Scheme 1.
Synthetic approaches for isatins
Copper-Catalyzed Aerobic Oxidation Strategy: A Concise Route to Isatin
Muhammad Siddique Ahmad , Yamin Zhu , Yunlong Guo , Saisai Zhang , Zengming Shen

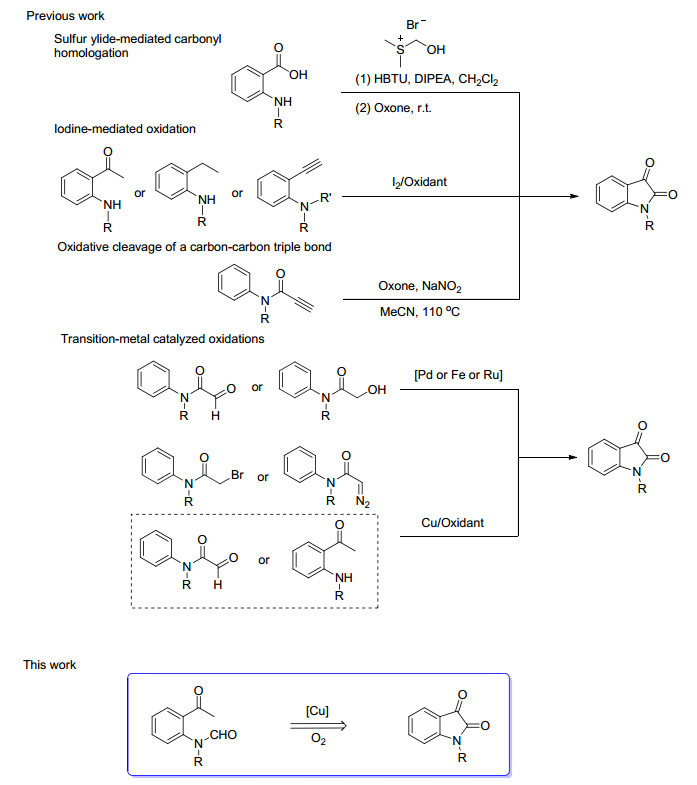
Isatins and their derivatives are a group of important structural units which have been found in many bioactive molecules.[1] Isatins are also powerful building blocks for the construction of heterocyclic systems and spirocyclic compounds.[2] Therefore, many methods have been developed for the synthesis of isatins. Traditionally, the synthesis of isatins was achieved through the methods developed by Sandmeyer, [3] Stolle, [4] and Martinet.[5] However, these methods suffer from harsh conditions, poor yields and limited substrate choices. In view of this, new synthetic methods to overcome these drawbacks are still highly required. Inspiringly, some efficient methods have been found, such as aryne-based methods, [6] transition-metal catalyzed oxidation, [7] oxidative cleavage of carbon-carbon triple bond, [8] sulfur ylide-mediated carbonyl homologation, [9] iodine-mediated oxidation, [10] and C—H amination (Scheme 1).[11] Although these methods showed some improvements, there are still many problems like the requirement of heavy metal catalysts and excess amount of bases or strong oxidants and relatively low yields of products. Thus, the development of a concise and environmentally benign process to access isatins remains highly desirable. In 2010, Li and co-workers[7c] reported a significant and elegant example concerning copper-catalyzed preparation of isatins through intramolecular aromatic C—H and aldehyde C—H dual activation to construct C—C bonds (Scheme 1). After that, Cheng and co-workers[7g] documented that 2-aminoacetophenones can undergo intramolecular cyclization to form isatins in the presence of a catalytic amount of copper salt under a balloon of O2 (Scheme 1). Inspired by these works, we envisioned a complementary way of C(sp3)—H bond functionalization catalyzed by copper to form β-ketoamide core structure. Herein, a copper-catalyzed decarbonylation cyclization to form isatins using oxygen as terminal oxidant is reported.
To optimize reaction conditions, N-(2-acetylphenyl)-N-methylformamide (1a) derived from readily available 1-(2-aminophenyl)ethanone was chosen as a model substrate. Pleasingly, by using catalytic amounts of CuCl2 in the presence of THF and oxygen at 100 ℃, the conversion of 1a into the desired N-methyl isatine 2a was smoothly performed (34% yield of 2a, Table 1, Entry 1). Next, a series of other copper catalysts was screened, such as CuCl, CuBr, CuI, CuSCN, CuCN, Cu2O and Cu(OAc)2. The results exhibited that activity of these copper salts is inferiors to CuCl2 or even ineffective (Table 1, Entries 2~8). Using CuCl2 as the catalyst, a variety of Lewis acids were examined. A significant Lewis acid effect was observed. CoCl2 was the most effective Lewis acid for promoting formation of α-ketoamide structure, affording 2a in 62% isolated yield (Table 1, Entry 16). ZnCl2 and CoSO4 slightly increased yield to 40% and 45%, respectively (Table 1, Entries 9 and 15). Other Lewis acids, such as AlCl3, BF3·Et2O, FeCl3, PdCl2, Cu(ClO4)2·6H2O, Yb(OTf)3, Ni(OAc)2·4H2O, Co(NO3)2·6H2O and Cu(OAc)2·4H2O suppressed reaction and even shut down the process (Table 1, Entries 10~14 and 17~20). Interestingly, when 1 equiv. of protonic acid, PhCOOH, combined with CoCl2 (1 equiv.) was added into the reaction, the yield of 2a was increased to 69% (Table 1, Entry 24). To our surprise, when the loading of CoCl2 and PhCOOH was decreased to 20 mol% catalytic amount, the efficiency of reaction was further improved and 78% yield of 2a was obtained (Table 1, Entry 25). Notably, control experiment showed that CoCl2 could not promote this transformation in the absence of CuCl2 (Entry 26). Consequently, the optimal conditions involved the use of CuCl2 (20 mol%), CoCl2 (20 mol%), PhCOOH (20 mol%) in O2 and THF at 100 ℃.
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entry | Cat. | Additive | Time/h | Yieldb/% |
| 1 | CuCl2 | None | 49 | 34 |
| 2 | CuCl | None | 110 | 23 |
| 3 | CuBr | None | 110 | 17 |
| 4 | CuI | None | 82 | N.R. |
| 5 | CuSCN | None | 82 | N.R. |
| 6 | CuCN | None | 82 | N.R. |
| 7 | Cu2O | None | 110 | Trace |
| 8 | Cu(OAc)2 | None | 66 | N.R. |
| 9 | CuCl2 | ZnCl2 (1 equiv.) | 49 | 40 |
| 10 | CuCl2 | AlCl3 (1 equiv.) | 49 | N.R. |
| 11 | CuCl2 | BF3·Et2O (1 equiv.) | 67 | N.R. |
| 12 | CuCl2 | FeCl3 (1 equiv.) | 67 | N.R. |
| 13 | CuCl2 | PdCl2 (1 equiv.) | 67 | N.R. |
| 14 | CuCl2 | Cu(ClO4)·6H2O (1 equiv.) | 67 | N.R. |
| 15 | CuI | CoSO4 (1 equiv.) | 59 | 45 |
| 16 | CuSCN | CoCl2 (1 equiv.) | 59 | 62 |
| 17 | CuCN | Yb(OTf)3 (1 equiv.) | 48 | N.R. |
| 18 | Cu2O | Ni(OAc)2·4H2O (1 equiv.) | 65 | 16 |
| 19 | Cu(OAc)2 | Co(NO3)2·6H2O (1 equiv.) | 65 | Trace |
| 20 | CuCl2 | Cu(OAc)2·4H2O (1 equiv.) | 65 | N.R. |
| 21 | CuCl2 | LiCl (1 equiv.) | 49 | N.R. |
| 22 | CuCl2 | CaCl2 (1 equiv.) | 49 | 23 |
| 23 | CuCl2 | CoCl2 (1 equiv.)+ PivOH (1 equiv.) |
58 | 47 |
| 24 | CuCl2 | CoCl2 (1 equiv.)+ PhCOOH (1 equiv.) |
57 | 69 |
| 25 | CuCl2 | CoCl2 (20 mol%)+ PhCOOH (20 mol%) |
84 | 78 |
| 26 | None | CoCl2 (1 equiv.)+ PhCOOH (20 mol%) |
77 | N.R. |
| a Conditions: 1a (50 mg, 0.28 mmol), CuCl2 (20 mol%), additive (1 equiv.), THF (1.2 mL), O2, 100 ℃. b Isolated yield. | ||||
With the optimized reaction conditions in hand, the generality and limitations of this Cu-catalyzed decarbonylation cyclization for forming isatins were explored (Table 2). A series of electron-donating and electron-withdrawing substitutents on the aromatic ring was examined. It was found that these substrates could be successfully converted to the corresponding isatins in moderate to good yields. Isatin derivatives 2f and 2g containing Br and F atoms were smoothly obtained in 71% and 56% yields, respectively, which had potential application for further functionalization into some bioactive compounds (Table 2). It is worth noting that electron-withdrawing CF3 and COOMe substitutents at the meta position on the aromatic rings were tolerated well to furnish the products 2h and 2i in moderate yields. To the best of our knowledge, the previous approach reported by Cheng et al.[7g] could not synthesize these isatins. Accordingly, a complementary access to isatin derivatives with electron-withdrawing groups was offered. In addition to examining the substituents on the aromatic ring, the substrates with different size chain on nitrogen atom were also investigated. As expected, substrates with Et, Bn, n-Bu on nitrogen efficiently underwent the Cu-catalyzed annulation to give the corresponding isatins in 54%~69% yields (Table 2). To our surprise, N-(2-acetylphenyl)formamide without methyl group on the nitrogen also worked well under the standard conditions and provided the desired product 2m in 48% yield (Table 2).
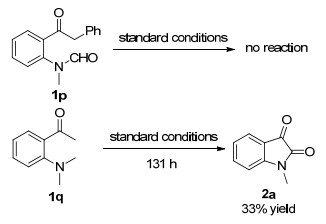
Substrates 1p and 1q were also subjected to the standard conditions. Substrate 1p did not work. Substrate 1q could give the desired product 2a in 33% yield after 131 h. These substrates did not show good reactivity (Scheme 2).
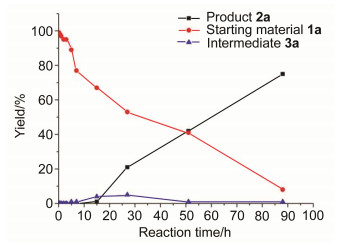
To gain insight into the reaction mechanism, some mechanistic studies were conducted. Compound ln bearing 13C isotope on aldehyde group was prepared, and 1n was subjected to the standard conditions (Eq. 1). It was found that the carbon of amide in 2a came from methyl group, not aldehyde. In addition, it was found that the substrate 1o without aldehyde on nitrogen was performed sluggishly under Cu catalytic conditions to furnish the corresponding product 2a with low 29% yield (Eq. 2). It was worth noting that an intermediate was discovered during the course of reaction, which was determined as a dichloronated compound 3a by the analysis of 1H/13C NMR and HRMS. To our surprise, compound 3a smoothly converted into the deserted product 2a under the standard conditions (75% yield, Eq. 3), which revealed that compound 3a was a key intermediate in this reaction. Consequently, the reaction profile using 1a as the model substrate was obtained. It was found that the desired product 2a was produced with the formation of intermediate 3a (Figure 1). It further supported dichloronated compound 3a as a key intermediate in this reaction. When a radical trapping agent such as 2, 2, 6, 6-tetramethyl-1-piperidinyloxy free radical (TEMPO) (20 mol%) was added into the reaction, no reaction occurred (Eq. 4). However, although the mechanism for the present catalytic reaction is not clear, a radical process for this reaction can be possible.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
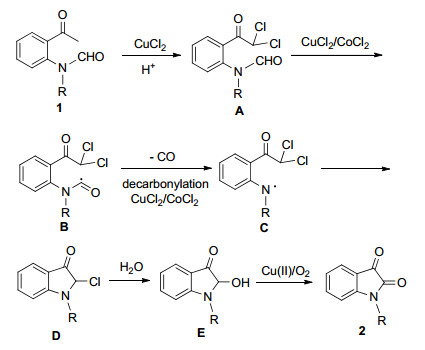
On the base of the above mechanistic studies and previous reports, [7] a tentative reaction mechanism was proposed (Scheme 3). First, substrate reacts with CuCl2 under the acidic conditions to undergo the halogenation of ketone, forming a dichloronated intermediate A, [12] followed by decarbonyaltion in Cu/Co system to give a secondary amine radical intermediate C.[7i, 7j] The secondary amine radical attacks one of C—Cl bonds to yield compound D which undergo substitution reaction with H2O to provide the cyclic α-keto alcohol E. Finally, intermediate E is oxidized by Cu(Ⅱ)/O2 to form isatin 2.
In summary, a copper-catalyzed decarbonylation cyclization to form isatins using oxygen as a terminal oxidant was developed. This complementary way offers a new protocol for the synthesis of isatins through C(sp3)—H bond functionalization in Cu/O2/Co system. This system shows good reactivity and compatibility. Both electron-rich and electron-deficient functional groups can be tolerated. Further exploration of the generality and application of this approach is ongoing in our laboratory.
All experiments were carried out under an oxygen atmosphere unless otherwise noted. Reactions were monitored using thin-layer chromatography (TLC). 1H NMR and 13C NMR spectra were obtained at 400 and 100 MHz, respectively on a Mercury Plus-400. NMR spectra were run in a solution of deuterated chloroform (CDCl3). High-resolution mass spectra (HRMS) were recorded using electrospray ionization (ESI) with a Q-TOF MS Spectrometer. The substrates were prepared according to the reported literatures.[13, 14]
An oven-dried Schlenk tube equipped with a magnetic stir bar was evacuated and backfilled with oxygen three times. Under oxygen, substrates 1 (0.3 mmol), CuCl2 (20 mol%, 0.06 mmol, 8.1 mg), CoCl2 (20 mol%, 0.06 mmol, 7.8 mg), PhCOOH (20 mol%, 0.06 mmol, 7.3 mg) and tetrahydrofuran (THF) (1.2 mL) were added into the tube. Then the reaction was stirred at 100 ℃ for specific time. THF was removed under reduced pressure. Then, the mixture was extracted with EtOAc (10 mL×3). The combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography to give the corresponding isatin products. All of products are known compounds.[14~18]
1-Methylindoline-2, 3-dione (2a):[15] 78% yield. 1H NMR (400 MHz, CDCl3) δ: 3.26 (s, 3H), 6.91 (d, J=8.0 Hz, 1H), 7.16 (td, J=1.2, 7.6 Hz, 1H), 7.62~7.59 (m, 2H); HRMS (ESI) calcd for C9H8NO2 162.0555, found 162.0544.
1, 6-Dimethylindoline-2, 3-dione (2b):[15] 62% yield. 1H NMR (400 MHz, CDCl3) δ: 2.45 (s, 3H), 3.23 (s, 3H), 6.69 (s, 1H), 6.93 (d, J=7.6 Hz, 1H), 7.50 (d, J=7.6 Hz, 1H); HRMS (ESI) calcd for C10H10NO2 176.0712, found 176.0704.
6-Methoxy-1-methylindoline-2, 3-dione (2c):[16] 44% yield. 1H NMR (400 MHz, CDCl3) δ: 3.22 (s, 3H), 3.93 (s, 3H), 6.37 (d, J=2.4 Hz, 1H), 6.57 (dd, J=8.4, 2.0 Hz, 1H), 7.59 (d, J=8.4 Hz, 1H); HRMS (ESI) calcd for C10H10NO3 192.0661, found 192.0647.
1, 5-Dimethylindoline-2, 3-dione (2d):[15] 61% yield; 1H NMR (400 MHz, CDCl3) δ: 2.33 (s, 3H), 3.22 (s, 3H), 6.80 (d, J=7.6 Hz, 1H), 7.41~7.38 (m, 2H); HRMS (ESI) calcd for C10H10NO2 176.0712, found 176.0700.
5-Methoxy-1-methylindoline-2, 3-dione (2e):[15] 65% yield; 1H NMR (400 MHz, CDCl3) δ: 3.24 (s, 3H), 3.82 (s, 3H), 6.84 (d, J=8.4 Hz, 1H), 7.19~7.15 (m, 2H); HRMS (ESI) calcd for C10H10NO3 192.0661, found 192.0651.
5-Bromo-1-methylindoline-2, 3-dione(2f):[15] 71% yield; 1H NMR (400 MHz, CDCl3) δ: 3.25 (s, 3H), 6.82 (dd, J=7.2, 1.6 Hz, 1H), 7.71 (s, 1H), 7.74 (dd, J=7.6, 2.0 Hz, 1H); HRMS (ESI) calcd for C9H7NO3Br 239.9660, found 239.9640.
5-Fluoro-1-methylindoline-2, 3-dione (2g):[15] Orange solid, 56% yield. m.p. 150.4~151.3 ℃(lit. 150.7~152.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 3.26 (s, 3H), 6.88~6.85 (m, 1H), 7.36~7.31 (m, 2H); HRMS (ESI) calcd for C9H7NO3F 180.0461, found 180.0454.
1-Methyl-6-(trifluoromethyl)indoline-2, 3-dione (2h):[17] 45% yield. 1H NMR (400 MHz, CDCl3) δ: 3.32 (s, 3H), 7.11 (s, 1H), 7.43 (d, J=8.0 Hz, 1H), 7.73 (d, J=8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 26.4, 106.9 (d, J=3.5 Hz, 1C), 119.4, 120.8 (d, J=3.7 Hz, 1C), 124.1, 125.5, 139.2 (d, J=33.3 Hz, 1C), 151.5, 157.4, 182.4; HRMS (ESI) calcd for C10H7NO2F3 230.0429, found 230.0425.
1-Methyl-2, 3-dioxoindoline-6-carboxylate (2i):[18] 50% yield. 1H NMR (400 MHz, CDCl3) δ: 3.32 (s, 3H), 3.98 (s, 3H), 7.54 (d, J=1.2 Hz, 1H), 7.68 (dd, J=8.0, 0.8 Hz, 1H), 7.84 (dd, J=7.6, 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 26.4, 52.9, 110.6, 120.0, 125.2, 138.5, 151.1, 157.6, 165.2, 183.1; HRMS (ESI) calcd for C11H10NO4 220.0610, found 220.0604.
1-Ethylindoline-2, 3-dione (2j):[19] 69% yield. 1H NMR (400 MHz, CDCl3) δ: 1.34 (t, J=7.2 Hz, 1H), 3.82 (q, J=7.2 Hz, 2H), 6.92 (d, J=7.6 Hz, 1H), 7.13 (td, J=0.8, 7.2 Hz, 1H), 7.62~7.57 (m, 2H); HRMS (ESI) calcd for C10H10NO2 176.0712, found 176.0700.
1-Butylindoline-2, 3-dione (2k):[20] 54% yield. 1H NMR (400 MHz, CDCl3) δ: 0.99 (t, J=7.2 Hz, 3H), 1.46~1.37 (m, 2H), 1.72~1.65 (m, 2H), 3.74 (t, J=7.2 Hz, 2H), 6.91 (d, J=8.0 Hz, 1H), 7.13 (dt, J=7.6, 0.8Hz, 1H), 7.61~7.56 (m, 2H); HRMS (ESI) calcd for C12H14NO2 204.1025, found 204.1011.
1-Benzylindoline-2, 3-dione (2l):[15] 62% yield. 1H NMR (400 MHz, CDCl3) δ: 4.93 (s, 2H), 6.78 (d, J=8.0 Hz, 1H), 7.11 (t, J=7.6 Hz, 1H), 7.37~7.30 (m, 5H), 7.50 (dt, J=7.6, 0.8 Hz, 1H), 7.62 (d, J=7.6 Hz, 1H); HRMS (ESI) calcd for C15H12NO2 238.0868, found 238.0858.
Indoline-2, 3-dione (2m):[15] 48% yield; 1H NMR (400 MHz, CDCl3) δ: 6.93 (d, J=8.0 Hz, 1H), 7.15 (td, J=0.8, 7.6 Hz, 1H), 7.59 (td, J=1.2, 7.6 Hz, 1H), 7.63(d, J=7.6 Hz, 1H), 8.09 (br s, 1H); HRMS (ESI) calcd for C8H6NO2 148.0399, found 148.0386.
N-(2-Acetylphenyl)-N-methylformamide-13C (1n): Major:Minor=3:1; 1H NMR (400 MHz, CDCl3) δ: Major: 2.51 (s, 3H), 3.26 (s, 3H), 7.25 (dd, J=1.2, 7.2 Hz, 1H), 7.43 (td, J=1.2, 7.6 Hz, 1H), 7.57 (td, J=1.6, 7.6 Hz, 1H), 7.70 (dd, J=7.6, 1.6 Hz, 1H), 8.38 (s, 1H); Minor: 2.56 (s, 3H), 3.37 (s, 3H), 7.24 (d, J=7.2 Hz, 1H), 7.38 (td, J=1.2, 7.6 Hz, 1H) 7.53 (td, J=1.6, 7.6 Hz, 1H), 7.65 (dd, J=1.2, 7.6 Hz, 1H), 8.40 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 29.5, 33.8, 128.0, 129.8, 132.7, 136.4, 162.6, 199.6; HRMS (ESI) calcd for C10H12NO2 178.0902, found178.0891.
N-(2-(2, 2-Dichloroacetyl)phenyl)-N-methylformamide (3a): Major:Minor=3:1; 1H NMR (400 MHz, CDCl3) δ: Major: 3.25 (s, 3H), 6.48 (s, 1H), 7.33 (d, J=7.6 Hz, 1H), 7.52 (t, J=7.6 Hz, 1H), 7.67 (t, J=7.6 Hz, 1H), 7.84 (d, J=7.6 Hz, 1H), 8.19 (s, 1H); Minor: 3.39 (s, 3H), 6.51 (s, 1H), 7.30 (d, J=8.0 Hz, 1H), 7.44 (t, J=8.0 Hz, 1H), 7.64 (t, J=8.0 Hz, 1H), 7.91 (d, J=8.0 Hz, 1H), 8.18 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: Major: 34.6, 69.3, 128.7, 129.5, 130.4, 132.1, 134.5, 141.8, 162.8, 188.1; Minor: 38.0, 69.0, 127.9, 128.1, 129.3, 132.7, 134.0, 139.1, 163.1, 188.3; HRMS (ESI) calcd for C10H9Cl2NO2Na (M+Na+) 267.9908, found 267.9903.
Supporting Information 1H NMR spectra of all products and data of intermediate 3a. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Sumpter, W. C. Chem. Rev. 1944, 34, 393.
(b) da Silva, J. F. M.; Garden, S. J.; Pinto, A. C. J. Braz. Chem. Soc. 2001, 12, 273.
(a) Ratan, B. T.; Anand, B.; Yogeeswari, P.; Sriram, D. Bioorg. Med. Chem. Lett. 2005, 15, 4451.
(b) Raj, A.; Raghunathan, R.; Sridevikumaria, M. R.; Raman, N. Bioorg. Med. Chem. 2003, 11, 407.
(c) Verma, M.; Pandeya, S. N.; Singh, K. N.; Stables, J. P. Acta Pharm. 2004, 54, 49.
(d) Jiang, T.; Kuhen, K. L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntlad, T.; Zhang, K.; Karanewsky, D.; He, Y. Bioorg. Med. Chem. Lett. 2006, 16, 2109.
(e) Aboul-Fadl, T.; Bin-Jubair, F. A. S. Int. J. Res. Pharm. Sci. 2010, 1, 113.
(f) Sharma, S.; Gupta, M. K.; Saxena, A. K.; Bedi, P. M. S. Bioorg. Med. Chem. 2015, 23, 7165.
(g) Harbinder, S.; Jatinder, V. S.; Gupta, M. K.; Sharma, S.; Nepali, K.; Bedi, P. M. S. Bioorg. Med. Chem. Lett. 2017, 27, 3974.
Sandmeyer, T. Helv. Chim. Acta 1919, 2, 234. doi: 10.1002/hlca.19190020125
(a) Stollé, R. Ber. Dtsch. Chem. Ges. 1913, 46, 3915.
(b) Stollé, R. J. Prakt. Chem. 1922, 106, 137.
(a) Martinet, J. Compt. Rend. 1918, 166, 85.
(b) Bonnefoy, J.; Martinet, J. Compt. Rend. 1921, 172, 220.
Xie, Y. Chem. Commun. 2016, 52, 12372. doi: 10.1039/C6CC05769A
(a) Sun, J.; Liu, B.; Xu, B. RSC Adv. 2013, 3, 5824.
(b) Liu, T.; Yang, H.; Jiang, Y.; Fu, H. Adv. Synth. Catal. 2013, 355, 1169.
(c) Tang, B.-X.; Song, R.-J.; Wu, C.-Y.; Liu, Y.; Zhou, M.-B.; Wei, W.-T.; Deng, G.-B.; Yin, D.-L.; Li, J.-H. J. Am. Chem. Soc. 2010, 132, 8900.
(d) Liu, T.; Yang, H.; Jiang, Y.; Fu, H. Adv. Synth. Catal. 2013, 355, 1169.
(e) Sun, J.; Liu, B.-X.; Xu, B. RSC Adv. 2013, 3, 5824.
(f) Ilangovan, A.; Satish, G. Org. Lett. 2013, 15, 5726.
(g) Huang, P. C.; Gandeepan, P.; Cheng, C. H. Chem. Commun. 2013, 49, 8540.
(h) Li, J.; Zheng, Y.; Yu, X. L.; Lv, S. Y.; Wang, Q. T.; Hai, L.; Wu Y. RSC Adv. 2015, 5, 103280.
(i) Liu, Z.; Zhang, J.; Chen, S.; Shi, E.; Xu, Y.; Wan, X. Angew. Chem., Int. Ed. 2012, 51, 3231.
(j) Meng, Q.; Wang, F.; Li, M. J. Mol. Model. 2013, 19, 2225.
Liao, Y.-Y.; Gao, Y.-C.; Zheng, W.; Tang, R.-Y. Adv. Synth. Catal. 2018, 360, 3391. doi: 10.1002/adsc.201800592
Lollar, C. T.; Krenek, K. M.; Bruemmer, K. J.; Lippert, A. R. Org. Biomol. Chem. 2014, 12, 406. doi: 10.1039/C3OB42024H
(a) Satish, G.; Polu, A.; Ramar, T.; Ilangovan, A. J. Org. Chem. 2015, 80, 5167.
(b) Reddy, M. R.; Rao, N. N.; Ramakrishna, K.; Meshram, H. M. Tetrahedron Lett. 2014, 55, 4758.
(c) Ilangovan, A.; Satish, G. J. Org. Chem. 2014, 79, 4984.
(d) Gao, F. F.; Xue, W. J.; Wang, J. G.; Wu, A. X. Tetrahedron 2014, 70, 4331.
(a) Huang, P. C.; Gandeepan, P.; Cheng, C. H. Chem. Commun. 2013, 49, 8540.
(b) Wu, H.; Zhang, Z. G.; Liu, Q. F.; Liu, T. X.; Ma, N. N.; Zhang, G. S. Org. Lett. 2018, 20, 2897.
(c) Salvanna, N.; Reddy, L. M.; Kumar, R. A.; Das, B. ChemistrySelect 2018, 3, 8019.
Nobrega, J. A.; Goncalves, S. M. C.; Peppe, C. Synth. Commun. 2002, 32, 3711. doi: 10.1081/SCC-120015383
Shekhar, A. C.; Kumar, A. R.; Sathaiah, G.; Paul, V. L.; Sridhar, M.; Rao, P. S. Tetrahedron Lett. 2009, 50, 7099. doi: 10.1016/j.tetlet.2009.10.006
Kirincich, S. J.; Xiang, J.; Green, N.; Tam, S.; Yang, H. Y.; Shim, J.; Clark, J. D.; McKew, J. C. Bioorg. Med. Chem. 2009, 17, 4383. doi: 10.1016/j.bmc.2009.05.027
Luo, J. F.; Gao, S. S.; Ma, Y. R.; Ge, G. P. Synlett 2018, 29, 969. doi: 10.1055/s-0036-1591904
Ji, H. H.; Zhu, Y. Z.; Shao, Y.; Liu, J.; Yuan, Y.; Jia, X. D. J. Org. Chem. 2017, 82, 9859. doi: 10.1021/acs.joc.7b01480
Wang, H. Y.; Wang, K. Y.; Man, Y. Q.; Gao, X. N.; Yang, L. M.; Ren, Y. F.; Li, N.; Tang, B.; Zhao, G. Adv. Synth. Catal. 2017, 359, 3934. doi: 10.1002/adsc.201700649
Bredenkampa, A.; Mohrb, F.; Kirsch, S. F. Synthesis 2015, 47, 1937. doi: 10.1055/s-0034-1380517
Zhang, C.; Li, S.; Filip, B.; Richmond, L.; Ye, X.; Jiang, Z. ACS Catal. 2016, 6, 10, 6853. doi: 10.1021/acscatal.6b01969
Salvanna, N.; Ramesh, P.; Kumarc, K. S.; Das, B. New J. Chem. 2017, 41, 13754. doi: 10.1039/C7NJ02441J
Table 1. Optimization of reaction conditions for the synthesis of isatinsa, b
| ||||
| Entry | Cat. | Additive | Time/h | Yieldb/% |
| 1 | CuCl2 | None | 49 | 34 |
| 2 | CuCl | None | 110 | 23 |
| 3 | CuBr | None | 110 | 17 |
| 4 | CuI | None | 82 | N.R. |
| 5 | CuSCN | None | 82 | N.R. |
| 6 | CuCN | None | 82 | N.R. |
| 7 | Cu2O | None | 110 | Trace |
| 8 | Cu(OAc)2 | None | 66 | N.R. |
| 9 | CuCl2 | ZnCl2 (1 equiv.) | 49 | 40 |
| 10 | CuCl2 | AlCl3 (1 equiv.) | 49 | N.R. |
| 11 | CuCl2 | BF3·Et2O (1 equiv.) | 67 | N.R. |
| 12 | CuCl2 | FeCl3 (1 equiv.) | 67 | N.R. |
| 13 | CuCl2 | PdCl2 (1 equiv.) | 67 | N.R. |
| 14 | CuCl2 | Cu(ClO4)·6H2O (1 equiv.) | 67 | N.R. |
| 15 | CuI | CoSO4 (1 equiv.) | 59 | 45 |
| 16 | CuSCN | CoCl2 (1 equiv.) | 59 | 62 |
| 17 | CuCN | Yb(OTf)3 (1 equiv.) | 48 | N.R. |
| 18 | Cu2O | Ni(OAc)2·4H2O (1 equiv.) | 65 | 16 |
| 19 | Cu(OAc)2 | Co(NO3)2·6H2O (1 equiv.) | 65 | Trace |
| 20 | CuCl2 | Cu(OAc)2·4H2O (1 equiv.) | 65 | N.R. |
| 21 | CuCl2 | LiCl (1 equiv.) | 49 | N.R. |
| 22 | CuCl2 | CaCl2 (1 equiv.) | 49 | 23 |
| 23 | CuCl2 | CoCl2 (1 equiv.)+ PivOH (1 equiv.) |
58 | 47 |
| 24 | CuCl2 | CoCl2 (1 equiv.)+ PhCOOH (1 equiv.) |
57 | 69 |
| 25 | CuCl2 | CoCl2 (20 mol%)+ PhCOOH (20 mol%) |
84 | 78 |
| 26 | None | CoCl2 (1 equiv.)+ PhCOOH (20 mol%) |
77 | N.R. |
| a Conditions: 1a (50 mg, 0.28 mmol), CuCl2 (20 mol%), additive (1 equiv.), THF (1.2 mL), O2, 100 ℃. b Isolated yield. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们