4对产物的区域选择性也有影响作用, Ψ4接近0°, 过渡态稳定性变差, 在过渡态中在碳氢活化步骤中活化邻位和间位得到的过渡态分别为1°和-44°, 因此主要得到间位产物.模板中t-Bu使模板的构象接近过渡态的构象, 对反应活性和选择性也起到了重要作用.总之, 过渡态中底物的变形程度影响了反应的区域选择性.
|
|
(1) |
与此同时, Tan课题组[10]报道了以硅氧基连接氰基模板与苄醇类衍生物, Pd(OAc)2催化苄醇间位烯烃化反应(Eq. 2), 该模板合成方法简单, 容易与苄醇反应, 而且容易脱除, 在考察范围内, 各种取代苄醇有很好的间位选择性, 但是邻位取代的底物会得到一定比例的二取代产物, 苯环上强吸电子的取代基导致烯烃化产物产率降低, 由于位阻效应, 对位取代的苄醇间位选择性略有降低; 该体系对一系列缺电子的烯烃都有好的反应活性和选择性.氰基与苯环之间的夹角α对区域选择性有很大的影响, 通过调整苄基位取代基可改变α, 当取代基是s-Bu时, 得到最好的间位选择性和最高产率.
|
|
(2) |
由于氮原子的给电子效应, 二氢吲哚类化合物苯环电子云密度增大, 邻位和对位碳氢键更容易被活化, Yu等[11]以硫酰基连接氰化苄模板和二氢吲哚, Pd(OAc)2作催化剂, 实现了二氢吲哚类化合物间位烯烃化(Eq. 3)、芳基化(Eq. 4)和乙酰氧化(Eq. 5), 反应底物适用范围广且选择性好, 以较高的选择性得到间位取代的产物.硫酰基能降低苯环邻位和对位电子云的密度, 使苯环的间位容易活化.另外, 可能由于模板苄基位偕二甲基效应的存在, 模板中氰基α位偕二烷基的引入可以调整模板的构象, 使模板的构象更接近间位活化过渡态的构象, 有利于导向基辅助间位碳氢键的活化.
|
|
(3) |
|
|
(4) |
|
|
(5) |
2017年, Zhou等[12]以硅氧键连接氰化苄模板和苯酚, 实现了苯酚及其衍生物间位烯烃化反应(Eq. 6), 该体系底物适用范围广, 对一系列的烯烃及苯酚衍生物都有很高的催化活性和间位选择性. 2018年, 该小组[13]又将此氰基模板应用于Rh催化的苯酚衍生物间位烯烃化反应中(Eq. 7), 该体系底物适用范围广, 对一系列的烯烃及苯酚衍生物都有很高的催化活性和间位选择性.当苯酚含有一个取代基时, 无论取代基在苯环上的位置还是取代基的空间效应如何, 都能高选择性得到间位产物, 丙烯酸酯、丙烯腈、乙烯基膦酸酯、丙烯醛和苯基乙烯基砜都可与苯酚反应, 高选择性得到间位取代产物.
|
|
(6) |
|
|
(7) |
Yu小组[8]将氨基苯腈模板T5应用于3-苯基丙酸及其衍生物间位烯烃化反应中, 反应以N-乙酰基甘氨酸为配体, 得到了很好的结果(Eq. 8). Houk等[14]对N-乙酰基甘氨酸配体在反应中的作用进行了研究, 认为碳氢键活化决定反应速率以及产物的区域选择性, Pd(OAc)2L是反应的活性物种, 氨基酸配体既能稳定钯催化剂, 又能作为碱, 在碳氢活化步骤脱除苯环中的氢. α1(Pd1-C2-O3-C4)影响产物的区域选择性, α1=0时, 四个原子在同一个平面上, 有利于氢原子的脱除.底物中苯环与Pd1-C2-O3-C4所在平面的夹角α2对产物的区域选择性也有影响作用, 两个平面垂直对反应有利, 当烯烃化分别发生在邻、间和对位时α1和α2分别为17°、81°、0°和88°、-6°和84°.此外, O5=C6-C7-C8顺式共平面对反应有利(二面角α3=0), 在反应物中α3=-7°, 在邻、间和对位取代的过渡态中, α3分别是-60°、-27°和-42°(图 3), 也就是说活化间位底物的变形程度小.综合以上因素, 过渡态的变形程度和模板的变形程度决定了产物的区域选择性.
|
|
(8) |

随后, Yu等[15]又将T5模板应用到富电子苯酚类衍生物的间位烯烃化反应中(Eq. 9), 该反应以Pd(OAc)2为催化剂、AgOAc为氧化剂、六氟异丙醇(HIFP)作溶剂, 并加入一定量的配体, 此反应体系的底物适用范围广, 选择性好, 与苯酚环上的各种电子效应的取代基都有很好的兼容性; α, β-不饱和羧酸酯、膦酸酯、酰胺以及苯环上含有吸电子取代基的苯乙烯都可作为烯烃化试剂发生烯烃化反应.
|
|
(9) |
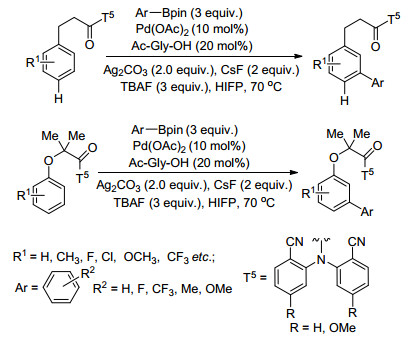
Yu等[16]以T5为模板首次实现3-苯基丙酸和苯酚及其衍生物的芳基化反应, 反应以Pd(OAc)2为催化剂、碳酸银为氧化剂、Ac-Gly-OH为配体、CsF和四丁基六氟磷酸铵(TBAPF6)分别为碱和添加物, 芳基硼酸作芳基化试剂(Scheme 1), 以42%~85%产率得到芳基化产物, 各种一取代的3-苯基丙酸衍生物都高选择性地得到间位芳基化产物, 但是由于位阻效应, 二取代3-苯基丙酸衍生物基本不发生反应, 该反应体系为合成芳基取代的3-苯基丙酸和酚衍生物提供了新的合成方法.
随后, Yu等[17]又将模板T5应用于Pd(OAc)2催化苯乙酸衍生物间位的烯烃化反应(Eq. 10), 该反应适用范围广, 考察范围内的各种单取代苯乙酸、α-甲基取代苯乙酸都能与丙烯酸乙酯发生反应, 高选择性地得到间位取代产物, 但是对位取代的苯乙酸间位烯烃化产率要低一些, 而溴代和碘代苯乙酸不能发生反应, 二取代苯乙酸也能高选择性地得到间位烯烃化产物; 甲基乙烯基酮、N, N-二甲基丙烯酰胺、乙烯基膦酸二乙酯和丙烯醛都能与间甲基苯乙酸发生反应, 高产率和高选择性地得到间位烯烃化产物, 配体和KH2PO4对反应至关重要.
|
|
(10) |
Rh催化剂在邻位碳氢官能化反应中有很高的活性. 2017年, Yu课题组[18]首次报道了[RhCp*Cl2]2催化3-苯基丙酸衍生物间位烯烃化反应(Eq. 11), 该反应以氨基苯腈T6为模板、Cu(CO2CF3)2•xH2O为氧化剂、CF3COOH作添加剂.在考察的底物范围内, 各种取代的1-苯基丙酸都有很高的间位选择性, 而且没有间位二取代产物生成, 但是该反应对1-苯基丙酸对位取代基的电子效应和位阻效应敏感, 当取代基是4-NO2或4-tBu时, 反应基本不发生.催化体系对于烯烃底物适用范围广, 丙烯酸酯、N, N-二甲基丙烯酰胺、乙基乙烯基酮、丙烯醛、β-取代烯酮、乙烯基二乙基膦酸酯、苯基乙烯基砜以及苯环上含有吸电子取代基的苯乙烯都能高产率地得到烯烃化产物.最近, Yu课题组[19]又将该模板应用到以二取代炔烃为烯烃化试剂的1-苯基丙酸间位烯烃化反应中(Eq. 12), 反应以[RhCp*Cl2]2为催化剂, 当底物是邻、间、对位一取代的1-苯基丙酸衍生物时都只得到间位一取代产物, 炔丙基醇也能很好地发生烯烃化反应, 此反应为合成间烯基取代3-苯丙酸提供了一种方法.这是Rh催化炔烃与1-苯基丙酸间位烯烃化的首例报道.
|
|
(11) |
|
|
(12) |
羟基苯腈可直接购买得到, 不需要合成, 作为模板与底物容易反应, 且结构简单, 反应后模板易于脱除.
2014年, Maiti等[20]以2-羟基苯腈或2-羟基-5-甲氧基苯腈为模板, 以羰基连接模板和底物苯乙酸, 实现了苯乙酸及其衍生物间位烯烃化, 反应以Pd(OAc)2为催化剂, Ag2CO3为氧化剂, 该反应体系能够容忍多种邻、间、对位的取代基, 对于各种取代的苯乙酸都能以中等到良好的产率得到间位选择性产物(Eq. 13), 但是该模板对取代苯乙酸间位乙酰氧化和芳基化基本没有作用, 而且对于间位烯烃化产物会与溶剂发生酯交换反应.
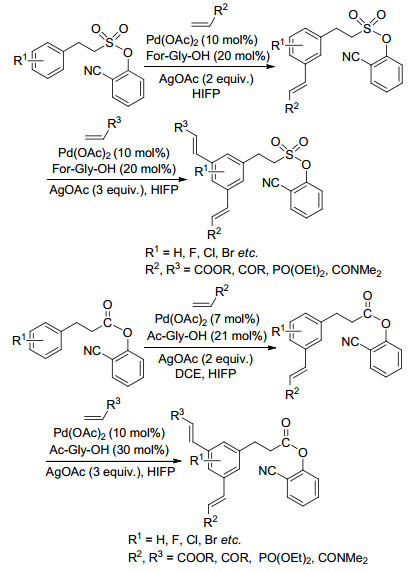
硫酰基有强的吸电子效应, 能降低苄磺酸酯邻位和对位电子云密度, 与羰基相比硫酰基可自由旋转, 能增强氰基的导向能力, 而且硫酰基可转化为多种官能团. 2015年, Maiti等[21]以2-羟基苯腈为模板, 以硫酰基连接底物与模板, Pd(OAc)2为催化剂, AgOAc为氧化剂, Ac-Gly-OH为配体, 在优化的条件下实现了苄磺酸间位的烯烃化, 在考察范围内苯环上分别含有吸电子和给电子的邻、间、对取代基时都有很好的间位选择性.烯烃可以是α, β-不饱和酮、醛、羧酸酯、膦酸酯、磺酸酯和酰胺等, 在优化的条件下, 二取代反式烯烃作为烯烃化试剂主要得到E构型产物, 甲基丙烯酸甲酯和环状三取代烯烃得到间位烯丙基取代苄磺酸.间位一取代产物可以继续与不同的烯烃反应, 得到两个间位同时被不同取代基取代的产物.当增加反应体系中烯烃的量时, 可得到两个间位同时被相同取代基取代的产物, 二烃基取代的苄磺酸酯脱除模板后得到三取代烯烃, 为合成1, 3, 5-三取代烯烃提供了一种重要的方法(Scheme 2).随后, 他们[22]改变反应条件, 以Pd(OAc)2为催化剂、For-Gly- OH为配体, 在PhI(TFA)2的作用下, 得到了苄磺酯间位羟基化的产物(Eq. 14).当配体变为Boc-Ala-OH时, 在PhI(OAc)2的作用下, 得到是间位乙酰氧化的产物(Eq. 15).
|
|
(13) |
|
|
(14) |
|
|
(15) |
Maiti等[23]还将2-羟基苯腈模板应用到2-苯基乙烷磺酸和3-苯基丙酸间位烯烃化反应中, 高选择性地得到间位产物.得到的间位取代产物可以继续与烯烃发生反应, 得到间位带相同或不同取代基的二取代2-苯基乙烷磺酸以及3-苯基丙酸(Scheme 3).
2017年, Maiti等[24]以2-羟基-4-甲氧基苯腈为模板, 实现了苄磺酸(Eq. 16)和苯乙酸(Eq. 17)间位烯烃化, 该反应以[{Rh(COD)Cl}2]为催化剂, XPhos为配体, V2O5为氧化剂, Cu(CO2CF3)2作共氧化剂, 共氧化剂对反应起到了重要作用, 吸电子和供电子取代基的邻、间、对位苄磺酸都有很好的间位选择性和反应活性, 位阻对该反应活性略有影响, 当苯磺酸对位被甲基取代时, 间位产物的产率是49%;烯烃可以是α, β-不饱和酮、α, β-不饱和羧酸酯、乙烯基砜、丙烯醛和甲基乙烯基酮等.他们对反应机理进行了研究, 认为反应也是经过脱质子碳氢键活化、烯烃插入、β-氢消除和还原消除四个步骤.
|
|
(16) |
|
|
(17) |
同年, Maiti课题组[25]首次报道了氰基模板导向钯催化的苄磺酸酯类衍生物间位的硅烷化(Eq. 18)和硒烷化(Eq. 19)反应, CH3, F, Cl和CF3取代的苄磺酸、二氟代和三氟代苄磺酸以中等到良好产率和较高的选择性得到间位硅烷化产物, 2-苯基乙烷磺酸和3-苯基-1-丙烷磺酸同样可以发生硅烷化反应.
|
|
(18) |
|
|
(19) |
2016年, Maiti等[26]报道了2-氰基苯酚导向的苄基膦酸酯间位的烯烃化反应(Eq. 20), 这是关于此类反应可在室温下进行的首例报道, 在优化的条件下, Br和CH3邻位或间位取代的苄基磷酸酯都高产率地得到间位取代产物, 当对位取代基为Br, CH3, OCH3和Cl时同样得到间位烯烃化产物, 但对位叔丁基取代苄基膦酸酯只得到了15%间位烯烃化产物.乙烯基膦酸酯、丙烯酸酯、烯酮、乙烯基砜及苯环上含有乙酰基和硝基的苯乙烯都可作为烯烃化试剂.当改变反应体系中加入的配体, 并提高反应体系的温度时, 可得到间位相同或不同取代基的二取代膦酸酯.间位二取代膦酸酯脱去模板后可得到1, 3, 5-三烃基取代的芳香化合物.当用PhI(OAc)2或PhI(TFA)2代替烯烃时可得到间位乙酰氧化(Eq. 21)或羟基取代(Eq. 22)的产物.反应后的产物膦酸酯容易脱除模板.
|
|
(20) |
|
|
(21) |
|
|
(22) |
2016年, Maiti课题组[27]报道了氰基模板导向苄基硅烷间位烯烃化反应, 在考察范围内该催化体系对各种取代苄基硅烷和烯烃都有良好的兼容性, 高选择性地得到间位取代产物, 间位一取代产物可继续发生间位烯烃化, 从而得到间位二取代产物.间位一取代苄基硅烷可以先脱除模板, 再发生邻位烯烃取代反应, 得到2, 5-二烃基取代产物, 也可以继续发生间位烯烃化反应, 再脱除模板, 从而得到3, 5-二烃基取代产物(Scheme 4).在脱除模板的反应中可采取不同的反应条件, 从而得到甲苯、苯甲醛和苄醇等产物.
2016年, Maiti等[28]以羰基连接2-羟基苯腈模板和二苯基羧酸以及二苯基酚及其衍生物, 实现了Pd(OAc)2催化二苯基羧酸和二苯基酚及其衍生物间位烯烃化反应(Scheme 5), 丙烯酸酯、乙烯基酮、乙烯基膦酸酯、乙烯基砜、β-取代丙烯醛以及丁烯酸二甲酯都可以作为烯烃化试剂. CH3和Cl取代的二苯基乙酸与催化体系有好的兼容性, 而OMe和OPh会使反应的选择性大大降低, 杂环及萘环底物不能发生反应.
2017年, Xu课题组[29]报道了2-羟基-5-甲氧基苯腈模板导向的2-苯乙醇、3-苯丙醇以及具有相似结构长链同系物的苯环间位碳氢键的烯烃化(Eq. 23), 反应以Pd(OAc)2为催化剂, AgOAc为氧化剂, 催化体系的活性高, 底物适用范围广, 区域选择性好, 产物中二取代烯烃的比例低, 不同碳原子数的醇和酚氧基醚等化合物都可以与丙烯酸乙酯发生反应, 以较高的选择性得到间位烯烃化产物.作为烯烃化试剂, N, N-二甲基丙烯酰胺、苯基乙烯基砜和4-氟代苯乙烯等都可以与3-苯丙醇和间位甲基取代的3-苯丙醇发生反应, 得到间位取代产物.在优化的条件下二肽衍生物(l-Phe-Ser和l-Phg-Ser)也能发生间位烯烃化反应.通过机理研究, 他们认为PdAg(OAc)3是反应的活性物种, 当苯环上的链比较短时, 大环过渡态中C-N-Ag之间的角度对间位选择性起重要作用, 最优角度是180°, 与邻位和对位产物相比, 间位产物碳氢活化过渡态中此夹角接近180°.当链比较长时, 苯基醚键的邻位交叉构象导致的扭转张力也起到了重要作用(图 4), 在碳氢活化过渡态中二面角Ψ越接近180°越稳定.
2014年, Yu小组[30]以T12为模板, 氰基为导向基、Pd(OAc)2为催化剂实现了四氢喹啉和苯并吗啉等化合物及其衍生物7-位碳氢键的烯烃化(Eq. 24).模板中F的电子效应对区域选择性影响不大, F取代的模板的构象对反应的区域选择性起到了重要的作用, 由于F的存在, 模板中羰基远离C(8)位, 从而避免C(8)位的烯烃化, 有利于-CN导向活化C(7), 从而得到间位产物.他们还将此模板应用到N-甲基苯胺及苄胺类化合物间位的烯烃化(Eq. 25)和乙酰氧化反应(Eq. 26)中, 该反应体系对各种化学性质的取代基都有良好的兼容性, 但是苯环上无取代基的底物二取代产物的比例略高于一取代产物.
|
|
(24) |
|
|
(25) |
|
|
(26) |
2015年, Li小组[31]报道了氰基导向Pd(OAc)2催化苯乙胺衍生物烯烃化反应(Eq. 27), 苯乙胺邻位和对位含有给电子的CH3和OCH3以及吸电子的Br和Cl等取代基时都可以高选择性地得到间位取代产物, 但是对位和邻位取代苯乙胺会生成间位二取代产物, 邻位取代苯乙胺还会生成两个间位各自被取代的间位一取代产物, 组成较复杂.模板中酰胺氮原子上的甲基对产物的选择性至关重要, 当酰胺氮原子上没有甲基取代基时得到的是邻位烯烃化的产物.
|
|
(27) |
2016年, Li小组[32]报道了模板T13导向、Pd(OAc)2催化的苯甲酸衍生物间位选择性烯烃化(Eq. 28)及乙酰氧化反应(Eq. 29), 反应体系以氧气作氧化剂, 苯甲酸邻位上给电子的取代基CH3和OAc以及吸电子的取代基F和Cl时都可以高选择性得到间位产物, 但是空间位阻小的F, Cl和OAc主要得到间位二取代产物.对位以及间位取代基的电子效应和空间效应对选择性和产率基本没有影响, 都以高产率和选择性得到间位一取代的产物.二取代以及三取代的苯甲酸都能高产率和选择性地得到间位取代的产物.苯甲酸的乙酰氧化活性虽然没有烯烃化高, 但是也以中等到良好产率得到目标产物.
|
|
(28) |
|
|
(29) |
2017年, Houk等[33]通过氰基模板导向实现了苯甲酸间位烯烃化(Eq. 30), 与苯乙酸、3-苯基丙酸和苄醇等底物相比, 苯甲酸是具有刚性结构的底物, 模板结构灵活, 通过一系列的模板筛选发现, 与底物中模板的构象相比, 过渡态中模板的变形程度对产物的区域选择性影响很大, 在间位选择性过渡态中模板的变形程度最小.
|
|
(30) |
2017年, Li课题组[34]以氨基甲酸酯连接氰基模板和苯胺衍生物, 实现了苯胺衍生物间位烯烃化(Eq. 31).在考察范围内邻位和间位取代的苯胺反应活性高, 区域选择性好, 对位取代的苯胺以中等到良好产率得到目标产物, 该反应体系同样适用于苯胺间位的乙酰氧化反应, 相比间位烯烃化反应, 活性略有降低(Eq. 32).
|
|
(31) |
|
|
(32) |
最近, Yu课题组[35]又报道了双官能团氰基模板T15并将其应用于钯催化的苯酚及其衍生物间位烯烃化反应中(Eq. 33), 吸电子和给电子的取代基都可以高选择性地得到间位产物.烯烃化反应后在Ni(Xantphos)Cl2催化下与ArB(OH)2反应, 原位芳烃化反应脱除模板, 得到1, 3-二取代芳香化合物(Eq. 34).该模板合成方法简单, 原料廉价易得, 既可辅助间位碳氢活化, 又可作为官能团发生C—O断裂, 引入芳基.该方法可由酚制备1, 3-二取代芳香化合物.
|
|
(33) |
|
|
(34) |
Xu课题组[36]以T16为模板, 通过芳香季铵盐苯环间位烯烃化反应实现了芳香叔胺及其衍生物苯环的间位烯烃化(Eq. 35), 该反应体系以Pd(OAc)2为催化剂, AgOAc为氧化剂, 苯环上各种电子效应取代基的邻、间、对-N, N-二甲基苯胺衍生物都高选择性地得到了间位产物, 烷基、烷氧基、氟和氯取代基都与催化体系有很好的兼容性, 但是氟和氯取代基导致间位产物产率降低.
|
|
(35) |
由于氰基模板的配位效应弱, 当有其他配位性强的试剂或反应物存在时, 氰基不能与催化剂钯有效配位, 因此限制了底物的范围.
2015年, Yu小组[37]报道了基于配位能力强的吡啶模板导向的苄醇和苯乙醇间位烯烃化(Eq. 36)和碘化反应(Eq. 37).通过巧妙设计模板的几何结构成功实现了间位活化反应, 考察范围内对各种取代的苄醇底物都取得了满意的结果, 邻位取代的底物在位阻小的一侧间位反应生成单取代产物, 间位取代底物高产率和选择性地生成间位一取代产物, 对位F和COOCH3取代的苄醇高选择性地得到间位一取代和二取代的混合产物, 二级苄醇和苯乙醇也能发生间位烯烃化反应.在优化的条件下, 邻、间、对位取代的苄醇以64%~85%的产率高间位选择性地得到碘代产物.
|
|
(36) |
|
|
(37) |
2018年, Yu课题组[38]合成了U型吡啶模板, 实现了钯催化苯乙酸间位的烯烃化(Eq. 38), 他们还以ArBF3K和1, 3-二碘-5, 5-二甲基海因(DIH)作芳基化和碘化试剂, 得到了间位芳基化(Eq. 39)以及碘化产物(Eq. 40).该反应体系的底物适用范围广, 对各类吸电子和给电子的取代苯乙酸都有好的兼容性, 萘乙酸、杂环羧酸和α-取代苯基乙酸都能发生烯烃化、芳基化和碘化反应, 并高选择性地得到间位取代产物.
|
|
(38) |
|
|
(39) |
|
|
(40) |
与乙烯基膦酸酯、丙烯酸酯、烯酮和乙烯基砜等烯烃化试剂相比, 烯丙基醇双键上电子云密度大, 在烯烃的插入过程中, 金属中心的电正性越强越有利于烯烃的插入.与吡啶相比, 嘧啶中两个氮原子的存在会使电子云密度减小, 与金属配位时, 金属中心的电正性增强.嘧啶的结构与吡啶相似, 作为导向基团替代吡啶保持底物的U型构象.此外, 嘧啶环可绕碳碳单键旋转, 增加了氮原子与间位碳氢键接触的机率. 2017年, Maiti课题组[39]报道了嘧啶模板导向Pd(OAc)2催化的苄磺酸酯及其衍生物间位活化反应, 该反应分别以烯丙基醇和苄基烯丙基醚为底物, 在优化的条件下分别得到了β-芳基醛酮和α, β-不饱和醛, 他们还将该反应拓展到了3-苯基丙酸酯、2-苯基乙基醚和苄基磷酸酯等物质的间位活化反应中(Scheme 6), 同样得到了满意的结果.
随后, 该课题组[40]又将该嘧啶模板用于苄基硅烷的间位氰化反应中(Eq. 41), 反应以Pd(OAc)2为催化剂、CuCN作氰化试剂, 苄基硅烷苯环上吸电子取代基和给电子取代基都具有良好的反应兼容性, 对位取代基位阻大的苄基硅烷也有较高的间位选择性, 该反应体系同样适用于苄磺酸酯、苄基磷酸酯(Eq. 41)、苯乙烷磺酸酯和苯乙醚(Eq. 42).
|
|
(41) |
|
|
(42) |
2018年, Maiti小组[41]又用嘧啶模板导向3-苯丙醇以及具有相似结构的长链同系物的苯环间位碳氢键的活化, 碳链的长度使间位碳氢键与导向基参与配位的原子间间隔20个原子以上, 反应以Pd(OAc)2为催化剂, 在不同的反应条件下可高选择性地得到间位烷基化(Eq. 43)、烯烃化(Eq. 44)、乙酰氧化(Eq. 45)和氰化的产物(Eq. 46), 成功实现了C—C及C—O的构建.通过对n为3, 5和10的底物的烯烃化反应速率的研究, 他们认为随着n值的增加, 反应速率减小.
|
|
(43) |
|
|
(44) |
|
|
(45) |
|
|
(46) |
最近, Wang课题组[42]以嘧啶模板T19为导向基实现了3-苯基-1, 2-丙二醇及其衍生物苯环间位烯烃化反应(Eq. 47), 反应体系以Pd(OAc)2为催化剂, AgF为氧化剂, Na2CO3为添加物, 催化体系适用范围广, 苯环上各种电子效应的邻、间、对位取代二醇都选择性得到间位取代产物, 丙烯酸酯、N, N-二甲基丙烯酰胺、乙烯基膦酸二乙酯、乙烯基砜和五氟代苯乙烯都可作为烯烃化试剂.
|
|
(47) |
2017年, Maiti课题组[43]以硫酰基连接8-硝基喹啉模板, 实现了苄磺酸酯间位烯烃化(Eq. 48)和乙酰氧化(Eq. 49)反应, 反应体系适用范围广, α, β-不饱和酮、丙烯酸酯、N, N-二甲基丙烯酰胺、乙烯基膦酸二乙酯、乙烯基砜和丁烯二酸二甲酯等一系列烯烃和苄磺酸酯都得到了较高的产率和选择性, 含有不同化学性质取代基的苄磺酸酯衍生物与烯烃反应也高产率地得到间位烯烃化产物.间位单取代产物可以继续与烯烃发生反应, 得到间位带相同或不同取代基的二烃基取代的苄磺酸酯.反应之所以有高的反应活性和选择性原因如下: (1)硫酰基氧原子间的电子效应和空间效应以及硫原子sp3杂化方式增强了导向基间位导向能力; (2)硝基与金属钯之间的作用力以及硝基氧原子与底物中芳基碳氢键之间的次级效应有利于间位产物的生成.
|
|
(48) |
|
|
(49) |
虽然U-型模板在间位选择性活化中表现出优越的性能, 但此策略需要设计特定模板, 模板以共价键与底物结合, 反应后需借助化学方法脱除模板, 限制了该方法的应用.
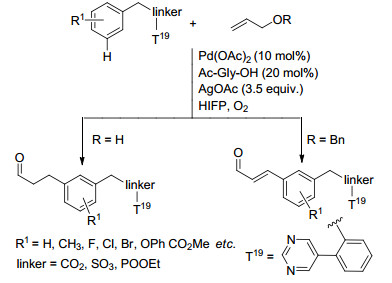
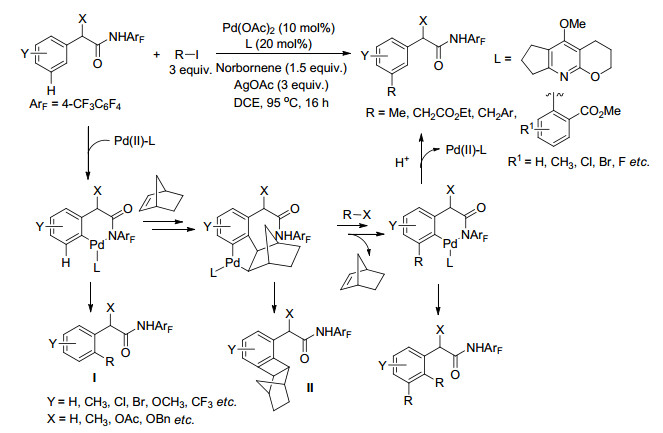
Yu课题组采用降冰片烯作为瞬态介质, 使用简单的邻位导向取代基来实现苯环间位选择性碳氢键活化. 2015年, Yu等[44]报道了Pd(OAc)2催化、降冰片烯(NBE)瞬态介导、具有给电子效应取代基的吡啶类配体作用下下苯乙酰胺苯环间位的烷基化以及芳基化反应(Scheme 7), 在优化的条件下该反应对各种酰胺的衍生物都得到了较好的结果, 但是该反应体系适用范围有限, 对于酰胺的乙基化反应只得到了21%的产物, 对于芳基化反应, 当芳基化试剂邻位无配位基团时也不能发生反应.他们推测反应机理为:酰胺基团诱导金属钯活化苯环邻位碳氢键, 之后降冰片烯双键与钯进行配位, 随后发生Catellani反应使钯对苯环间位进行活化, 随后降冰片烯离去, 钯插入碳卤键中发生异构化反应, 还原消除得到间位活化产物.反应过程中副产物I和II的生成会导致无法得到间位活化产物, 配体和降冰片烯的加入能够抑制副产物I的生成, 对反应非常重要.随后他们[45]通过优化降冰片烯结构和配体, 使用NBE-CO2Me作瞬态介质, 成功克服了上述缺点, 拓展了底物适用范围, 苯乙酰胺衍生物可以与各种长链的烷基碘和芳基碘发生反应(Eq. 50). Shi课题组[46]通过在苯乙胺底物中引入含有N, O二齿配位的乙二酰基导向基, 也成功克服了上述缺点, 实现了苯乙胺衍生物间位芳烃化反应, 该反应体系以降冰片烯作为瞬态介质, 对各种性质取代基的碘苯以及苯乙胺衍生物都有很好的间位选择性(Eq. 51).
|
|
(50) |
|
|
(51) |
Dong课题组[47]以Pd/NBE/AsPh3催化体系实现了苯乙胺及其衍生物间位的芳基化反应, 产率最高可达80%, 底物中胺基作为导向基活化邻位, NBE发挥导向基团作用, AsPh3可能作为配体来稳定过渡态(Eq. 52).
|
|
(52) |
Yu课题组[48]首次报道了Pd(OAc)2催化、NBE- CO2Me瞬态介导苯胺衍生物(Eq. 53)和酚衍生物(Eq. 54)间位胺基化和炔基化反应(Eq. 55), 配体和NBE-CO2Me对反应起到了重要作用, 吲哚、二氢吲哚、吲唑及其衍生物都以良好到优秀产率得到间位氨基化和炔基化产物.
|
|
(53) |
|
|
(54) |
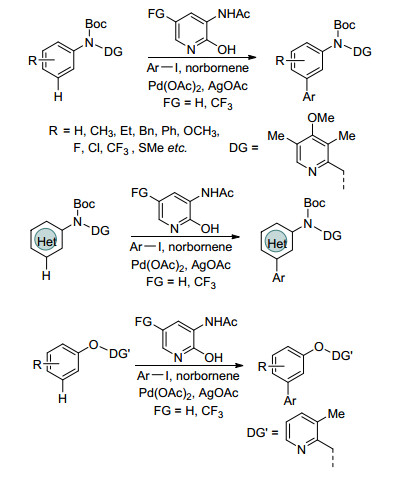
Yu课题组[49]首次以吡啶、呋喃、噻吩、喹啉、苯并吡唑等杂环碘代物作为芳基化试剂, 实现了苯胺、苯酚、杂环芳胺(Scheme 8)、2-苄基杂环芳胺及其衍生物(Eq. 56)间位芳基化反应.催化体系以降冰片烯作瞬态介质, 以3-乙酰氨基-2-羟基吡啶作配体, 在底物和芳基化试剂考察范围内, 得到了较好的结果.
|
|
(55) |
|
|
(56) |
2017年, Yu课题组[50]以Pd(OAc)2催化、NBE- CO2Me瞬态介导实现了苯乙酸及其衍生物间位芳基化反应(Eq. 57), 反应以乙酸底物中羧基作导向基团, 吡啶类化合物作配体, 对各种电子性质的芳基碘都有很好的反应活性, 但是杂环芳基碘不能作为芳基化试剂.
|
|
(57) |
Yu课题组[51]将瞬态介导催化体系应用到对硝基苯磺酰基保护的苯胺、苄胺以及2-芳基苯胺(Eq. 58)的间位芳基化反应中, 首次以催化剂量(20 mol%)的降冰片烯作瞬态介质, 以4-乙酰基吡啶作配体, 考察范围内的各种苯胺和2-芳基苯胺均有好的反应活性, 各种性质取代基取代的芳基碘、邻位含有配位基团的芳基溴都可作为芳基化试剂, 这是芳基溴作芳基化试剂的首例报道, 但是对于苄胺只有苯基甘氨酸能与邻位含有配位基团的芳基碘发生反应.随后他们[52]改变反应的配体和瞬态介质, 以NBE-CO2Me为瞬态介质, 吡啶酮为配体, 该催化体系对各种苄胺衍生物芳基化都有好的催化效果, 该反应体系对苄胺衍生物的胺基化、氯化同样有好的催化效果, 而且杂环芳基碘也可作为芳基化试剂(Eq. 59). 2018年, Ding课题组[53]报道了对硝基磺酰基保护苄胺的烷基化反应, 反应以NBE5为瞬态介质, 吡啶为配体, 当CH3I和ICH2COOEt作烷基化试剂时, 以中等到优秀产率得到间位烷基化产物, 而CH3CH2I作烷基化试剂, 当R1是Me和F时分别得到了24%和27%的间位烷基化产物(Eq. 60).
|
|
(58) |
2017年, Yu课题组[54]报道了苄磺酰胺及其衍生物间位烷基化以及芳基化反应(Eq. 61).该体系以Pd(OAc)2为催化剂NBE-CO2Me为瞬态介质, 异喹啉为配体, 该催化体系适用范围广, 对各种性质取代基取代的苄磺酰胺都有高的催化活性, 芳基化试剂可以是各种取代的芳基碘和杂环芳基碘.
|
|
(59) |
|
|
(60) |
|
|
(61) |
Ferreira课题组[55]首次以瞬态介导催化体系实现了苄醇及其衍生物间位芳基化(Scheme 9), 该体系以Pd-(TFA) 2为催化剂, NBE-CO2Me为瞬态介质, TFA-Gly- OH为配体, 考察范围内各种取代苄醇和取代芳基碘都有较高的反应活性和选择性.
Yu课题组[56]以Pd(OAc)2为催化剂, NBE-CO2Me为瞬态介质, 喹啉酮为配体, 实现了苯甲醛及其衍生物间位芳基化, 苯甲醛首先与烷基锂试剂试剂反应, 同时以叔丁基二甲基氯硅烷(TBSCl)捕获羟基, 在醛分子中引入导向基, 然后再与芳基碘反应(Eq. 62).
|
|
(62) |
瞬态介导间位活化反应主要是围绕配体和瞬态介质的优化展开的, 以实现不同底物的间位碳氢键活化.
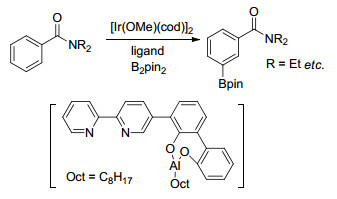
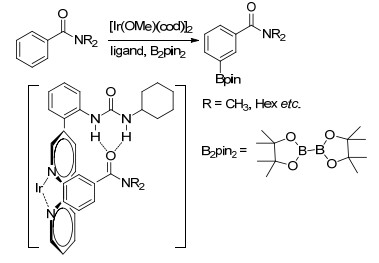
Kanai小组[57]报道了另一策略来实现苯环间位碳氢活化, 利用具有次级作用的双官能配体, 将具有较强成氢键能力的脲结构与联吡啶配体巧妙结合在一起, 在温和的条件下, 脲结构较易与底物中的氧原子形成氢键, 配体联吡啶与催化剂Ir配位, 通过调整连接联吡啶与脲结构的桥使金属中心靠近间位(Scheme 10), 成功实现了具有多种酰氧官能团取代的芳香族化合物的间位硼化反应.该小组发现通过调整配体中的取代基、脲中的取代基可加速硼化反应的进行, 当R1是给电子的取代基t-Bu时, 金属Ir中心的电子云密度增加; 由于反应是通过扭曲结构进行的, 因此, R2=CH3时联吡啶和苯环之间会保持一定角度, 有利于反应进行; 而R3会导致脲结构域底物形成氢键能力的改变.底物氮原子上的取代基的位阻效应以及与脲结构发生配位的原子发生变化会导致产物发生变化(图 5)[58]. Sunoj等[59]对该反应体系的机理进行了研究, 他们认为除了配体中脲结构与底物中原子间的氢键作用力, 底物中C—H与吡啶氮原子、吡啶配体π、B2pin2中氧原子间的非共价键作用对产物的区域选择性也起到了重要作用.
Nakao等[60]通过路易斯酸碱的相互作用力, 实现了苯甲酰胺以及吡啶环间位硼化反应, 通过路易斯酸Al识别底物酰胺或吡啶环中的碱氮原子, 联吡啶与催化剂Ir配位, 使Ir活化苯甲酰胺和吡啶环的间位(Scheme 11).
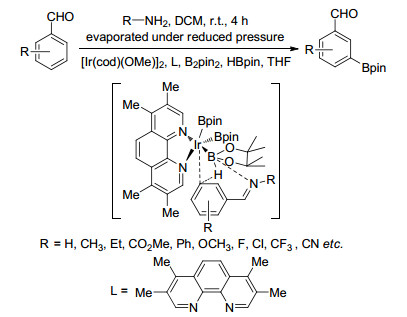
Chattopadhyay等[61]利用配合物与底物的静电作用力以及配合物中硼原子与底物氮原子的次级作用力实现了苯甲醛间位硼化反应.首先反应中苯甲醛与胺生成亚胺, 借助静电作用和次级效应使催化剂活化亚胺的间位发生硼化反应(Scheme 12).
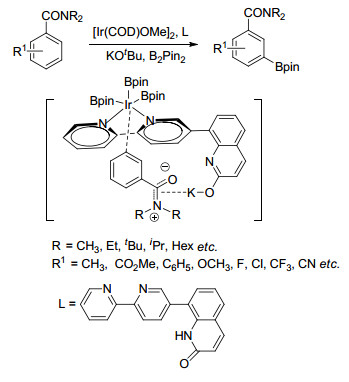
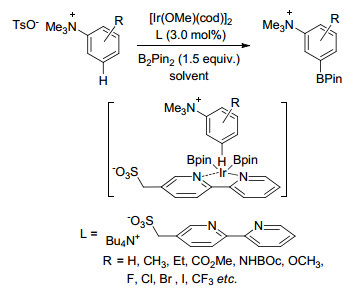
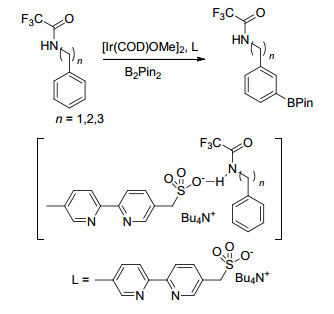
Phipps课题组[62]借助离子对相互作用实现了芳香季铵盐苯环间位硼化反应, 该反应中底物是含有正电荷的季铵盐, 配体则含有磺酸根负离子, 磺酸根与季铵盐相互作用, 使催化剂Ir接近苯环间位碳氢, 从而得到间位硼化产物(Scheme 13).他们[63]还将该配体应用到三氟乙酰苄胺、三氟乙酰苯乙胺和三氟乙酰苯丙胺衍生物间位硼化反应中, 通过研究发现, 该反应的区域选择性与磺酸根与酰胺氮原子上氢原子之间的氢键作用力有关(Scheme 14).
Chattopadhyay小组[64]通过配体中阳离子与底物π键之间的非共价键作用力实现了苯甲酰胺间位硼化反应, 对各种取代的苯甲酰胺衍生物都以高的产率得到间位硼化产物(Scheme 14).
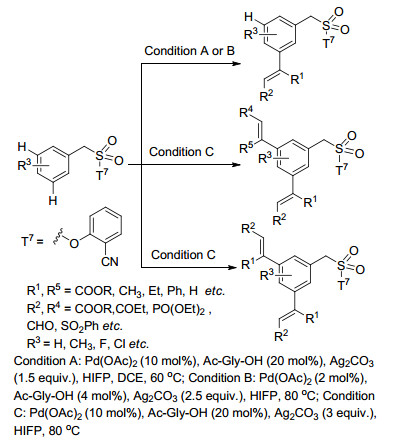
虽然U-型模板在间位选择性活化中表现出优越的性能, 但此策略需要设计特定模板, 模板以共价键与底物结合, 反应后需借助化学方法脱除模板.配体次级作用力导向的间位活化缺点是次级作用力较弱, 在较高温度条件下不易形成, 目前催化体系比较单一, 都是Ir催化下的硼化反应.降冰片烯介导碳氢间位活化导向基具有一定的优势, 但是降冰片烯介导间位活化多为芳基化和烷基化, 芳基化和烷基化试剂多为碘试剂.因此, 开发催化体系多样化的瞬态介导间位活化的未来的发展方向.
Mkhalid, I.-A.-I.; Barnard, J.-H.; Marder, T.-B.; Murphy, J.-M.; Hartwig, J.-F. Chem. Rev. 2010, 110, 890. doi: 10.1021/cr900206p
Zhang, Y.-H.; Shi, B.-F.; Yu, J.-Q. J. Am. Chem. Soc. 2009, 131, 5072. doi: 10.1021/ja900327e
Hofmann, N.; Ackermann, L. J. Am. Chem. Soc. 2013, 135, 5877. doi: 10.1021/ja401466y
Madalina, T.-M.; Genov, G.-R.; Phipps, R.-J. Chem. Soc. Rev. 2018, 47, 149. doi: 10.1039/C7CS00637C
Dey, A.; Sinha, S.-K.; Achar, T.-K.; Maiti, D. Angew. Chem., Int. Ed. 2019, 58, 2. doi: 10.1002/anie.201813331
Yang, J. Org. Biomol. Chem. 2015, 13, 1930. doi: 10.1039/C4OB02171A
Chen, Z.-K.; Wang, B.-J.; Zhang, J.-T.; Yu, W.-L.; Liu, Z.-X.; Zhang, Y.-H. Org. Chem. Front. 2015, 2, 1107. doi: 10.1039/C5QO00004A
Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Nature 2012, 486, 518. doi: 10.1038/nature11158
Yang, Y.-F.; Cheng, G.-J.; Liu, P.; Leow, D.; Sun, T.-Y.; Chen, P.; Zhang, X.-H.; Yu, J.-Q.; Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 344. doi: 10.1021/ja410485g
Lee, S.; Lee, H.; Tan, K. L. J. Am. Chem. Soc. 2013, 135, 18778. doi: 10.1021/ja4107034
Yang, G.-Q.; Lindovska, P.; Zhu, D.-J.; Kim, J.; Wang, P.; Tang, R.-Y.; Movassaghi, M.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 10807. doi: 10.1021/ja505737x
Mi, R.-J.; Sun, J.; Kϋhn, F.-T.; Zhou, M.-D.; Xu, Z.-Q. Chem. Commun. 2017, 53, 1320.
Mi, R.-J.; Sun, Y.-Z.; Wang, J.-Y.; Sun, J.; Xu, Z.-Q.; Zhou, M.-D. Org. Lett. 2018, 20, 5126. doi: 10.1021/acs.orglett.8b01995
Cheng, G.-J.; Yang, Y.-F.; Liu, F.; Chen, P.; Sun, T.-Y.; Li, G.; Zhang, X.-H.; HouK, K.-N.; Yu, J.-Q.; Wu, Y.-D. J. Am. Chem. Soc. 2014, 136, 897
Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A.-F.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 7567 doi: 10.1021/ja400659s
Wan, L.; Dastbaravardeh, N.; Li, G.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 18056. doi: 10.1021/ja410760f
Deng, Y.-Q.; Yu, J.-Q. Angew. Chem., Int. Ed. 2015, 54, 888. doi: 10.1002/anie.201409860
Xu, H.-J.; Lu, Y.; Farmer, M.-E.; Wang, H.-W.; Zhao, D.; Kang, Y.-S.; Sun, W.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 2200. doi: 10.1021/jacs.6b13269
Xu, H.-J.; Kang, Y.-S.; Shi, H.; Zhang, P.; Chen, Y.-K.; Zhang, B.; Liu, Z.-Q.; Zhao, J.; Sun, W.-Y.; Yu, J.-Q.; Lu, Y. J. Am. Chem. Soc. 2019, 141, 76. doi: 10.1021/jacs.8b11038
Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760. doi: 10.1021/ol502823c
Bera, M.; Maji, A.; Sahoo, S.-K.; Maiti, D. Angew. Chem., Int. Ed. 2015, 54, 8515. doi: 10.1002/anie.201503112
Maji, A.; Bhaskararao, B.; Singha, S.; Sunoj, R.-B.; Maiti, D. Chem. Sci. 2016, 7, 3147. doi: 10.1039/C5SC04060D
Modak, A.; Mondal, A.; Watile, R.; Mukherjee, S.; Maiti, D. Chem. Commun. 2016, 52, 13916. doi: 10.1039/C6CC08302A
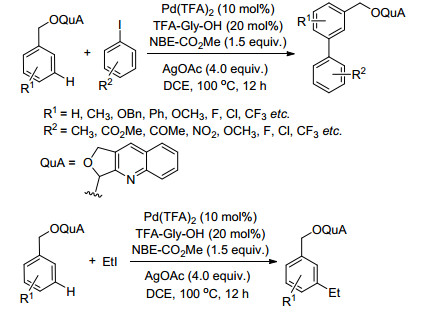
Bera, M.; Agasti, S.; Chowdhury, R.; Mondal, R.; Pal, D.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 5272. doi: 10.1002/anie.201701579
Modak, A.; Patra, T.; Chowdhury, R.; Raul, S.; Maiti, D. Organometallics 2017, 36, 2418. doi: 10.1021/acs.organomet.7b00309
Bera, M.; Sahoo, S.-K.; Maiti, D. ACS Catal. 2016, 6, 3575. doi: 10.1021/acscatal.6b00675
Patra, T.; Watile, R.; Agasti, S.; Naveen, T.; Maiti, D. Chem. Commun. 2016, 52, 2027. doi: 10.1039/C5CC09446A
Maity, S.; Hoque, E.; Dhawa, U.; Maiti, D. Chem. Commun. 2016, 52, 14003. doi: 10.1039/C6CC07861C
Zhang, L.-L.; Zhao, C.-Y.; Liu, Y.; Xu, J.-C.; Xu, X.-F.; Jin, Z. Angew. Chem., Int. Ed. 2017, 56, 12245. doi: 10.1002/anie.201705495
Tang, R.-Y.; Li, G.; Yu, J.-Q. NATURE 2014, 507, 215. doi: 10.1038/nature12963
Li, S.-D.; Ji, H.-F.; Cai, L.; Li, G. Chem. Sci. 2015, 6, 5595. doi: 10.1039/C5SC01737H
Li, S.-D.; Cai, L.; Ji, H.-F.; Yang, L.; Li, G. Nat. Commun. 2016, 7, 1.
Fang, L.-Z.; Saint-Denis, T.-G.; Taylor, B.-L.; Ahlquist, S.; Hong, K.; Liu, S.-S.; Han, L.-L.; Houk, K.-N.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 10702. doi: 10.1021/jacs.7b03296
Yang, L.; Fu, L.; Li, G. Adv. Synth. Catal. 2017, 359, 2235. doi: 10.1002/adsc.201700261
Xu, J.-C.; Chen, J.-J.; Gao, F.; Xie, S.-G.; Xu, X. H.; Jin, Z.; Yu, J.-Q. J. Am. Chem. Soc. 2019, 141, 1903. doi: 10.1021/jacs.8b13403
Wang, B.; Zhou, Y.; Xu, N.-N.; Xu, X.-F.; Xu, X.-H.; Jin, Z. Org. Lett. 2019, 21, 1885. doi: 10.1021/acs.orglett.9b00499
Chu, L.; Shang, M.; Tanaka, K.; Chen, Q.-H.; Pissarnitski, N.; Streckfuss, E.; Yu, J.-Q. ACS Cent. Sci. 2015, 1, 394. doi: 10.1021/acscentsci.5b00312
Jin, Z.; Chu, L.; Chen, Y.-Q.; Yu, J.-Q. Org. Lett. 2018, 20, 425. doi: 10.1021/acs.orglett.7b03336
Bag, S.; Jayarajan, R.; Mondal, R.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 3182. doi: 10.1002/anie.201611360
Bag, S.; Jayarajan, R.; Dutta, U.; Chowdhury, R.; Mondal, R.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 12538. doi: 10.1002/anie.201706360
Jayarajan, R.; Das, J.; Bag, S.; Chowdhury, R.; Maiti, D. Angew. Chem., Int. Ed. 2018, 57, 7659. doi: 10.1002/anie.201804043
Fang, S.-Q.; Wang, X.-B.; Yin, F.-C.; Cai, P.; Yang, H.-L.; Kong, L.-Y. Org. Lett. 2019, 21, 1841. doi: 10.1021/acs.orglett.9b00433
Dutta, U.; Modak, A.; Bhaskararao, B.; Bera, M.; Bag, S.; Mondal, A.; Lupton, D.-W.; Sunoj, R.-B.; Maiti, D. ACS Catal. 2017, 7, 3162. doi: 10.1021/acscatal.7b00247
Wang, X.-C.; Gong, W.; Fang, L.-Z.; Zhu, R.-Y.; Li, S.-H.; Engle, K.-N.; Yu, J.-Q. Nature 2015, 519, 334. doi: 10.1038/nature14214
Shen, P.-X.; Wang, X.-C.; Wang, P.; Zhu, R.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 11574. doi: 10.1021/jacs.5b08914
Han, J.; Zhang, L.; Zhu, Y.; Zheng Y.-X.; Chen, X.-L.; Huang, Z.-B.; Shi, D.-Q.; Zhao, Y.-S. Chem. Commun. 2016, 52, 6903. doi: 10.1039/C6CC02384C
Dong, Z.; Wang, J.-C.; Dong, G.-B. J. Am. Chem. Soc. 2015, 137, 5887. doi: 10.1021/jacs.5b02809
Wang, P.; Li, G.-C.; Jain, P.; Farmer, M.-E.; He, J.; Shen, P.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 14092. doi: 10.1021/jacs.6b08942
Wang, P.; Farmer, M.-E.; Huo, X.; Jain, P.; Shen, P.-X.; Ishoey, M.; Brandner, J.-E.; Wisniewski, S.-R.; Eastgate, M.-D.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 9269. doi: 10.1021/jacs.6b04966
Li, G.-C.; Wang, P.; Farmer, M.-E.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 6874. doi: 10.1002/anie.201702686
Ding, Q.-P.; Ye, S.-Q.; Cheng, G.-L.; Wang, P.; Farmer, M.-E.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 417. doi: 10.1021/jacs.6b11097
Wang, P.; Farmer, M.-E.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 5125. doi: 10.1002/anie.201701803
Liu, J.-Y.; Ding, Q.-P.; Fang, W.-J.; Wu, W.-H.; Zhang, Y.-G.; Peng, Y.-Y. J. Org. Chem. 2018, 83, 13211. doi: 10.1021/acs.joc.8b01933
Cheng, G.-L.; Wang, P.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 8183. doi: 10.1002/anie.201704411
Li, Q.-K.; Ferreira, E. M. Chem.-Eur. J. 2017, 23, 11519. doi: 10.1002/chem.201703054
Farmer, M.-E.; Wang, P.; Shi, H.; Yu, J.-Q. ACS Catal. 2018, 8, 7362. doi: 10.1021/acscatal.8b01599
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712. doi: 10.1038/nchem.2322
Lu, X.; Yoshigoe, Y.; Ida, H.; Nishi, M.; Kanai, M.; Kuninobu, Y. ACS Catal. 2019, 9, 1705. doi: 10.1021/acscatal.8b05005
Unnikrishnan, A.; Sunoj, R.-B. Chem. Sci. 2019, 10, 3826. doi: 10.1039/C8SC05335A
Yang, L.-C.; Uemura, N.; Nakao, Y. J. Am. Chem. Soc. 2019, 141, 7912.
Bisht, R.; Chattopadhyay, B. J. Am. Chem. Soc. 2016, 138, 84. doi: 10.1021/jacs.5b11683
Davis; H.-J.; Mihai, M.-T.; Phipps, R.-J. J. Am. Chem. Soc. 2016, 138, 12759. doi: 10.1021/jacs.6b08164
Davis, H.-J.; Genov, G.-R.; Phipps, R.-J..Angew. Chem., Int. Ed. 2017, 56, 13351. doi: 10.1002/anie.201708967
Bisht, R.; Hoque, M.-E.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2018, 57, 15762. doi: 10.1002/anie.201809929

图 2 苄基醚间位碳氢活化过渡态和Ψ3
Figure 2 Transition state of benzyl ether derivatives of meta- C—H activation and Ψ3

图 3 3-苯基丙酸间位碳氢活化过渡态
Figure 3 meta-C—H activation transition state of 3-phenyl- propanoic acid derivatives

图式 1 苯酚和3-苯基丙酸间位芳基化反应
Scheme 1 Meta-arylation of 3-phenylpropanoic acid and phenolic derivatives

图式 3 2-苯基乙磺酸和3-苯基丙酸间位二烯烃化反应
Scheme 3 meta-Diolefination of 2-phenyl ethanesulfonic acid and 3-phenylpropanoic acid derivatives

图式 5 二苯基羧酸和二苯基酚间位烯烃化
Scheme 5 Meta-olefination of biphenyl phenol and biphenyl carboxylic acid derivatives

图式 6 嘧啶模板导向间位碳氢键活化反应
Scheme 6 meta-C—H activation directed by pyrimidine-based template

图式 8 苯胺、苯酚和杂环芳胺间位芳基化反应
Scheme 8 meta-C—H arylation of phenylamine, phenol and heterocycle phenylamine

图式 9 苄醇及其衍生物间位芳基化和乙基化
Scheme 9 meta-C—H arylation and ethylation of benzyl alcohol derivatives

图式 11 路易斯酸碱作用力促进间位活化
Scheme 11 Interaction between Lewis acid and base accelerated meta-C—H activation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





























































 下载:
下载:









