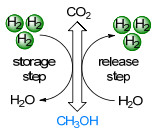
图 1.
基于甲醇/CO2的可逆储氢循环
Figure 1.
Reversible hydrogen storage cycle based on the methanol/CO2 couple

全球能源80%以上是化石燃料[1], 如果继续维持这样的能源供给, 石油资源将会在本世纪末消耗殆尽.氢气是最理想的清洁能源, 燃烧会释放大量的热, 其唯一产物是无污染的水[2].风能、太阳能和水电能技术虽然已经发展了50多年, 但由于时间和地理位置的局限性, 不能持续性供能[3], 而“氢经济”概念[4]的提出弥补了这些不足, 并提供了一个有希望的解决方案.然而, 氢气在室温下是易燃易爆气体, 极易溢出容器, 体积能量密度极低, 这使得其在运输和存储时必须经过高压压缩(35~70 MPa)或低温(-253 ℃)液化, 所以氢气本身并不是一种理想的能源载体, 这是发展“氢经济”的一大难题.
现如今储氢主要有高压气态储氢技术[5]、低温液化储氢技术[6]、吸附储氢技术[7]、水合物储氢技术[8]和化学储氢技术[9]等[10].前两种物理储氢技术成本低、易放氢, 但安全性较差.吸附储氢技术受吸附剂影响较大[11], 其优点是速度快、完全可逆和循环性好[12], 但通常需要在低温和高压下才能达到合适的存储容量[13].水合物储氢技术易脱氢、成本低、能耗低, 但其储氢密度较低[10].化学储氢方法是固态或液态化合物通过适当的脱氢反应(最好是完全可逆的)生成稳定的化合物来释放氢气.作为氢载体必须要考虑以下几个标准:含氢量(H2的质量分数w)、氢体积容量(H2/载体:单位为kg/m3)和脱氢效率[14].现已报道的固体载体有纯金属氧化物、金属氧化物合金[15]、金属硼氢化物[16]、氨硼烷[17]等.液态有机氢载体(LOHCs)主要包括环己烷衍生物和杂环化合物[18]、氨[19]、肼[20]和氨硼烷[17]、2-甲基噻吩[21]、甲酸[22]和醇类[23].其中甲醇的含氢量为12.6%, 室温下1 m3甲醇可容纳99.77 kg氢气, 脱氢效率约90%以上.此外甲醇价格便宜, 在室温下为稳定的液体, 环境条件下挥发性低、溶解性好, 具有使用现有基础设施存储和运输的可能性, 备受人们的关注, 是一种很有应用潜力的储氢燃料.在二十世纪末期, Asinger[24]提出了甲醇经济的概念(图 1), 即二氧化碳加氢生成甲醇以及甲醇脱氢为二氧化碳的碳平衡系统.根据使用底物分子中不同类型的氢原子, 甲醇脱氢反应可以分为三类:第一类为仅利用甲醇的两个氢原子, 脱氢生成甲醛和氢气(表 1, 反应式1);第二类为甲醇的四个氢原子全部放出, 1 mol甲醇脱氢为1 mol一氧化碳和2 mol氢气(表 1, 反应式2和3[25]); 第三类为水中的氢原子共同参与反应, 这使得1 mol甲醇脱氢为1 mol CO2和3 mol H2(表 1, 反应式4和5), 其中1 mol H2来自水.以上所有反应都是吸热反应.通过比较表 1中甲醇的五种脱氢反应, 可以看出反应式1的氢气利用率低, 反应式2和3会产生有毒性的CO, 所以反应1~3都是不可取的.反应式4和5是人们研究甲醇经济的焦点.
 下载:
导出CSV
下载:
导出CSV
| No. | Eq. | ∆H0/(kJ•mol-1) |
| (1) | CH3OH (l)→HCHO (g)+H2 (g) | +129.8 |
| (2) | CH3OH (g)→CO (g)+2H2 (g) | +94.6 |
| (3) | CH3OH (l)→CO (g)+2H2 (g) | +127.9 |
| (4) | CH3OH (g)+H2O (g)→3H2 (g)+CO2 (g) | +53.3 |
| (5) | CH3OH (l)+H2O (l)→3H2 (g)+CO2 (g) | +130.7 |
目前, 工业上已经实现了使用多相催化剂体系催化甲醇重整脱氢工艺.以CuO/ZnO/Al2O3多相催化剂为代表的铜基催化剂[27]已商业化生产, 其优点是活性高、选择性高, 以及成本较低, 但反应需要300 ℃以上的高温, 这会使金属烧结失活并自燃.以钯-锌合金催化剂为代表的金属基催化剂[28]比铜基催化剂稳定, 但是活性较低. Dumesic等[29]首次报道了使用多相催化剂Pt/ Al2O3在温度为200~225 ℃时催化甲醇水相重整. 2017年, Lin等[30]研发出一种Pt/α-MoC双功能催化剂, 该催化剂可以使甲醇水相产氢在150~190 ℃下发生, 且转化频率达到18046 h-1, α-MoC诱导水解离.多相催化剂虽然可以催化甲醇大量产氢, 但是催化条件仍需要高温、高压[31], 这对甲醇的稳定性是极其不利的, 还可能产生大量有毒的CO[32], 因此不适用于燃料电池, 所以当下人们想将高温系统转化为低温系统(<150 ℃).近年来, 在温和条件下利用均相催化剂催化甲醇脱氢的研究取得了很大的进展.本文将主要介绍钌、铑、铱、铁、锰基均相催化剂在甲醇脱氢反应方面的应用.
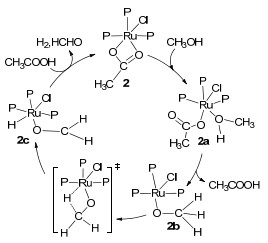
对于甲醇直接脱氢, Ru基催化剂是目前研究最广泛、最成熟的.相比于其他贵金属而言, 钌基催化剂价格低廉、催化性能高[33].早在1985年, Maitlis等[34]首次使用[Ru(PPh3)3Cl2] (1)催化甲醇直接脱氢合成甲酸甲酯, 这对甲醇脱氢领域的发展具有开创性的历史意义.在150 ℃时反应18 h后氢气的转化数为65(表 2).其缺点是催化剂失活为[(PPh3)2Ru(CO)(μ-H)(μ-Cl)2(CO)-(PPh3)2]使反应结束, 另外150 ℃的温度不利于催化剂的稳定.在同一年, Saito等[35]证实了由[Ru2(OAc)4Cl]和PEt-PPh2反应生成的[Ru(OAc)Cl(PEtPPh2)3] (2)可以催化甲醇直接脱氢为甲醛, 该反应无副产物.在66 ℃下, 加入2 equiv.乙酸, 反应90 h后总转化数为34(表 2), 初始转化频率为0.96 h-1, 平均转化频率为0.38 h-1, 如不加乙酸, 初始转化频率则为0.60 h-1.随后, 该课题组[36]提出了[Ru(OAc)Cl(PEtPPh2)3] 2催化甲醇脱氢的反应机理(Scheme 1).首先, 甲醇与中心金属钌原子结合, 使得乙酸双齿配体变为单齿构型2a.随后, 甲醇配体上的氢转移到乙酸配体上, 使得乙酸解离.然后, 甲氧基上C—H键断裂, 氢与中心金属钌结合, 生成钌氢化物2c.最后, 钌氢化物与解离的乙酸结合, 生成甲醛并释放氢气再生初始催化剂.由此可见, 该反应不会产生一氧化碳和甲烷等气体污染物, 为后续甲醇脱氢领域向节能环保方向发展奠定了坚实的基础.尽管[Ru(OAc)Cl(PEtPPh2)3] 2能够在较低温度下使甲醇脱氢, 但其转化效率很低, 反应速率很慢.
下载:
导出CSV
| Catalyst (mmol) | MeOH/mL | Additive (mmol) | T/℃ | Time/h | TOFa/h-1 | TONb | Ref. |
| [RuCl2(PPh3)3] (1) (0.08) | 10 | — | 150 | 18 | 3.60 | 65 | [34] |
| [Ru(OAc)Cl(PEtPPh2)3] (2) (0.1) | 400 | CH3COOH (0.2) | 66 | 90 | 0.38 | 34 | [35] |
| [Ru(H)2(N2)(PPh3)3] (3) [(1~5)×10-3] | 5 | NaOH (5) | 150 | 2 | 6.40 | 12.8 | [37] |
| [Ru(H)2(PPh3)4] (4) [(1~5)×10-3] | 5 | NaOH (5) | 150 | 2 | 7.50 | 15 | [37] |
| RuCl3•3H2O (5) (1) | 200 | MeONa (610) | 79 | n.r. c | 1.68 d | — | [38] |
| a Average TOF referred to overall reaction time. b TON is referred to hydrogen production. c n.r. not reported. d Initial TOF. | |||||||
人们致力于开发转化频率高、产氢速率快的催化体系.在1988~1989年间, Cole-Hamilton课题组[37]发现使用[Ru(H)2(N2)(PPh3)3] (3)或[Ru(H)2(PPh3)4] (4)在150 ℃下催化甲醇脱氢时加入十倍的碱可以大幅度提高产氢速率(表 2).随后Saito等[38]发现RuCl3•3H2O 5不需要辅助配体也可以催化甲醇脱氢.在反应体系中加入20%甲醇钠, 转化频率由最初无碱时候的0.02 h-1提高到1.68 h-1(表 2), 增加了84倍. 2010年, Nicolas和Michael[39]的理论研究指出碱可以充当一种“辅助配体”, 促进活性物种之间的质子转移, 这说明碱在催化甲醇脱氢中起非常重要的作用.控制碱的添加量对反应效率有很大的影响, 反应速率及产量会因碱的添加有所提升, 但高浓度的碱会使催化剂失活.从表 2中我们可以看到, 大多数早期报道的甲醇脱氢系统的活性和选择性相对较低.
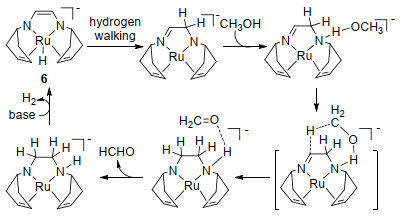
近年来, 随着国内外对过渡金属配合物催化甲醇直接脱氢反应的大量研究, 终于在甲醇水相重整脱氢方面取得了突破性进展. 2013年Grützmacher[40]和Beller[41]课题组利用甲醇的水相重整克服了一氧化碳使催化剂中毒的问题, 实现了甲醇的选择性脱氢. Grützmacher课题组[40]使用催化剂6 (10×10-3 mmol, 0.5 mol%), 在90 ℃时, 加入甲醇(2 mmol、81 μL)、水(2.6 mmol、47 μL)、苯(1 mmol、110 μL作为内标)和THF(1 mL), 在敞开系统中, 反应10 h以上, 转化数TOF可达到540, 转化频率TON可达到54 h-1 (1 mol催化剂催化生成1 mol氢气为一个转化过程)(表 3).该反应溶液中没有检测到甲醛或乙缩醛, 这表明甲醇脱氢生成甲醛后水会继续攻击甲醛, 进一步脱氢.另外催化剂6可以在90 ℃催化甲酸分解, 1 min内转化频率可高达24000 h-1.经过一系列的实验表征以及机理研究, 提出了该反应可能的催化循环, 如Scheme 2左环所示.第一步, 催化剂6不与甲醇直接反应, 而是先与水反应得到一个活性适中的钌化合物6a, 期间会产生1分子氢气和碱.第二步, 底物分子甲醇与钌化合物6a中的Ru—N键结合, 从而形成烯胺基化合物6b.第三步, 化合物6b配体的α-C—H键被活化, 使得化合物6b释放出脱氢产物而形成亚胺-氨基Ru(0)配合物6c.第四步, 配合物6c重复上一步活性位点的过程形成二氨基Ru(0)配合物6d并释放出等摩尔氢气.第五步, 中间体6d与碱反应使得初始催化剂6再生, 并有等摩尔氢气放出.其中用X射线表征了生成的中间体6d.化合物6a和6c可以催化甲醇、谐二醇或甲酸脱氢(化合物6a可以催化甲酸脱氢释放二氧化碳并形成化合物6c).脱氢过程时配体既具有催化活性也具有氧化性, 可使金属钌的氧化态在2和0之间变化.
下载:
导出CSV
| Cat. (10-3 mmol; 0.001‰ to MeOH) | MeOH/mmol | H2O/mmol | Solvent (mL) | Additive (mmol) | T/℃ | Time/h | TOFa/h-1 | TONb | Ref. |
| [K(dme)2][RuH(trop2dad)] (6) (10; 500) | 2 | 2.6 | THF (1) | — | 90 | 10 | 54 | 540 | [40] |
| [Ru(H)Cl(CO)(PNP-iPr)] (8) (0.88; 1) | 890 | 222 | — | KOH (320) | 91 | 576 | 613 | 353409 | [41] |
| [Ru(H)(BH4)(CO)(PNP-Ph)] (9a) (5; 22.5)/[Ru(H)2(dppe)2] (9b) (5; 22.5) | 222 | 55.5 | Triglyme (4) | — | 93.5 | 192 | 22 | 4286 | [50] |
| [Ru(H)Cl(CO)(NNP-tBu)] (10) (5; 250) | 20 | 111 | Toluene (2) | KOH (40) | 100~105 | 648 | 44 | 28661 | [51] |
| a Average TOF referred to overall reaction time. b TON is referred to hydrogen production. c Initial TOF. | |||||||||
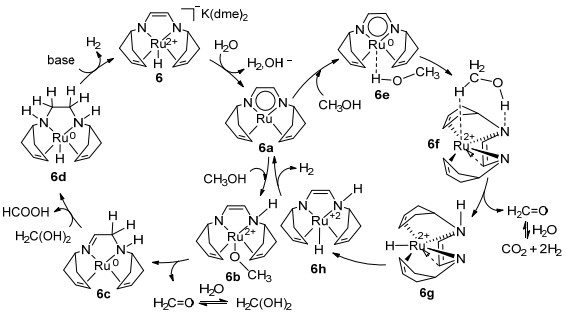
2015年, Hall等[42]使用密度泛函理论(DFT)方法报道了6催化甲醇脱氢的另一种反应机制, 如Scheme 3所示.首先Ru上的氢转移到烯基碳位, 随后甲醇不与钌中心结合直接脱氢, 而是与N形成氢键, 这是一种基于配体-配体双功能理论机理, 与Scheme 2中Grützmacher课题组最先提出的甲醇与钌中心原子结合的机理路径不同, 钌在此过程中只是“旁观者”. 2016年杨新征等[43]使用DFT方法研究了Grützmacher提出机理的可行性, 发现在反应过程中, trop2dad配体中的咪唑部分可以将甲醇和水中的氢原子转化为氢气释放出来, 起到储氢的作用.此外, 溶剂对该系统的反应能垒有很大的影响, 这说明溶剂有可能改进该系统的反应活性. 2018年Grützmacher课题组[44]用DFT方法验证了Ru中心金属原子并不仅仅是“旁观者”, dad配体部分与Ru原子共同作用降低了反应路径过渡态的能垒(Scheme 2, 右环), 这种机理更可行.该计算使用的模型是真实结构而不是简化模型, 说明简化模型存在一定的非系统误差, 应尽量避免.
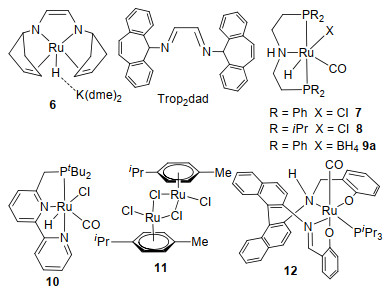
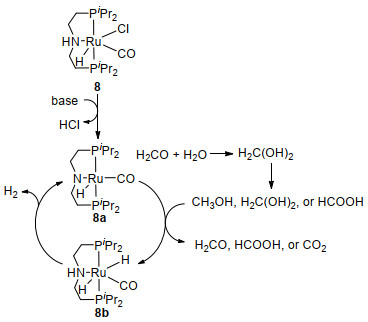
基于金属与配体结合的协同催化模式, 钳形Ru- PNP催化剂7和8被合成并用于催化酯加氢(7)[45]以及除甲醇以外的无受体醇脱氢反应(8)[46], Beller课题组[41]发现这两种钌配合物也可以催化甲醇水相重整, 但需要在碱性条件下进行.使用49.3 μmol、250 mg/L催化剂7, 在72 ℃下, 加入8 mL MeOH、2 mL H2O、0.1 mol/L NaOH, 催化甲醇脱氢可以释放出H2和CO2.使用1.58μmol、6.4 mg/L催化剂8, 在95 ℃下, 加入10 mL甲醇、8 mol/L KOH, 反应3 h后, 催化甲醇脱氢的平均转化频率可高达4723 h-1.由于该反应放出的CO2会被碳酸盐吸收, 且几乎不产生CO污染物, 所以可以得到很纯的氢气.催化剂8连续催化甲醇脱氢反应3个星期之后, 氢气的转化数可高达350000, 在最后的24 h测得其平均转化频率为200 h-1(表 3).
随着碱的消耗和沸点的降低, 催化剂活性逐渐下降.由此看出, 催化剂8只是一个催化前体, 雷鸣[47]、Beller[48]、杨新征[49]等课题组的密度泛函理论研究表明, 与等摩尔的碱反应才可以使其转化为具有催化活性的酰胺配合物8a.然后甲醇羟基上的氢与酰胺配合物8a上的N结合, α-C—H键上的氢与中心金属钌原子结合使得反应生成二氢化物8b (Scheme 4).由此可见, 酰胺配合物8a和二氢化物8b是甲醇、甲酸和谐二醇脱氢的关键化合物[41].
使用催化剂7和8进行甲醇脱氢的最大缺点是都需要高浓度的碱.由于在溶液中检测到了游离的甲酸盐, 所以高浓度的碱有可能减慢甲醇产氢速率.因此可以设想, 加大催化剂的用量, 使用对甲酸分解有效的第二种催化剂8或是高沸点溶剂就可以在无碱条件下进行甲醇脱氢. Beller等[50]经过对催化剂和溶剂的筛选, 报道了一种双催化系统, Ru-MACHO-BH4 (9a)和[Ru(H)2-(dppe)2] (dppe=1, 2-bis(diphenylphosphino)ethane) (9b)在三甘醇二甲醚溶液中共同作用可以不需要碱去激活活性位点来催化甲醇脱氢.如果两种催化剂单独催化甲醇脱氢, 活性极低, 但是当它们共同加入反应体系时, 甲醇脱氢产率迅速提高.利用5 μmol催化剂9a和5 μmol催化剂9b, 在93.5 ℃时, 加入9 mL MeOH、1 mL H2O、4 mL三甘醇二甲醚, 催化甲醇脱氢反应7 h后, 转化频率为93 h-1, 8 d之后反应结束, 总转化数为4286(表 3).
在2014年, Milstein课题组[51]证实一种带有PNN钳形配体的钌催化剂10可以催化甲醇水蒸气制氢.在100 ℃下, 加入2 mol KOH, 使用0.025 mol%催化剂10催化甲醇脱氢反应7 d后, 氢气产率可达70%; 9 d后, 产率升到90%, 平均转化频率分别为50和45 h-1(表 3)[51].上述反应在有机溶液中才可以检测到催化活性, 所以2 mL甲苯的加入是用来增加催化剂的溶解性.该催化剂不需要离析和纯化就可以重复多次使用, 寿命长达一个多月.要想让甲醇脱氢生成甲酸中间体, 就必须使催化剂10在碱性环境下生成具有真正催化活性的物种10a, 化合物10a与底物分子反应, 使得金属钌原子的另一侧上也有氢原子, 从而得到钌二氢化物10b, 同时释放出氢气(Scheme 5).
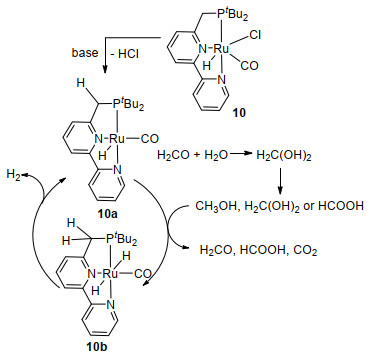
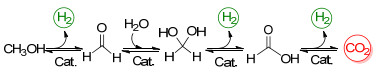
上述三类催化剂(图3)的共同特点是具有多齿配体, 这使得金属配合物的热稳定性有了显著提高.多齿配体可能具有阻挡金属活性位点的作用, 防止甲醇脱氢后甲醛进一步脱羰, 这对于甲醇脱氢的选择性是非常重要的.总的来说, 它们的反应机制为金属与配体协同作用, 如Scheme 6所示, 主要包括以下四步: (1)甲醇脱氢生成甲醛和氢气; (2)甲醛与水生成谐二醇[52]; (3)谐二醇脱氢为甲酸同时释放氢气; (4)甲酸脱羧分解为二氧化碳和氢气, 这些参与氧化还原反应的多功能配体[53]被称为“非单一(non-innocent)”协同配体[54a].
通过比较甲醇热分解(表 2)和甲醇水相重整脱氢(表 3)数据, 可以得出带有“非单一”配体的钌催化剂远比没有辅助配体的钌催化剂活性高.这可能是因为前者中催化剂与底物结合或是C—H键活化只发生在金属中心, 需要更高的活化能, 而后者采用的是金属与配体协同作用的反应机制, 反应活化能垒较低, 使得反应更容易进行, 从而活性相对更高[54b].随着甲醇脱氢均相催化剂在60~100 ℃范围内的发展, 生物酶催化体系也得到了探索, 其目的是实现室温下甲醇水相重整脱氢[55].在2015年, Prechtl等[56]报道了一种室温下生物诱导甲醇水相脱氢的方法.这种方法是利用[RuCl2(p-cymene)]2 11与氧化酶和过氧化氢酶共同作用, 在碳酸盐的缓冲溶液中以3.2 μmol/mL的速率进行甲醇脱氢.酶首先催化甲醇生成二甲醇, 然后利用催化剂11进一步脱氢, 这是反应能在室温下进行的关键.
2016年Olah和Prakash等[57]阐述了一种甲醇脱氢的新路径, 即利用二级胺与甲醇反应释放出氢气, 二级胺可以吸收甲醇脱氢的副产物二氧化碳, 然后还会生成N-甲酰胺和N, N'-二甲酰胺, 这些产物也可以可逆加氢为甲醇和二级胺.这个反应使用的催化剂是Ru-PNP 8, 该催化剂既可以用于脱氢反应也可以用于加氢反应.在120 ℃下, 使N, N'-二甲基乙二胺和甲醇以体积比为1:4的比例混合, 加入5 mol%添加剂K3PO4, 并将甲苯作为溶剂, 利用1 mol%催化剂8, 催化脱氢反应24 h后, 得到氢气的产率为90% (Eq. 6).仅给系统加压到40 bar就可以使其进行逆反应, 得到92%的N, N'-二甲基乙二胺.此系统可以避免产生CO2, 所以不需要再加气体提纯过程就可以应用于氢气燃料电池.
|
|
(6) |
2016年Reek课题组[58]利用Ru(salbinapht)(CO)(P-iPr3) [salbinapht=(R)-2'2'-bis(salicylideneamino)-1, 1'-binaphtyl] 12催化75%甲醇和水(其中醇水体积比为9:1)与25% 1, 4-二氧己环反应, 加入8 mol/L KOH, 在82 ℃时可以得到的最高转化频率为55 h-1.该系统除了释放出氢气, 还会生成甲酸盐和碳酸盐.催化剂上的羰基配体会被亲核试剂(KOH或H2O)进攻生成甲酸盐和氢气.作者猜想此羰基络合物12仅是一种催化前体, 在强碱环境中才可以形成真正具有催化活性的物种.在催化反应之后检测出微量的同样具有催化甲醇脱氢活性的[RuH2(CO)2(P-iPr3)2] (12a) (TOF=50 h-1).
除以上单核Ru-基催化剂以外, Shinoda和Yama- kawa等[59]报道了两种双金属催化体系.他们利用Ru(Ⅱ)-Sn(Ⅱ)双金属体系[Ru(SnCl3)5(PPh3)]3- 13成功催化甲醇脱氢为乙酸甲酯.此反应是在65 ℃下N2环境下进行的, MeOH和MeNO2以1:1等体积混合, 反应100 h后的转化数为15.7.虽然活性低, 但是该反应不需要任何添加剂, 催化成本较低.随后该课题组对此反应的机理研究表明, 第一步甲醇脱氢生成甲醛是决速步骤, 此外, 有可能是Ru(Ⅱ)−Sn(Ⅱ)双金属系统使得甲酸甲酯异构化为乙酸[60].另一种双金属系统是利用[CpRu(PPh3)2(SnF3)] (Cp=η5-C5H5) 14, 在140 ℃ MeOH和MeNO2以1:1体积混合的溶液中, 反应40 h后转化数可达120[61].含氟配体比含氯和溴配体的活性更高, 原因在于Sn—F键高电负性增加了Ru带正电荷的能力, F与甲醇的β-H易形成氢键, 有利于C—H键的解离.
近年来, 铱基络合物由于在甲醇脱氢方面具有高活性与高选择性, 受到了人们的关注. 2015年, Crabtree等[62]报道了一系列能稳定催化甲醇脱氢的双氮杂环羰基铱催化剂15, 16和17.因为这三种铱催化剂N杂环上的取代基不同, 所以催化活性也不同, 它们的催化活性以R=Me>Et>nBu的顺序降低.使用0.0013 mol%催化剂15, 在91 ℃时, 加入6.7 mol/L KOH溶液和3 ml甲醇(其中甲醇既是反应物又是溶剂), 反应40 h后, 甲醇脱氢的转化数可以高达8000[63].而且, 在该反应中, 几乎不产生CO.
在同一年, Yamaguchi等[64]成功实现了利用铱催化剂从甲醇水溶液中脱氢.他们使用的是带双功能吡啶酮盐配体的阴离子铱配合物18作为催化剂.相比于之前的反应体系, 这个系统只需要加入0.046 mol/L NaOH, 甲醇和水体积比为1:4, 反应15 h后, 产氢率可达64%, 转化数为10510.需要注意的是, 该反应pH值需要控制在8~12之间, 以确保阴离子催化剂的稳定性, 在pH值较低的情况下, 阴离子转变为中性离子和阳离子, 使得催化剂失活.在类似的条件下, 反应得到的甲醛和甲酸钠作为底物也可以大量产氢.
|
|
在2017年, Beller课题组[65]报道了一种Ir-PNP络合物19可以使甲醇水相重整脱氢.与Ru-PNP和Fe-PNP体系相比, 配合物19在低浓度碱中活性更高, 高浓度碱中稳定性更好.使用Ir-PNP催化剂, 加入0.5 mol/L KOH以及10 mL体积比为9:1的MeOH和水, 在70 ℃时, 反应1 h后TOF为326 h-1, 反应可持续16 h, TON为1400.把KOH的浓度降到0.1, 反应1 h后的TOF为525 h-1; 把KOH的浓度升高到8.0 mol/L, 反应可持续60 h, TON为1900.由此可看出, 碱浓度对催化剂活性和稳定性均有显著影响.
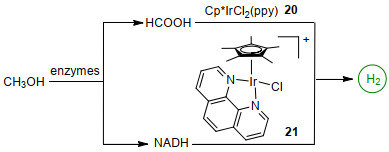
基于生物诱导方法, 2017年周小春等[66]报道了铱催化剂和酶诱导催化体系可以共同作用促使甲醇重整脱氢.该系统主要包括两个过程: CH3OH→HCOOH→H2和CH3OH→NADH→H2.先使用脱氢酶将甲醇同时氧化为甲酸和NADH, 随后利用催化剂[Cp*IrCl2(ppy)] (ppy=聚吡咯)20和[Cp*IrCl(phen)]Cl 21催化甲酸和NADH释放出氢气(Scheme 7).该反应可使400 mmol/L甲醇溶液(pH=8.05)在30 ℃时反应32 h后生成4 µmol氢气.
1987年, Cole-Hamilton课题组[67]使用[Rh(bipy)2]Cl (bipy=2, 2'-联吡啶) 22最先成功催化甲醇与水反应生成二氧化碳和氢气(表 1中Eq. 4).由于这条反应路线吸收热量较少, 使得反应更容易进行.利用5×10-6 mol [Rh(bipy)2]Cl, 在120 ℃以及体积比为95:5的醇/水混合溶液中, 再加0.25~5×10-3 mol氢氧化钠, 在封闭系统中反应3 h后得到的转化频率为7 h-1.当时作者认为加水在热力学上更有利于醇脱氢, 然而直到25年后才有了水相醇脱氢的突破.在上述催化剂报道之后, 仅出现了一种铑基催化剂催化甲醇重整脱氢. 2017年周小春等[68]利用均相催化剂[Cp*Rh(NH3)(H2O)2]3+ 23在70 ℃下脱氢, 其转化频率可达83 h-1.该反应不会产生使催化剂失活的CO, 并且将反应溶液的pH值控制在6左右催化剂活性最高.该催化剂进一步推动了低温甲醇脱氢领域的发展.
除了可以用贵金属钌、铑、铱催化剂催化甲醇脱氢, 诸如铁、锰这样地表产量丰富、价格低廉、毒性小或无毒性的过渡金属同样可以用于甲醇脱氢[69].在2013年, Beller等[70]首次报道了低温下使用铁螯合配体化合物24催化甲醇产氢.使用催化剂24, 体积比为9:1的MeOH和H2O混合溶液中加入8 mol/L KOH, 在91 ℃, 第1个小时内, 转化频率为702 h-1, 43 h后总转化数为6270.若加入过量的HN(CH2CH2P-iPr2)2配体可以进一步增强催化剂的稳定性, 延长催化剂的使用寿命, 使得转化数可以达到9184 (111 h). 2015年, Holthausen等[71]报道了另外几种铁螯合物25、26、27和28.在4:1 MeOH和H2O混合溶液中加入LiBF4, 使用催化剂27在无碱条件下催化甲醇脱氢, 经过回流, 反应94 h后转化数可达51000, 这是迄今为止第一过渡系金属均相催化甲醇重整所达到的最高产量.
|
|
2016年Chang等[72]使用tran-[FeⅡ(apH)2(MeOH)2](apH=o-aminophenolato ligand) 29光催化剂在室温下实现了催化无水甲醇重整的反应, 此反应并不需要添加任何光敏剂就可以释放出氢气并生成甲醛产物.
虽然铁基催化剂成本低、产量高, 但是活性不如钌基催化剂好.然而如果将Crabtree等[73]提出的每单位转化数成本作为未来绿色催化剂的评判标准, 那么铁基催化剂就会成为首选.这激励人们对其它第一过渡系金属催化剂进行研究. Beller课题组[74]报道了首例锰催化剂30在甲醇水相脱氢体系中的应用.虽然其单独使用活性比不上铁基催化剂, 但在加入过量HN(CH2CH2P- iPr2)2后, 脂肪族锰配合物30的稳定性会更好, 可以持续一个月之久, 并且实现了高达20000的转化数. 2018年, 焦海军课题组[75]关于配合物30催化甲醇水相脱氢反应的机理研究表明在强碱条件下需要克服的反应能垒低于无碱条件, 这又一次说明碱在甲醇脱氢反应中的促进作用.此外, 该催化剂也可以用于催化乙醇、多聚甲醛和甲酸脱氢.
为了满足全球能源需求的不断增长, 迫切需要加快甲醇脱氢催化剂的发展和应用研究进程.本文着重从多种过渡金属催化剂的结构、反应条件、产氢转化频率、反应机理等方面进行了综述.最初甲醇热脱氢使用的钌基催化剂是经典络合物, 为后续一系列研究奠定了基础.带有“非单一”辅助配体催化剂使得甲醇脱氢在产量和活性上有了大大的提高.然而, 这类反应通常需要高浓度的强碱来激活催化剂的活性位点, 从而有效地释放氢气, 随着反应的进行会有固体沉淀, 这就需要重新不断地添加碱, 因而使得产氢效率有所限制, 所以下一步需要重点研究的方向是不加入任何添加剂直接脱氢.低温均相过渡金属催化甲醇水相重整是相对较有前景的领域, 该领域的开创性工作仅开展了5年左右, 虽然报道的最高转化数超过了300000, 但仍需要更有效、稳定、便宜的金属催化剂来使这项工艺最终应用在工业上.目前, 醇脱氢工作研究的方向已经慢慢从之前的贵金属钌、铑、铱向相对便宜的铁、锰等金属转变, 虽然活性不如贵金属, 但是产量更高、成本更低、更为绿色环保.尽管用于甲醇脱氢的均相催化剂的活性和产率还远远达不到实际应用的要求, 但很明显这些年关于这一领域的均相催化剂增加很多.经过研究工作者们的努力, 相信在不久的未来终会取得更大的进步.
Armaroli, N.; Balzani, V. Chem. Asian J. 2011, 6, 768. doi: 10.1002/asia.201000797
Behrendt, F.; Schüth, F. Chem. Ing. Tech. 2011, 83, 1984. doi: 10.1002/cite.201100147
Cook, T. R.; Dogutan, D. K.; Reece, S. Y.; Surendranath, Y.; Teets, T. S.; Nocera, D. G. Chem. Rev. 2010, 110, 6474. doi: 10.1021/cr100246c
Armaroli, N.; Balzani, V. ChemSusChem 2011, 4, 21. doi: 10.1002/cssc.201000182
Zheng, J. Y.; Liu, X. X.; Xu, P.; Liu, P. F.; Zhao, Y. Z.; Yang, J. Int. J. Hydrogen Energy 2012, 37, 1048. doi: 10.1016/j.ijhydene.2011.02.125
许炜, 陶占良, 陈军, 化学进展, 2006, 18, 200. doi: 10.3321/j.issn:1005-281X.2006.02.008Xu, Y.; Tao, Z. -L.; Chen, J. Prog. Chem. 2006, 18, 200(in Chinese). doi: 10.3321/j.issn:1005-281X.2006.02.008
Dalebrook, A. F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Chem. Commun. 2013, 49, 8735. doi: 10.1039/c3cc43836h
陈俊, 陈秋雄, 陈运文, 樊栓狮, 王燕鸿, 杨亮, 郎雪梅, 张雯翔, 黄怡, 熊文涛, 储能科学与技术, 2015, 4, 131. doi: 10.3969/j.issn.2095-4239.2015.02.002Chen, J.; Chen, Q. -X.; Chen, Y. -W.; Fan, S. -S.; Wang, Y. -H.; Yang, L.; Lang, X. -M.; Zhang, W. -X.; Huang, Y.; Xiong, W. -T. Energy Storage Sci. Tech. 2015, 4, 131(in Chinese). doi: 10.3969/j.issn.2095-4239.2015.02.002
陈卓, 杨运泉, 包建国, 王威燕; 蒋新民, 化工进展, 2010, 29, 484. http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201003021.htmChen, Z.; Yang, Y. -Q.; Bao, J. -G.; Wang, W. -Y.; Jiang, X. -M.; Chem. Ind. Eng. Prog. 2010, 29, 484(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201003021.htm
李璐伶; 樊栓狮; 陈秋雄; 杨光; 温永刚, 储能科学与技术, 2018, 7, 586. http://www.cnki.com.cn/Article/CJFDTotal-CNKX201804011.htmLi, L. -L.; Fan, S. -S.; Chen, Q. -X.; Yang, G.; Wen, Y. -G. Energy Storage Sci. Tech. 2018, 7, 586(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-CNKX201804011.htm
Niaz, S.; Manzoor, T.; Pandith, A. H. Renew. Sust. Energ. Rev. 2015, 50, 457. doi: 10.1016/j.rser.2015.05.011
Ares, J. R. Int. J. Hydrogen Energy 2014, 39, 9824. doi: 10.1016/j.ijhydene.2014.05.096
Sordakis, K.; Tang, C.; Vogt, L. K.; Junge, H.; Dyson, P. J.; Beller, M.; Laurenczy, G. Chem. Rev. 2018, 118, 372. doi: 10.1021/acs.chemrev.7b00182
Alberico, E.; Nielsen, M. Chem. Commun. (Camb.) 2015, 51, 6714. doi: 10.1039/C4CC09471A
He, T.; Pachfule, P.; Wu, H.; Xu, Q.; Chen, P. Nat. Rev. Mater. 2016, 1, 16059. doi: 10.1038/natrevmats.2016.59
Li, H. -W.; Yan, Y.; Orimo, S. -I.; Züttel, A.; Jensen, C. M. Energies 2011, 4, 185. doi: 10.3390/en4010185
Peng, B.; Chen, J. Energ. Environ. Sci. 2008, 1, 479. doi: 10.1039/B809243P
Zhu, Q. -L.; Xu, Q. Energ. Environ. Sci. 2015, 8, 478. doi: 10.1039/C4EE03690E
Klerke, A.; Christensen, C. H.; N rskov, J. K.; Vegge, T. J. Mater. Chem. 2008, 18, 2304. doi: 10.1039/b720020j
Moury, R.; Moussa, G.; Demirci, U. B.; Hannauer, J.; Bernard, S.; Petit, E.; van der Lee, A.; Miele, P. Phys. Chem. Chem. Phys. 2012, 14, 1768. doi: 10.1039/C2CP23403C
Zhao, H. Y.; Oyama, S. T.; Naeemi, E. D. Catal. Today 2010, 149, 172. doi: 10.1016/j.cattod.2009.02.039
(a) Grasemann, M.; Laurenczy, G. Energ. Environ. Sci. 2012, 5, 8171.
(b) Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Chem. Soc. Rev. 2016, 45, 3954.
Johnson, T. C.; Morris, D. J.; Wills, M. Chem. Soc. Rev. 2010, 39, 81. doi: 10.1039/B904495G
Asinger, F. Methanol-Chemie und Energierohstoff, Springer, Berlin, Heidelberg, 1986, pp. 1~9.
Smith, T. A.; Maitlis, P. M. J. Organomet. Chem. 1984, 269, c7. doi: 10.1016/0022-328X(84)80283-1
Wang, W. -H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; Fujita, E. Chem. Rev. 2015, 115, 12936. doi: 10.1021/acs.chemrev.5b00197
Sá, S.; Silva, H.; Brand o, L.; Sousa, J. M.; Mendes, A. Appl. Catal. B-Environ. 2010, 99, 43. doi: 10.1016/j.apcatb.2010.06.015
Iulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Renew. Sust. Energ. Rev. 2014, 29, 355. doi: 10.1016/j.rser.2013.08.032
Cortright, R. D.; Davda, R. R.; Dumesic, J. A. Nature 2002, 418, 964. doi: 10.1038/nature01009
Lin, L.; Zhou, W.; Gao, R.; Yao, S.; Zhang, X.; Xu, W.; Zheng, S.; Jiang, Z.; Yu, Q.; Li, Y. -W.; Shi, C.; Wen, X. -D.; Ma, D. Nature 2017, 544, 80. doi: 10.1038/nature21672
Palo, D. R.; Dagle, R. A.; Holladay, J. D. Chem. Rev. 2007, 107, 3992. doi: 10.1021/cr050198b
Navarro, R. M.; Pe a, M. A.; Fierro, J. L. G. Chem. Rev. 2007, 107, 3952. doi: 10.1021/cr0501994
吴松; 熊晓东; 王胜国, 稀有金属, 2007, 31, 237. doi: 10.3969/j.issn.0258-7076.2007.02.022Wu, S.; Xiong. X. -D.; Wang, S. -G. Rare Metals 2007, 31, 237(in Chinese). doi: 10.3969/j.issn.0258-7076.2007.02.022
Smith, T. A.; Aplin, R. P.; Maitlis, P. M. J. Organomet. Chem. 1985, 291, c13. doi: 10.1016/0022-328X(85)80213-8
Shinoda, S.; Itagaki, H.; Saito, Y. J. Chem. Soc.; Chem. Commun. 1985, 13, 860. doi: 10.1039/C39850000860
Itagaki, H.; Shinoda, S.; Saito, Y.; Bull. Chem. Soc. Jpn. 1988, 61, 2291. doi: 10.1246/bcsj.61.2291
Morton, D.; Cole-Hamilton, D. J. J. Chem. Soc. Chem. Commun.; 1988, 1154. doi: 10.1002/chin.198904276
Fujii, T.; Saito, Y. J. Mol. Catal. 1991, 67, 185. doi: 10.1016/0304-5102(91)85045-4
Sieffert, N.; Bühl, M. J. Am. Chem. Soc. 2010, 132, 8056. doi: 10.1021/ja101044c
Rodríguezlugo, R. E.; Trincado, M.; Vogt, M.; Tewes, F.; Santisoquinones, G.; Grützmacher, H. Nat. Chem. 2013, 5, 342. doi: 10.1038/nchem.1595
Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H. -J.; Junge, H.; Gladiali, S.; Beller, M. Nature 2013, 495, 85. doi: 10.1038/nature11891
Li, H.; Hall, M. B. J. Am. Chem. Soc. 2015, 137, 12330. doi: 10.1021/jacs.5b07444
Jing, Y.; Chen, X.; Yang, X. J. Organomet. Chem. 2016, 820, 55. doi: 10.1016/j.jorganchem.2016.07.020
Sinha, V.; Trincado, M.; Grützmacher, H.; de Bruin, B.; J. Am. Chem. Soc. 2018, 140, 13103. doi: 10.1021/jacs.8b09011
Kuriyama, W.; Matsumoto, T.; Ogata, O.; Ino, Y.; Aoki, K.; Tanaka, S.; Ishida, K.; Kobayashi, T.; Sayo, N.; Saito, T. Org. Process. Res. Dev. 2012, 16, 166. doi: 10.1021/op200234j
(a) Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G. Organometallics 2011, 30, 3479.
(b) Nielsen, M.; Kammer, A.; Cozzula, D.; Junge, H.; Gladiali, S.; Beller, M. Angew. Chem.; Int. Ed. Engl. 2011, 50, 9593.
Lei, M.; Pan, Y.; Ma, X. Eur. J. Inorg. Chem. 2015, 2015, 794. doi: 10.1002/ejic.201403027
Alberico, E.; Lennox, A. J.; Vogt, L. K.; Jiao, H.; Baumann, W.; Drexler, H. J.; Nielsen, M.; Spannenberg, A.; Checinski, M. P.; Junge, H.; Beller, M. J. Am. Chem. Soc. 2016, 138, 14890. doi: 10.1021/jacs.6b05692
Yang, X. ACS Catal. 2014, 4, 1129. doi: 10.1021/cs500061u
Monney, A.; Barsch, E.; Sponholz, P.; Junge, H.; Ludwig, R.; Beller, M. Chem. Commun. 2014, 50, 707. doi: 10.1039/C3CC47306F
Hu, P.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. ACS Catal. 2014, 4, 2649. doi: 10.1021/cs500937f
Heim, L. E.; Schl rer, N. E.; Choi, J. H.; Prechtl, M. H. Nat. Commun. 2014, 5, 3621. doi: 10.1038/ncomms4621
Crabtree, R. H. New J. Chem. 2011, 35, 18. doi: 10.1039/C0NJ00776E
(a) Crabtree, R. H. Chem. Rev. 2017, 117, 9228.
(b) Grützmacher, H.; Angew. Chem.; Int. Ed. 2008, 47, 1814.
Kothandaraman, J.; Kar, S.; Goeppert, A.; Sen, R.; Prakash, G. K. S. Top. Catal. 2018, 61, 542. doi: 10.1007/s11244-018-0963-9
Heim, L. E.; Thiel, D.; Gedig, C.; Deska, J.; Prechtl, M. H. G. Angew. Chem.; Int. Ed. 2015, 54, 10308. doi: 10.1002/anie.201503737
Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G. A.; Prakash, G. K. S. J. Am. Chem. Soc. 2016, 138, 778. doi: 10.1021/jacs.5b12354
Van de Watering, F. F.; Lutz, M.; Dzik, W. I.; de Bruin, B.; Reek, J. N. H. ChemCatChem 2016, 8, 2752. doi: 10.1002/cctc.201600709
Shinoda, S.; Yamakawa, T. J. Chem. Soc.; Chem. Commun. 1990, 1511. doi: 10.1002/chin.199115049
Shinoda, S.; Ohnishi, T.; Yamakawa, T. Catal. Sur. Asia 1997, 1, 25. doi: 10.1023/A:1019060526478
Robles-Dutenhefnera, P. A.; Mourab, E. M.; Gamac, G. J. J. Mol. Catal. A Chem. 2000, 164, 39. doi: 10.1016/S1381-1169(00)00202-8
Campos, J.; Sharninghausen, L. S.; Manas, M. G.; Crabtree, R. H. Inorg. Chem. 2015, 54, 5079. doi: 10.1021/ic502521c
Manas, M. G.; Campos, J.; Sharninghausen, L. S.; Lin, E.; Crabtree, R. H. Green Chemistry 2015, 17, 594. doi: 10.1039/C4GC01694G
Fujita, K.; Kawahara, R.; Aikawa, T.; Yamaguchi, R. Angew. Chem. 2015, 127, 9185. doi: 10.1002/ange.201502194
Prichatz, C.; Alberico, E.; Baumann, W.; Junge, H.; Beller, M. ChemCatChem 2017, 9, 1891. doi: 10.1002/cctc.201700015
Shen, Y.; Zhan, Y.; Li, S.; Ning, F.; Du, Y.; Huang, Y.; He, T.; Zhou, X. Chem. Sci. 2017, 8, 7498. doi: 10.1039/C7SC01778B
Morton, D.; Cole-Hamilton, D. J. J. Chem. Soc. Chem. Commun. 1987, 0, 248. doi: 10.1039/C39870000248
Zhan, Y. L.; Shen, Y. B.; Li, S. P.; Yue, B. H.; Zhou, X. C. Chin. Chem. Lett. 2017, 28, 1353. doi: 10.1016/j.cclet.2017.03.038
柴会宁, 刘波, 刘爱芹, 喻琨, 分子催化, 2018, 32, 481. doi: 10.16084/j.cnki.issn1001-3555.2018.05.008Chai, H. -N.; Liu, B.; Liu, A. -Q.; Yu, K. J. Mol. Catal 2018, 32, 481(in Chinese). doi: 10.16084/j.cnki.issn1001-3555.2018.05.008
Alberico, E.; Sponholz, P.; Cordes, C.; Nielsen, M.; Drexler, H. J.; Baumann, W.; Junge, H.; Beller, M. Angew. Chem.; Int. Ed. Engl. 2013, 52, 14162. doi: 10.1002/anie.201307224
Bielinski, E. A.; F rster, M.; Zhang, Y.; Bernskoetter, W. H.; Hazari, N.; Holthausen, M. C. ACS Catal. 2015, 5, 2404. doi: 10.1021/acscatal.5b00137
Wakizaka, M.; Matsumoto, T.; Tanaka, R.; Chang, H. -C. Nat. Commun. 2016, 7, 12333. doi: 10.1038/ncomms12333
Eisenstein, O.; Crabtree, R. H. New J. Chem. 2013, 37, 21. doi: 10.1039/C2NJ40659D
Andérez-Fernández. M; Vogt, L. K.; Fischer, S.; Zhou, W.; Jiao, H.; Garbe, M.; Elangovan, S.; Junge, K.; Junge, H.; Ludwig, R.; Beller, M. Angew. Chem.; Int. Ed. Engl. 2017, 56, 559. doi: 10.1002/anie.201610182
Wei, Z.; de Aguirre, A.; Junge, K.; Beller, M.; Jiao, H. J. Catal. Sci. Technol. 2018, 8, 3649. doi: 10.1039/C8CY00746B

图 1 基于甲醇/CO2的可逆储氢循环
Figure 1 Reversible hydrogen storage cycle based on the methanol/CO2 couple


图式 2 Grützmacher课题组提出的反应机理
Scheme 2 Proposed reaction mechanism by Grützmacher's group

图式 4 [Ru(H)Cl(CO)(PNP-iPr)] (8)催化甲醇水相脱氢的简化循环机理
Scheme 4 Simplified catalytic cycle for the aqueous dehydrogenation of MeOH catalyzed by [Ru(H)Cl(CO)(PNP-iPr)] (8)


图式 6 均相催化甲醇水相重整过程
Scheme 6 Crucial steps in the homogeneously catalyzed aqueous-phase methanol reforming

图式 7 酶和铱催化剂共同作用的生物诱导甲醇重整方法
Scheme 7 Bioinduced approach for methanol reforming based on a combination of enzymes and iridium catalyst
表 1 基于甲醇/CO2可逆储氢的热力学数据[26]
Table 1. Thermodynamic data for reversible H2 storage based on methanol/CO2
| No. | Eq. | ∆H0/(kJ•mol-1) |
| (1) | CH3OH (l)→HCHO (g)+H2 (g) | +129.8 |
| (2) | CH3OH (g)→CO (g)+2H2 (g) | +94.6 |
| (3) | CH3OH (l)→CO (g)+2H2 (g) | +127.9 |
| (4) | CH3OH (g)+H2O (g)→3H2 (g)+CO2 (g) | +53.3 |
| (5) | CH3OH (l)+H2O (l)→3H2 (g)+CO2 (g) | +130.7 |
 下载: 导出CSV
下载: 导出CSV
表 2 用于甲醇热脱氢的均相钌催化剂活性
Table 2. Active homogeneous ruthenium catalysts for methanol thermal dehydrogenation.
| Catalyst (mmol) | MeOH/mL | Additive (mmol) | T/℃ | Time/h | TOFa/h-1 | TONb | Ref. |
| [RuCl2(PPh3)3] (1) (0.08) | 10 | — | 150 | 18 | 3.60 | 65 | [34] |
| [Ru(OAc)Cl(PEtPPh2)3] (2) (0.1) | 400 | CH3COOH (0.2) | 66 | 90 | 0.38 | 34 | [35] |
| [Ru(H)2(N2)(PPh3)3] (3) [(1~5)×10-3] | 5 | NaOH (5) | 150 | 2 | 6.40 | 12.8 | [37] |
| [Ru(H)2(PPh3)4] (4) [(1~5)×10-3] | 5 | NaOH (5) | 150 | 2 | 7.50 | 15 | [37] |
| RuCl3•3H2O (5) (1) | 200 | MeONa (610) | 79 | n.r. c | 1.68 d | — | [38] |
| a Average TOF referred to overall reaction time. b TON is referred to hydrogen production. c n.r. not reported. d Initial TOF. | |||||||
下载: 导出CSV
表 3 用于甲醇水相重整的均相钌催化剂活性
Table 3. Active homogeneous ruthenium catalysts for methanol reforming
| Cat. (10-3 mmol; 0.001‰ to MeOH) | MeOH/mmol | H2O/mmol | Solvent (mL) | Additive (mmol) | T/℃ | Time/h | TOFa/h-1 | TONb | Ref. |
| [K(dme)2][RuH(trop2dad)] (6) (10; 500) | 2 | 2.6 | THF (1) | — | 90 | 10 | 54 | 540 | [40] |
| [Ru(H)Cl(CO)(PNP-iPr)] (8) (0.88; 1) | 890 | 222 | — | KOH (320) | 91 | 576 | 613 | 353409 | [41] |
| [Ru(H)(BH4)(CO)(PNP-Ph)] (9a) (5; 22.5)/[Ru(H)2(dppe)2] (9b) (5; 22.5) | 222 | 55.5 | Triglyme (4) | — | 93.5 | 192 | 22 | 4286 | [50] |
| [Ru(H)Cl(CO)(NNP-tBu)] (10) (5; 250) | 20 | 111 | Toluene (2) | KOH (40) | 100~105 | 648 | 44 | 28661 | [51] |
| a Average TOF referred to overall reaction time. b TON is referred to hydrogen production. c Initial TOF. | |||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们