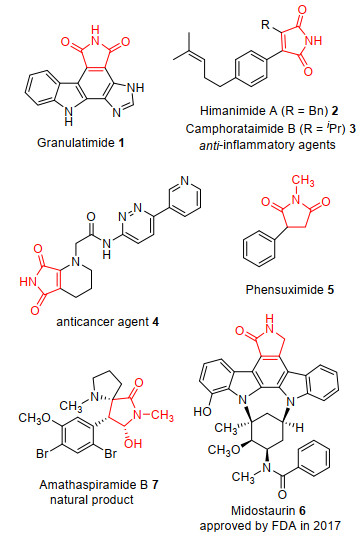
图 1.
具有重要生物活性的天然产物或生物活性分子的化学结构
Figure 1.
Selected examples of biologically relevant maleimide derivatives

马来酰亚胺是一类海洋天然生物碱和生物活性分子的重要结构母核, 例如海洋天然生物碱Granulatimide (1)、Himanimide A (2)、Camphorataimide B (3)以及化合物4等结构中含有马来酰亚胺的结构片段[1~3], 具有抗肿瘤和抗菌等多种生物活性.其中Granulatimide (1)对鼠P388白血病细胞的IC50为39 μmol/L[1], 化合物Himanimide A (2)对LPS诱导巨噬细胞因子IL-6抑制作用的IC50值达到10 μg/mL[2].化合物4为2017年Poulsen等[3]报道的一种特异性Porcupine(PORCN)抑制剂, 也可以有效抑制Wnt信号通路, IC50为0.4 nmol/L, 显示抗肿瘤活性, 具有较好的研究价值和应用前景.
另外, 马来酰亚胺经化学转化还可以合成琥珀酰亚胺、四氢吡咯和2-吡咯酮等化合物, 如抗癫痫药苯琥胺(Phensuximide, 5)、抗肿瘤药物米哚妥林(Midostaurin, 6)和天然产物(Amathaspiramide B, 7)等[4, 5](图 1), 其中米哚妥林为美国食品药品监督管理局(FDA)于2017年批准上市的一种新型口服多靶点激酶抑制剂, 用于携带FTL3突变的急性髓性白血病(AML)患者的治疗[4].
鉴于马来酰亚胺和琥珀酰亚胺类化合物具有广泛的应用价值, 研究简便有效、高选择性地构建含有马来酰亚胺和琥珀酰亚胺结构单元的杂环化合物具有重要的意义, 已经成为国内外有机合成化学的研究热点之一.
近年来, 有关马来酰亚胺类化合物的合成方法主要包括以下两种: (1)马来酰亚胺或琥珀酰亚胺环的合成, 如将2, 3-二取代丁二酸化合物脱水制备丁二酸酐, 随后胺解制得琥珀酰亚胺类化合物, 最后氧化得到马来酰亚胺[6]; 也可以采用过渡金属催化炔、CO和胺类化合物环合制备琥珀酰亚胺类化合物[7]. (2)以马来酰亚胺及其衍生物3-溴马来酰亚胺等为合成子构建马来酰亚胺类化合物, 特别是近年来马来酰亚胺及N-取代的马来酰亚胺的产业化, 为该类合成提供了廉价的原料.其官能化反应主要围绕马来酰亚胺的双键展开, 包括Michael加成反应、氧化偶联反应和Diels-Alder反应等, 并通过对马来酰亚胺分子结构中酰亚胺基团的还原, 也可以构建含四氢吡咯类及2-吡咯酮类化合物.本文对马来酰亚胺双键参与的Michael加成、氧化偶联反应和环加成反应等进行综述, 探讨其在有机合成领域中的应用.
马来酰亚胺参与的Michael加成反应是合成琥珀酰亚胺类化合物一种较为常用的方法.近年来由于手性合成技术的发展, 不对称Michael加成反应也成为众多科学家的研究热点之一.马来酰亚胺的Michael加成反应按照加成原子的不同, 可以分为马来酰亚胺与碳原子的Michael加成反应和马来酰亚胺与杂原子的Michael加成反应.
马来酰亚胺与碳原子化合物发生Michael加成反应是合成3-取代琥珀酰亚胺类化合物的主要合成方法.
2005年, Prudhomme等[8]报道了将取代吲哚和N-取代马来酰亚胺在醋酸中回流36 h制备吲哚琥珀酰亚胺类化合物, 收率为25%~74%.但该反应存在反应时间较长, 且在质子酸作用下, 马来酰亚胺类化合物容易聚合等缺点, 反应收率较低(Eq. 1).
|
|
(1) |
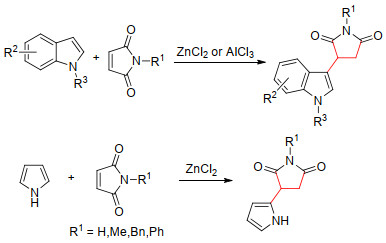
2013年, 本课题组安玉龙等[9]用Lewis酸ZnCl2或AlCl3作为催化剂, 实现了取代吲哚和吡咯对马来酰亚胺类化合物的Michael加成反应.与使用醋酸作催化剂相比, 该方法具有价格便宜、操作简便, 而且对空气和水很稳定、收率高等优点, 是一种制备吲哚琥珀酰亚胺类化合物的有效方法, 收率为82%~96% (Scheme 1).
通常, 在Lewis酸催化下, 吲哚3-位的富电子的氢会与马来酰亚胺发生加成反应. 2015年, Prabhu等[10]成功实现了吲哚-1-位苯甲酰基定位的吲哚-2-位与马来酰亚胺的区域选择性Michael加成反应, 得到了吲哚-2-位取代的琥珀酰亚胺类化合物, 收率为44%~78%, 但是该反应仅仅适用于N上取代的马来酰亚胺(Eq. 2).
|
|
(2) |
2017年, Prabhu等[11]又报道了采用吲哚-1-位N原子上嘧啶基团作为导向基, N-嘧啶取代的吲哚与马来酰亚胺在钴催化剂的作用下在三氟乙醇中可以顺利实现吲哚-2-位选择性Michael加成反应, 得到吲哚-2-取代的琥珀酰亚胺类化合物, 收率为30%~98% (Eq. 3).
|
|
(3) |
2017年, Li研究小组[12]同样以[Cp*Co(CO)I2]作为催化剂, N-(2-嘧啶基)吲哚和N-取代基马来酰亚胺为反应物, AgSbF6和NaOAc为添加剂, 三氟乙醇为溶剂, 在30 ℃下反应得到了一系列带有琥珀酰亚胺结构的吲哚类化合物, 收率为45%~95% (Eq. 4).
|
|
(4) |
2017年, Song等[13]报道了Mn2(CO)10催化的吲哚-2-位与马来酰亚胺在乙酸乙酯中化学选择性Michael加成反应.该反应以2-吡啶基为定位基团, 无需任何添加剂就可以得到2-取代吲哚琥珀酰亚胺类化合物, 收率为46%~96% (Eq. 5).
|
|
(5) |
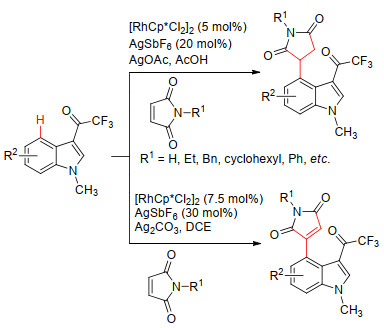
2018年, Prabhu等[14]报道了一种以[RhCp*Cl2]2为催化剂, 3位三氟乙酰基为定位基实现吲哚-4-位的官能团化反应.研究表明, 铑催化的马来酰亚胺与吲哚的反应在加入酸性添加剂醋酸时发生1, 4-加成反应, 生成琥珀酰亚胺类化合物, 收率为55%~80%;当加入碱性添加剂碳酸银时发生Heck型偶联反应, 生成马来酰亚胺类化合物, 收率为50%~76% (Scheme 2).
2018年, Yu等[15]报道了一种铑[RhCp*Cl2]2催化的选择性吲哚7位官能团化反应.该反应以吲哚-1-位的乙酰基为定位基, 在铑催化下吲哚-7-位与马来酰亚胺发生Michael加成得到吲哚-7-位取代的琥珀酰亚胺类化合物, 收率为55%~90% (Eq. 6).
马来酰亚胺双键亲电性较弱, 通常需要Lewis酸活化. 2006年Koltunov等[16]报道了苯与马来酰亚胺发生Michael加成反应, 该反应需要使用2~4 equiv.的AlCl3, 反应底物适应性较差, 仅仅报道了3个反应实例, 以苯甲醚为原料反应收率达到83%, 但是以3, 4-二氯苯和甲苯为原料, 反应收率分别为34%和66% (Eq. 7).
|
|
(6) |
|
|
(7) |
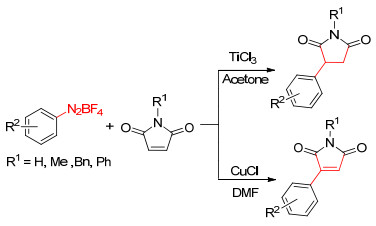
2015年, 本课题组杨振华等[17]报道了化学选择性调控芳基重氮四氟硼酸盐与马来酰亚胺的选择性加成反应.在三氯化钛的作用下发生还原芳基化反应生成3-芳基琥珀酰亚胺, 反应收率为60%~83%.然而, 在氯化亚铜的催化下可以发生氧化芳基化生成3-芳基马来酰亚胺, 反应收率为78%~91% (Scheme 3).
2015年, Prabhu等[18]报道了一种新型的铑催化苯乙酮与马来酰亚胺的1, 4-加成反应.该反应以乙酰基为定位基团促进苯环邻位C—H键与马来酰亚胺反应生成3-芳基琥珀酰亚胺类化合物, 收率为54%~90%.由于羰基的定位作用使得该反应具有良好的化学选择性, 并且反应体系中未检测到Heck偶联产物和烯醇化加成产物, 其缺点是只适用于N上有取代基的马来酰亚胺类化合物(Eq. 8).
2018年, Sundararaju等[19]报道了一种钴催化的芳香酮邻位C—H键与马来酰亚胺的芳基化反应.该反应通过利用酮羰基的协调定位作用, 实现多种芳香酮邻位C—H键与马来酰亚胺的加成反应, 反应收率为27%~96% (Eq. 9).
|
|
(8) |
|
|
(9) |
2018年, Zhang等[20]报道了钌催化的芳香醛邻位C—H键与马来酰亚胺的芳基化反应.反应体系中苯胺和醛反应生成亚胺产生定位作用, 产物收率为45%~85% (Eq. 10).
|
|
(10) |
2018年, Pan等[21]报道了铑催化的α, β-不饱和羰基定位的马来酰亚胺芳基化反应.由于羰基的定位作用, 芳基邻位的氢优先发生Michael加成反应生成3-芳基琥珀酰亚胺类化合物, 产物为33%~86% (Eq. 11).
|
|
(11) |
2016年, Kim等[22]报道了铑催化的色酮、1, 4-萘醌类、氧杂蒽酮与马来酰亚胺的Michael加成反应.该反应以羰基为定位基合成一系列含有色酮、萘醌类和氧杂蒽酮的琥珀酰亚胺, 收率为36%~95% (Eq. 12).
|
|
(12) |
2017年, Chatani等[23]报道了铑催化的酰胺邻位C—H键与马来酰亚胺的芳基化反应.该反应采用8-氨基喹啉为定位基, 无需其他任何添加剂, 收率为51%~100%, 且底物实用范围广, 甚至对于N上未保护的马来酰亚胺同样适用, 但反应温度较高是其缺点(Eq. 13).
|
|
(13) |
2018年, Wu课题组[24]使用了Cp*Co(CO)I2作催化剂, 实现了马来酰亚胺和芳基酮肟的芳环sp2-C—H键的共轭加成反应, 得到了一系列琥珀酰亚胺类产物.该反应不需要额外加入碱, 收率为41%~92%, 底物适用性也很好(Eq. 14).
|
|
(14) |
2017年, Prabhu等[25]报道了一种铑催化的苯甲酸邻位C—H键与马来酰亚胺的芳基化反应.该反应采用羧酸为定位基, 在生成芳基琥珀酰亚胺的同时发生脱羧反应, 收率为35%~82%, 但是该反应需要5 equiv.的乙酸为添加剂, 且不适用于呋喃、噻唑和吲哚等杂环类化合物(Eq. 15).
|
|
(15) |
2017年, Baidya等[26]报道了另一种钌催化的苯甲酸邻位C—H键与马来酰亚胺的芳基化反应.该反应通过加入三环己基氧化膦(Cy3PO)提高钌催化剂反应活性, 进一步提高反应收率至60%~95%, 且底物适用范围更广, 对各种芳环和芳杂环均具有良好的适用性(Eq. 16).
|
|
(16) |
2017年, Prabhu等[27]又报道了一种钴催化的偶氮苯与马来酰亚胺的Michael加成反应.该反应采用偶氮基团为定位基高化学选择性地合成了一系列的芳基琥珀酰亚胺类化合物, 收率为22%~94% (Eq. 17).
|
|
(17) |
2013年, Albini等[28]报道了十聚钨酸季铵盐(TBADT)光催化剂催化的马来酰亚胺与甲苯的苄基化反应.该反应以乙腈/水(V:V=5:1)的混合溶液为反应溶剂, 同时添加0.5 mol/L的高氯酸锂提高其离子浓度, 光照(310 nm)下反应24 h可以得到3-苄基琥珀酰亚胺, 反应收率为46%~55%.该反应对各种N取代的马来酰亚胺都具有良好的兼容性, 但是反应收率较低(Eq. 18).
|
|
(18) |
2016年, Ravelli等[29]也报道了在该催化剂催化下N-苯基马来酰亚胺与苯乙酸的脱羧加成反应.研究表明, 苯乙酸可以在光催化下发生脱酸反应产生苄基自由基, 随后与N-苯基马来酰亚胺发生加成反应生成3-苄基琥珀酰亚胺, 反应收率为72%(Eq. 19).
|
|
(19) |
2016年, Kim等[30]首次研究了8-甲基喹啉与马来酰亚胺的sp3-C—H官能化反应.他们使用AgSbF6和金刚烷甲酸(AdCOOH)作添加剂, 1, 2-二氯乙烷为溶剂, 70 ℃下反应24 h, 得到加成产物的最高收率可达96%.该反应具有良好的官能团耐受性和广泛的底物适用性, 在喹啉环上任意位置带有吸电子基或供电子基均可发生反应, 且具有较好的收率(Eq. 20).
|
|
(20) |
通常, 马来酰亚胺与含活泼亚甲基化合物发生Michael加成反应, 生成琥珀酰亚胺类化合物[31], 但近年来随着手性合成技术的发展, 其不对称Michael加成反应有了很大进展, 取得了较好的实验结果.
2007年, Zhao等[32]报道了手性二苯脯氨醇催化醛类对马来酰亚胺类化合物的不对称Michael加成反应.琥珀酰亚胺类产物收率为21%~96%, ee值为51%~99% (Eq. 21).
|
|
(21) |
2010年, Ye等[33]首次报道了由一种高效的双功能硫脲伯胺手性催化剂催化N-取代马来酰亚胺和α, α-二取代醛的不对称Michael加成反应.该方法是用10 mol%的手性二胺作为催化剂, 加入10 mol%的苯甲酸, 于二氯甲烷中室温下反应得到含有一个手性季碳中心的产物, 收率85%~99%, ee值为91%~99% (Eq. 22).
|
|
(22) |
同年, 该课题组还报道[34]了马来酰亚胺和酮的不对称Michael加成反应.他们将上述催化剂进行了稍微改变, 使用了10 mol%的(R, R)-N-(2, 6-二氯苯磺酰基)- 1, 2-二苯基乙二胺来催化反应, 同样以苯甲酸为添加剂, 甲苯为溶剂在室温下反应, 以62%~99%收率得到了一系列Michael加成产物, ee值为91%~99%.
2013年, Zhao等[35]报道了奎尼丁硫脲和L-2-氯苯基甘氨酸作为催化剂可以有效催化醛、酮与马来酰亚胺的不对称Michael加成反应, 收率为71%~99%, ee值为89%~99%.其缺点是该反应体系仅仅适用于N-取代的马来酰亚胺(Eq. 23).
|
|
(23) |
2015年, Gomez-Bengoa等[36]合成了手性的(S, S)-反式环己基胺-2-氨基嘧啶有机催化剂, 它可以在DMF/水溶液中与己二酸(HDA)共催化促进醛与马来酰亚胺的不对称Michael加成反应, 反应收率为70%~92%, ee值为60%~93%.其优点是NH-马来酰亚胺也能发生该反应(Eq. 24).
|
|
(24) |
2015年, Miura等[37]报道了一种新型二氨基亚甲基茚满二酮(DMI), 它能够有效催化酮与马来酰亚胺的不对称Michael加成反应.该反应收率为51%~92%, 且对映选择性较好, ee值为85%~99%, 但底物兼容性较差(Eq. 25).
|
|
(25) |
2011年, Yan课题组[38]报道了氰基乙酸酯和马来酰亚胺的不对称Michael加成反应.通过对几种金鸡纳碱类和氨基硫脲类手性催化剂的筛选, 发现对反应的催化效果最佳的还是上述手性叔胺硫脲类Takemoto催化剂, 以86%~99%收率得到了一系列加成产物, ee值为89%~99% (Eq. 26).
|
|
(26) |
2012年, Wang等[39]报道了一种手性的双官能化硫脲-叔胺催化的α-取代的异腈基乙酸酯与马来酰亚胺的不对称加成反应.该反应收率为63%~98%, 且具有良好的收率和高度的对映选择性, ee值为72%~94%, 生成的手性琥珀酰亚胺衍生物可以进一步转化为h5-HT1d受体拮抗剂(Eq. 27).
|
|
(27) |
除此之外, 2011年, Maruoka等[40]报道了α-取代的硝基乙酸酯与N-苄基马来酰亚胺的不对称Michael加成反应.该反应采用哌啶衍生物为催化剂, 在中性相转移条件下合成一系列不对称加成产物, 收率为42%~92%, ee值为83~91%, 再经锌粉还原可用于合成α, α-双取代的α-氨基酸衍生物(Eq. 28).
|
|
(28) |
2011年, Najera等[41]报道了手性C2-对称的双(2-氨基苯并咪唑)催化的1, 3-二羰基化合物与马来酰亚胺的共轭加成反应, 收率69%~100%, ee值为78%~99%.该方法成功实现克级规模Michael加成产物的合成, 并且手性有机催化剂可以循环使用, 部分原料收率可以实现定量转化(Eq. 29).
2010年, Cheng等[42]同样使用一种手性双功能硫脲叔胺类催化剂催化了N-取代马来酰亚胺和3-苯基苯并呋喃-2(3H)-酮的不对称Michael加成反应.该方法是以10 mol%的氨基硫脲为催化剂, 在4Å分子筛的存在下于-80 ℃的二氯甲烷中反应, 得到Michael加成产物, 收率为88%~99%, ee值为91%~99% (Eq. 30).
|
|
(29) |
|
|
(30) |
2010年, Yuan等[43]首次尝试以手性的双官能化硫脲-叔胺为催化剂, 实现了3-取代二氢吲哚酮衍生物和马来酰亚胺的不对称Michael加成反应.该反应收率为36%~92%, 具有良好的立体选择性和对映选择性, ee值为85%~98% (Eq. 31).但缺点是吲哚酮的1-位只有被Boc保护时才具有较好的反应活性, 限制了该反应的应用范围.
|
|
(31) |
2015年, Feng等[44]进一步研究了未保护的3-取代二氢吲哚衍生物和马来酰亚胺的不对称Michael加成反应.发现N, N'-二氧化物和三氟甲磺酸钪的络合物(L-RaPr3- Sc(OTf)3)能够有效催化未保护3-取代氧化吲哚衍生物和马来酰亚胺的不对称加成反应.该反应收率为88%~99%, 且具有较好的立体选择性, ee值为88%~99% (Eq. 32).
|
|
(32) |
2018年, Reddy等[45]报道了2-羧酸酯取代的氧化吲哚与马来酰亚胺的不对称Michael加成反应.该反应采取手性双官能团化的方酰胺为有机催化剂, 顺利实现了3-氧化吲哚2位与马来酰亚胺的不对称加成反应.该体系收率为55%~98%, ee值为60%~99%, 且具有反应条件温和、收率高和对映选择性好等优点, 同时适用于NH未保护的3-氧化吲哚类化合物(Eq. 33).
|
|
(33) |
2016年, Jiang等[46]报道了缩二氨酸的手性叔胺(DP-UAA)催化5H-噻唑烷-4-酮和噁唑烷-4-酮与马来酰亚胺的不对称共轭加成反应.该反应可高收率地合成一系列含杂环类化合物, 收率为82%~98%, 并且具有良好的立体选择性, ee值为98%~99% (Eq. 34).
2007年, Iyer等[47]报道了铑催化剂催化芳基硼酸与马来酰亚胺类化合物的Michael加成反应, 该反应在50 ℃反应3~6 h可以生成消旋体的3-芳基琥珀酰亚胺, 反应收率为34%~80%.后期经过改进, 将反应在微波条件下于100 ℃反应5~15 min, 得到3-芳基琥珀酰亚胺, 收率为55%~85% (Eq. 35).
|
|
(34) |
|
|
(35) |
2005年, Hayashi等[48]报道了在含有碳碳双键的手性磷二齿配体下, 铑催化剂催化芳基硼酸对马来酰亚胺类化合物的不对称Michael加成反应, 不同取代的芳基硼酸和马来酰亚胺类化合物反应性良好, 得到的芳基取代的琥珀酰亚胺类产物收率为88%~98%, ee值为88%~95% (Eq. 36).
|
|
(36) |
2011年, Ratovelomanana-Vidal等[49]报道了铑催化的芳香硼酸与马来酰亚胺的不对称Michael加成反应.该反应采用缺电子的二磷化物为配体, 对映选择性地合成3-芳基取代的马来酰亚胺类化合物, 收率大多为75%以上, ee值可达93%.但是以NH未保护的马来酰亚胺原料, 反应收率较低(Eq. 37).
|
|
(37) |
2013年, Korenaga等[50]报道了[RhOH(cod)]2催化的芳基硼酸与马来酰亚胺的不对称加成反应, 使用手性二苯基磷化合物为配体, 收率为86%~99%, ee值为72%~99%.此外, NH未取代的马来酰亚胺也可以获得良好的反应收率和对映选择性(Eq. 38).
|
|
(38) |
2015年, Wu等[51]报道了2, 5-二芳基取代的二环[2.2.1]二烯作为配体促进铑催化芳香硼酸和马来酰亚胺的对映选择性加成反应.该反应收率为72%~99%, 且具有良好的收率和较高的对映选择性, ee值为88%~98% (Eq. 39).此外, 所合成的琥珀酰亚胺还可以经氢化铝锂还原, 生成合成HSD-1抑制剂前体吡咯烷类化合物.
|
|
(39) |
通常在不加催化剂的条件下, 硫酚可以与马来西亚胺发生Michael加成, 而醇不与马来酰亚胺发生反应. 2011年, Nair等[52]报道了N-芳基马来酰亚胺和硫酚(醇)的Michael加成反应.该反应不需要任何催化剂或添加剂, 以水作溶剂, 在室温下反应15 min, 高收率地得到硫代琥珀酰亚胺类产物, 收率为80%~96% (Eq. 40). 2018年, Keillor等[53]对反应机理进行了深入研究, 对反应能量进行了计算.
|
|
(40) |
2012年, Xia等[54]报道了对甲基苯磺酸1-甲基咪唑盐([Hmim]Ots)离子液体催化含O或S杂原子的亲核试剂与马来酰亚胺类化合物的Michael加成反应, 反应在无溶剂条件下进行, 使用催化量的离子液体作为催化剂, 对甲氧基苯硫酚和甲醇分别与N-苯基马来酰亚胺反应得到取代琥珀酰亚胺类化合物的收率分别为91%和65% (Eq. 41).
|
|
(41) |
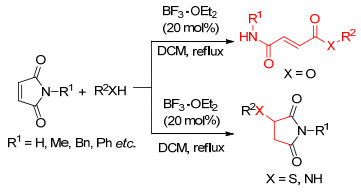
2015年, 我们课题组安玉龙等[55]研究了Lewis酸催化的马来酰亚胺和氧、硫、氮等杂原子的Michael加成反应.他们发现在BF3•OEt2催化下在1, 2-二氯乙烷中, 马来酰亚胺和硫醇或者胺类化合物发生Michael加成反应得到3-取代琥珀酰亚胺类化合物, 收率为34%~91%.当马来酰亚胺和烷基醇在二氯甲烷中回流反应时, 得到的产物为1, 2-Michael加成产物, 收率42%~95%, 这也是对Lewis酸催化马来酰亚胺的1, 2-加成反应的首次报道.与碱催化的方法相比, 该方法操作简单, 反应条件温和(Scheme 4).
2007年, Rogal等[56]报道了马来酰亚胺和芳伯胺的Michael加成反应, 在异丙醇中通过加热得到了3-取代马来酰亚胺产物(Eq. 42).
|
|
(42) |
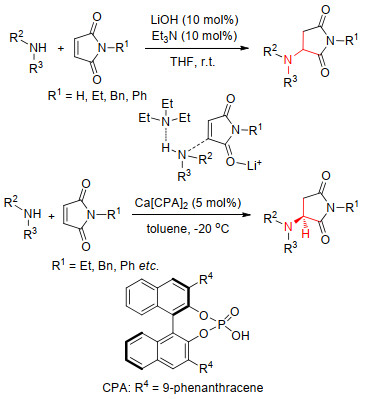
2018年, Scheidt等[57]报道了Lewis酸和胺碱共催化的胺类与马来酰亚胺类的共轭加成反应.该反应进一步提升了反应速率和转化率.并且被成功应用于合成克唑替尼-荧光探针.同年, 该课题组[58]报道了使用手性钙与磷酸络合物Ca(CPA)2催化胺与马来酰亚胺的不对称Michael加成反应, 构建手性3-氨基琥珀酰亚胺类化合物, 收率为49%~96% (Scheme 5).
2007年, Tan及其团队[59]研究了N-取代基马来酰亚胺分子上碳磷键的构造方法.在1, 5, 7-三氮杂二环- [4.4.0]癸-5-烯(TBD)的催化下, 以甲苯作溶剂, 室温下马来酰亚胺可以与二苯基膦酸酯或二苯氧膦反应, 得到加成产物, 收率73%~99%.该反应无需金属催化剂, 条件温和, 反应速度快, 且效率高(Eq. 43).
|
|
(43) |
2012年, Keglevich课题组[60]对上述反应进行了改进.他们在微波作用下N-甲基或N-苯基马来酰亚胺能与亚膦酸二烷基酯、二苯氧基膦等含磷化合物于175 ℃反应2.5~3 h, 得到了一系列的含磷琥珀酰亚胺类产物.该反应的优点是不需要溶剂和催化剂, 但是缺点是反应温度较高(Eq. 44).
|
|
(44) |
2016年, Kong等[61]报道了N-杂环膦酸酯-硫脲化合物作为磷基化试剂与马来酰亚胺发生硫杂-Michael加成反应, 合成了1-芳基-2, 5-二氧吡咯烷基膦酸酯类化合物, 收率为44%~92%.该反应无需任何催化和添加剂, 且反应条件温和(Eq. 45).
|
|
(45) |
马来酰亚胺为含有缺电子的环内烯烃, 与末端烯烃和脂肪族烯烃相比, 其偶联反应活性相对较低.通常采用3, 4-溴代的马来酰亚胺与芳基硼酸或末端炔烃发生偶联反应[62].但是, 由于众多天然有机药物分子中都含有马来酰亚胺骨架, 所以探究马来酰亚胺在偶联反应中的可能性具有重要意义.近年来, 有关马来酰亚胺的偶联反应已经取得了一些进展.
2008年, Roshchin等[63]报道了氯化钯催化取代的碘苯与马来酰亚胺类化合物的Heck反应, 反应收率较低, 为28%~40%.反应中使用了2.5 equiv.的HCOOK作为碱, 由于马来酰亚胺类化合物遇强碱会发生聚合, 这可能是导致收率低的原因. N-苯基取代的马来酰亚胺比N—H取代的马来酰亚胺的反应效果好, 吸电子取代的芳基碘苯反应收率较低, 例如当R1=4-CO2CH3, R2=Ph时, 收率只有28%.尽管如此, 这是第一个通过Heck反应使得马来酰亚胺类化合物的双键芳基化(Eq. 46).
|
|
(46) |
2015年, Zhou等[64]报道了碘苯与马来酰亚胺的Heck偶联反应.采用碳酸亚乙酯为反应溶剂, 反应收率为74%~99%.但是, 目前该反应仅适用于NH保护的马来酰亚胺(Eq. 47).
|
|
(47) |
2017年, Jafarpour等[65]报道了氯化钯催化碘苯与马来酰亚胺类化合物的Heck反应, 与Roshchin等[63]报道的Heck反应不同的是, 该反应得到的是双芳基取代的马来酰亚胺类化合物, 收率为28%~88%.当苯环上有强吸电子基时收率较低, 有NO2时反应不能进行(Eq. 48).
|
|
(48) |
2016年, Shamsianpour等[66]报道了钯催化的N-取代马来酰亚胺与芳环的直接脱氢偶联反应.该反应具有较高的原子经济性, 可以快速合成对称的双取代的芳基马来酰亚胺类化合物, 收率为21%~78% (Eq. 49).
|
|
(49) |
通常马来酰亚胺与胺和硫醇或硫酚化合物发生Michael加成反应得到琥珀酰亚胺类化合物.但在金属存在下可发生氧化加成反应, 得到3-取代的马来酰亚胺类化合物.
2016年, 我们课题组杨振华等[67]报道了铜催化的硫酚/硫醇和马来酰亚胺的氧化偶联反应.该反应通过加入氟硼酸促进铜的催化循环, 减少催化剂的用量.反应机理研究表明, 该反应涉及加成和氧化脱氢过程, 可以一步构建3-硫基马来酰亚胺, 反应收率为60%~91%.此外, 该反应同时适用于3-吲哚马来酰亚胺与硫酚的氧化偶联反应, 表现出良好的区域选择性(Eq. 50).
|
|
(50) |
2017年, Baidya等[68]报道了钌催化的马来酰亚胺与二苯基二硫(硒)醚或芳基甲酰胺的氧化偶联反应, 收率为44%~94%.该反应具有反应条件温和、操作简单和底物兼容性好等优点(Eq. 51).
2016年, 我们课题组安玉龙等[69]报道了碳酸银和醋酸铜共催化的马来酰亚胺和胺类化合物的氧化偶联反应.该反应可以一步构建3-氨基马来酰亚胺, 同时还适用于3-芳基取代的马来酰亚胺类底物, 反应收率为38%~95% (Eq. 52).
|
|
(51) |
|
|
(52) |
2018年我们课题组[70]又报道了在CuBr2催化下, 以NBS为溴源, 马来酰亚胺发生溴氨化反应得到3-溴-4-氨基取代的马来酰亚胺类化合物, 收率为62%~92% (Eq. 58.).
|
|
(53) |
2018年, 我们课题组杨振华等[71]报道了铜催化的硫酚/硫醇、马来酰亚胺和胺类的三组分氧化偶联反应.该反应首次实现一锅法构建3-氨基-4-硫基马来酰亚胺和噻嗪类化合物, 具有操作简单、原子经济性高等优点, 反应收率为27%~85% (Eq. 54).由于甲胺为气体, 在上述反应条件下, 甲胺容易溢出, 需要在封管中进行.为此, 我们用DMF作为甲胺的来源, 以CuI为催化剂, 以HBF4为添加剂使DMF缓慢分解为二甲胺, 发生马来酰亚胺的硫胺化反应[71b].
|
|
(54) |
2018年, 我们课题组杨振华等[72]报道了银催化的吲哚/吡咯、马来酰亚胺和有机磷化合物的三组分偶联反应.该反应可以一步实现马来酰亚胺的C—P双官能团化, 反应收率为52%~92%.该反应仅发生在吲哚的3位和吡咯的2位, 表现出良好的区域选择性(Eq. 55).
|
|
(55) |
马来酰亚胺中含有不饱和双键可以发生Diels-Alder反应以及[2+2]和[3+2]等环加成反应, 在有机合成、材料和生物医药领域得到较为广泛的应用[73].
2012年, Baker等[74]报道了光照下烯烃与3-硫基取代的马来酰亚胺类化合物发生[2+2]环加成反应, 收率为41%~80% (Eq. 56).
|
|
(56) |
2015年, Sun等[75]报道了可见光诱导异吲哚啉化合物形成异吲哚中间体, 再与马来酰亚胺发生[4+2]环化反应, 收率为61%~84%, 且反应条件温和, 无需过渡金属催化, 具有较高的原子经济性(Eq. 57).
|
|
(57) |
2017年, Xie等[76]报道了异吲哚酮与二苯基硅氢反应得到异吲哚, 而后与N-苯基马来酰亚胺发生Diels- Alder反应, 收率为24%~91% (Scheme 6).
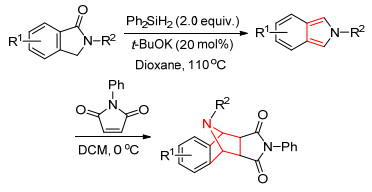
2016年, Jiang等[77]报道了使用Takemoto’s催化剂催化5H-噁唑-4-酮与N-取代的马来酰亚胺的[4+2]环化反应, 该反应收率为68%~90% (Eq. 58).
|
|
(58) |
2016年, Hara等[78]报道了一种手性吡啶磷酰胺作为双布朗斯特酸催化剂催化1-氨基二烯烃与马来酰亚胺的对映选择性Diels-Alder反应.该反应收率为55%~99%, ee值为63%~98% (Eq. 59).
|
|
(59) |
2012年, Kumar等[79]报道了氯胺-T(CAT)促进的腙与N-芳基马来酰亚胺的[3+2]环加成反应.该方法将N-芳基马来酰亚胺、腙和氯胺-T在乙醇中回流3 h, 即可得到1, 3, 5-三芳基-4, 6-二氧代吡咯并[3, 4-d]-7, 8-二氢吡唑, 收率为38%~66%, 该文献仅报道了N-苯基马来酰亚胺为反应原料, 以其它的N-取代马来酰亚胺作为反应底物有待进一步研究(Eq. 60).
|
|
(60) |
2016年, Liu等[80]报道了无催化剂条件下N-取代马来酰亚胺与腙的[3+2]环加成反应. N-取代马来酰亚胺与腙在乙醇中回流24 h, 得到一系列3a, 6a-二氢- 1H-吡咯并[3, 4-c]吡唑-4, 6-二酮衍生物, 测定了它们对HIV-1酶的抑制作用(Eq. 61).
|
|
(61) |
除上述Michael加成、氧化偶联和环加成反应外, 马来酰亚胺还可以与双键发生偶联反应, 生成螺环或3-取代的琥珀酰亚胺类化合物.
2014年, Shi等[81]报道了一种新型三苯基膦催化的4, 4-二氰基-2-亚甲基-3-丁烯酸酯与N-取代马来酰亚胺的[4+1]环化反应.该反应可以用于合成五元螺环类化合物, 收率为73%~95% (Eq. 62).
|
|
(62) |
2016年, Kim等[82]报道了铑催化的丙烯酰胺与马来酰亚胺的偶联反应.该反应实现末端烯烃与马来酰亚胺的直接偶联反应, 同时伴随双键的迁移形成亚甲基取代的琥珀酰亚胺类化合物, 收率为20%~95%.但是, 3-甲基取代的马来酰亚胺不能发生反应(Eq. 63).
|
|
(63) |
2016年, Miura等[83]报道了钌催化的N-取代马来酰亚胺与缺电子的烯烃(丙烯酸酯和丙烯酰胺)的偶联反应, 生成亚甲基取代的琥珀酰亚胺类化合物, 收率为35%~94%.但该反应并不适用于丙烯腈底物(Eq. 64).
|
|
(64) |
2017年, Zhang等[84]报道了钴催化的马来酰亚胺与烯胺的偶联反应生成乙烯基取代的琥珀酰亚胺类化合物.该反应收率为18%~91%, 且对NH未保护的马来酰亚胺具有良好的适用性(Eq. 65).
|
|
(65) |
2012年, Falck等[85]报道了一例铑催化苯甲酰基对甲苯磺酰胺和N-甲基马来酰亚胺的氧化偶联反应生成异吲哚啉酮衍生物, 收率为73% (Eq. 66).
|
|
(66) |
2015年, Miura等[86]报道了铜催化的苯甲酰胺与马来酰亚胺的氧化偶联反应, 生成含异吲哚酮的琥珀酰亚胺类化合物.其中8-氨基喹啉起辅助作用, 二环己基甲基胺为添加剂, 反应收率为19%~99% (Eq. 67).
|
|
(67) |
综上所述, 马来酰亚胺以其独特的化学结构和生物活性已成为有机化学研究热点之一.马来酰亚胺可以与多种亲核试剂发生Michael加成反应, 其中以碳-Michael加成反应为主, 氮杂、硫杂和磷杂-Michael加成反应还有待进一步研究.马来酰亚胺的氧化偶联和环加成反应, 特别是马来酰亚胺双键的双官能化反应, 在构建新型含氮杂环类化合物发挥着重要作用, 寻找高效、环保、可循环使用的催化剂, 用于合成具有不对称结构的天然产物和药物分子, 并进一步探索其在生物医药和新材料领域的应用是其研究方向, 将在今后将得到进一步的发展.
Lavrard, H.; Rodriguez, F.; Delfourne, E. Bioorg.Med.Chem.Lett.2014, 22, 1961. doi: 10.1002/cmdc.201500025
Chien, S.-C.; Chen, M.-L.; Kuo, H.-T.; Tsai, Y.-C.; Lin, B.-F.; Kuo, Y.-H. J.Agric.Food Chem.2008, 56, 7017. doi: 10.1021/jf801171x
(a) Ho, S.-Y.; Alam, J.; Jeyaraj, D.-A.; Wang, W.; Lin, G.-R.; Ang, S.-H.; Tan, E.-S.-W.; Lee, M.-A.; Ke, Z.; Madan, B.; Virshup, D.-M.; Ding, L.-J.; Manoharan, V.; Chew, Y.-S.; Low, C.-B.; Pendharkar, V.; Sangthongpitag, K.; Hill, J.; Keller, T.-H.; Poulsen, A. J.Med.Chem.2017, 60, 6678.
(b) Alam, J.; Poulsen, A.; Ho, S.-Y.; Wang, W.-L.; Duraiswamy, A. WO 2015094118, 2008[Chem.Abstr.2015, 163, 132777].
Kayser, S.; Levis, M.-J.; Schlenk, R.-F. Expert Rev.Clin.Pharmacol. 2017, 10, 1177. (b) Levis, M. Blood 2017, 129, 3403. doi: 10.1080/17512433.2017.1387051
Shimokawa, J.; Chiyoda, K.; Umihara, H.; Fukuyama, T. Chem.Pharm.Bull.2016, 64, 1239.
(b) Cai, S.-L.; Song, R.; Dong, H.-Q.; Lin, G.-Q.; Sun, X.-W. Org.Lett.2016, 18, 1996.
(a) Daly, M.-J.; Jones, G.-W.; Nicholls, P.-J.; Smith, H.-J.; Rowlands, M.-G.; Bunnett, M.-A. J.Med.Chem.1986, 29, 520.
(b) Sharma, D.-K.; Rajput, V.-S.; Singh, S.; Sharma, R.; Khan, I.A.; Mukherjee, D. ChemistrySelect 2016, 1, 1954.
(a) Driller, K.-M.; Klein, H.; Jackstell, R.; Beller, M. Angew.Chem., Int.Ed.2009, 48, 6041.
(b) Mathur, P.; Joshi, R.-K.; Rai, D.-K.; Jha, B.; Mobin, S.-M. Dalton. Trans.2012, 41, 5045.
Henon, H.; Messaoudi, S.; Hugon, B.; Anizon, F.; Pfeiffer, B.; Prudhomme, M. Tetrahedron 2005, 61, 5599. doi: 10.1016/j.tet.2005.03.101
An, Y.-L.; Shao, Z.-Y.; Cheng, J.; Zhao, S.-Y. Synthesis 2013, 45, 2719. doi: 10.1055/s-0033-1339515
Lanke, V.; Bettadapur, K.-R.; Prabhu, K.-R. Org.Lett.2015, 17, 4662. doi: 10.1021/acs.orglett.5b01809
Muniraj, N.; Prabhu, K.-R. ACS Omega 2017, 2, 4470. doi: 10.1021/acsomega.7b00870
Zhang, Z.; Han, S.; Tang, M.; Ackermann, L.; Li, J. Org.Lett.2017, 19, 3315. doi: 10.1021/acs.orglett.7b01480
Liu, S.-L.; Li, Y.; Guo, J.-R.; Yang, G.-C.; Li, X.-H.; Gong, J.-F.; Song, M.-P. Org.Lett.2017, 19, 4042. doi: 10.1021/acs.orglett.7b01795
Sherikar, M.-S.; Kapanaiah, R.; Lanke, V.; Prabhu, K.-R. Chem.Commun.2018, 54, 11200. doi: 10.1039/C8CC06264A
Pan, C.; Wang, Y.; Wu, C.; Yu, J.-T. Org.Biomol.Chem.2018, 16, 693. doi: 10.1039/C7OB03039H
Koltunov, K.-Y.; Prakash, G.-K.-S.; Rasul, G.; Olah, G.-A. Eur.J.Org.Chem.2006, 4861. doi: 10.1002/ejoc.200600486
Yang, Z.-H.; Chen, Z.-H.; An, Y.-L.; Zhao, S.-Y. RSC Adv.2016, 6, 23438. doi: 10.1039/C6RA00136J
Bettadapur, K.-R.; Lanke, V.; Prabhu, K.-R. Org.Lett.2015, 17, 4658. doi: 10.1021/acs.orglett.5b01810
Mandal, R.; Emayavaramban, B.; Sundararaju, B. Org.Lett.2018, 20, 2835. doi: 10.1021/acs.orglett.8b00761
Li, F.; Zhou, Y.; Yang, H.; Liu, D.; Sun, B.; Zhang, F.-L. Org.Lett.2018, 1, 146. doi: 10.1021/acs.orglett.7b03502
Yu, J.T.; Chen, R.; Jia, H.; Pan, C. J.Org.Chem.2018, 83, 12086. doi: 10.1021/acs.joc.8b02059
Han, S.-H.; Kim, S.; De, U.; Mishra, N.-K.; Park, J.; Sharma, S.; Kwak, J.-H.; Han, S.; Kim, H.-S.; Kim, I.-S. J.Org.Chem.2016, 81, 12416. doi: 10.1021/acs.joc.6b02577
He, Q.; Yamaguchi, T.; Chatani, N. Org.Lett.2017, 19, 4544. doi: 10.1021/acs.orglett.7b02135
Chen, X.; Ren, J.; Xie, H.; Sun, W.; Sun, M.; Wu, B. Org.Chem.Front.2018, 5, 184. doi: 10.1039/C7QO00687J
Bettadapur, K.-R.; Lanke, V.; Prabhu, K.-R. Chem.Commun.2017, 53, 6251. doi: 10.1039/C7CC02392H
Mandal, A.; Sahoo, H.; Dana, S.; Baidya, M. Org.Lett.2017, 19, 4138. doi: 10.1021/acs.orglett.7b01964
Muniraj, N.; Prabhu, K.-R. J.Org.Chem.2017, 82, 6913. doi: 10.1021/acs.joc.7b01094
Qrareya, H.; Ravelli, D.; Fagnoni, M.; Albinia, A. Adv.Synth.Catal.2013, 355, 2891. doi: 10.1002/adsc.201300598
Capaldo, L.; Buzzetti, L.; Merli, D.; Fagnoni, M.; Ravelli, D. J.Org.Chem.2016, 81, 7102. doi: 10.1021/acs.joc.6b00984
Han, S.; Park, J.; Kim, S.; Lee, S.-H.; Sharma, S.; Mishra, N.-K.; Jung, Y.-H.; Kim, I.-S. Org.Lett.2016, 18, 4666. doi: 10.1021/acs.orglett.6b02295
(a) Cunha, S.; Rodovalho, W.; Azevedo, N.R.; Mendonca, M.-D.-O.; Lariucci, C.; Vencato, I. J.Brazil.Chem.Soc. 2002, 13, 629.
(b) Gomez-Torres, E.; Alonso, D.-A.; Gomez-Bengoa, E.; Najera, C. Eur.J.Org.Chem.2013, 2013, 1434.
(c) Noeth, J.; Frankowski, K.-J.; Neuenswander, B.; Aube, J.; Reiser, O. J. Comb. Chem.2008, 10, 456.
Zhao, G.-L.; Xu, Y.-M.; Sunden, H.; Eriksson, L.; Sayah, M.; Cordova, A. Chem.Commun.2007, 7, 734. doi: 10.1039/b614962f
Yu, F.; Jin, Z.; Huang, H.; Ye.T.; Liang, X.; Ye, J.-X. Org.Biomol.Chem. 2010, 8, 4767. doi: 10.1039/c0ob00154f
Yu, F.; Sun, X.; Jin, Z.; Wen, S.; Liang, X.; Ye, J.-X. Chem.Commun.2010, 46, 4589. doi: 10.1039/c0cc00774a
Muramulla, S.; Ma, J.-A.; Zhao, J.-C.-G. Adv.Synth.Catal.2013, 355, 1260. doi: 10.1002/adsc.201300041
Vizcaino-Milla, P.; Sansano, J.-M.; Najera, C.; Fiser, B.; Gomez-Bengoa, E. Synthesis 2015, 47, 2199. doi: 10.1055/s-0034-1380718
Nakashima, K.; Kawada, M.; Hirashima, S.; Kato, M.; Koseki, Y.; Miura, T. Synlett 2015, 26, 1248. doi: 10.1055/s-0034-1380382
Wang, J.-J.; Dong, X.-J.; Wei, W.-T.; Yan, M. Tetrahedron: Asymmetry 2011, 22, 690. doi: 10.1016/j.tetasy.2011.04.009
Bai, J.-F.; Wang, L.-L.; Peng, L.; Guo, Y.-L.; Jia, L.-N.; Tian, F.; He, G.-Y.; Xu, X.-Y.; Wang, L.-X. J.Org.Chem.2012, 77, 2947. doi: 10.1021/jo2025288
Shirakawa, S.; Terao, S.J.; He, R.; Maruoka, K. Chem.Commun.2011, 47, 10557. doi: 10.1039/c1cc14043d
Gomez-Torres, E.; Alonso, D.A.; Gomez-Bengoa, E.; Najera, C. Org.Lett.2011, 13, 6106. doi: 10.1021/ol202599h
Li, X.; Hu, S.; Xi, Z.; Zhang, L.; Luo, S.; Cheng, J.-P. J.Org.Chem.2010, 75, 8697. doi: 10.1021/jo101832e
Liao, Y.-H.; Liu, X.-L.; Wu, Z.-J.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Org.Lett.2010, 12, 2896. doi: 10.1021/ol100822k
Feng, J.; Zhang, Y.; Lin, L.; Yao, Q.; Liu, X.; Feng, X. Chem.Commun.2015, 51, 10554. doi: 10.1039/C5CC03203B
Yarlagadda, S.; Reddy, C.-R.; Ramesh, B.; Kumar, G.-R.; Sridhar, B.; Reddy, B.-V.-S. Eur.J.Org.Chem.2018, 1364. doi: 10.1021/acs.orglett.6b03473
Li, J.; Qiu, S.; Ye, X.; Zhu, B.; Liu, H.; Jiang, Z. J.Org.Chem.2016, 81, 11916. doi: 10.1021/acs.joc.6b02384
Iyer, P.-S.; O'Malley, M.-M.; Lucas, M.-C. Tetrahedron Lett.2007, 48, 4413. doi: 10.1016/j.tetlet.2007.04.084
Shintani, R.; Duan, W.-L.; Nagano, T.; Okada, A.; Hayashi, T. Angew.Chem., Int.Ed.2005, 44, 4611. doi: 10.1002/anie.200501305
Berhal, F.; Wu, Z.; Genet, J.; Ayad, T.; Ratovelomanana-Vidal, V. J.Org.Chem.2011, 76, 6320. doi: 10.1021/jo201187c
Korenaga, T.; Ko, A.; Shimamda, K. J.Org.Chem.2013, 78, 9975. doi: 10.1021/jo4014707
Gopula, B.; Yang, S.-H.; Kuo, T.-S.; Hsieh, J.-C.; Wu, P.-Y.; Henschke, J.-P.; Wu, H.-L. Chem.Eur.J. 2015, 21, 11050. doi: 10.1002/chem.201501059
Kumar, V.; Mitra, R.; Bhattarai, S.; Nair, V.-A. Synth.Commun.2011, 41, 392. doi: 10.1080/00397910903576651
Raycroft, M.-A.-R.; Racine, K.-E.; Rowley, C.-N.; Keillor, J.-W. J.Org.Chem.2018, 83, 11674. doi: 10.1021/acs.joc.8b01638
Han, F.; Yang, L.; Li, Z.; Xia, C.-G. Org.Biomol.Chem.2012, 10, 346. doi: 10.1039/C1OB06346D
An, Y.-L.; Deng, Y.-X.; Zhang, W.; Zhao, S.-Y. Synthesis 2015, 47, 1581. doi: 10.1055/s-0034-1380404
Velchinskaya, E.; Petsushak, B.; Rogal, A. Chem.Heterocycl.Compd.2007, 43, 695. doi: 10.1007/s10593-007-0113-y
Uno, B.-E.; Deibler, K.-K.; Villa, C.; Raghuraman, A.; Scheidt, K.-A. Adv.Synth.Catal.2018, 360, 1719. doi: 10.1002/adsc.201800160
Uno, B.-E.; Dicken, R.-D.; Redfern, L.-R.; Stern, C.-M.; Krzywicki, G.-G.; Scheidt, K.-A. Chem.Sci.2018, 9, 1634. doi: 10.1039/C7SC05205G
Jiang, Z.; Zhang, Y.; Ye, W.; Tan, C.-H. Tetrahedron Lett.2007, 48, 51. doi: 10.1016/j.tetlet.2006.11.019
Balint, E.; Takacs, J.; Drahos, L.; Keglevich, G. Heteroat.Chem.2012, 23, 235. doi: 10.1002/hc.21007
Molleti, N.; Bjornberg, C.; Kong, J.-Y. Org.Biomol.Chem.2016, 14, 10695. doi: 10.1039/C6OB01987K
(a) Bourderioux, A.; Routier, S.; Beneteau, V.; Merour, J.-Y. Tetrahedron 2007, 63, 9465.
(b) Bouissane, L.; Sestelo, J.-P.; Sarandeses, L.A. Org.Lett.2009, 11, 1285.
(c) Awuah, E.; Capretta, A. J.Org.Chem.2011, 76, 3122.
(d) Souffrin, A.; Croix, C.; Viaud-Massuard, M.-C. Eur.J.Org.Chem.2012, 13, 2499
Roshchin, A.-I.; Polunin, E.-V. Mendeleev Commun.2008, 18, 332. doi: 10.1016/j.mencom.2008.11.016
Lim, L.-H.; Zhou, J. Org.Chem.Front.2015, 2, 775. doi: 10.1039/C5QO00015G
Jafarpour, F.; Shamsianpour, M.; Issazadeh, S.; Dorrani, M.; Hazrati, H. Tetrahedron 2017, 73, 1668. doi: 10.1016/j.tet.2017.01.069
Jafarpour, F.; Shamsianpour, M. RSC Adv.2016, 6, 103567. doi: 10.1039/C6RA24420C
Yang, Z.-H.; An, Y.-L.; Chen, Y.; Shao, Z.-Y.; Zhao, S.-Y. Adv.Synth.Catal.2016, 358, 3869. doi: 10.1002/adsc.201600812
Dana, S.; Mandal, A.; Sahoo, H.; Baidya, M. Org.Lett.2017, 19, 1902. doi: 10.1021/acs.orglett.7b00674
An, Y.-L.; Zhang, H.-H.; Yang, Z.-H.; Lin, L.; Zhao, S.-Y. Eur.J.Org.Chem. 2016, 2016, 5405.
Kong, D.-H.; An, Y.-L.; Shao, Z.-Y.; Zhao, S.-Y. J.Chem.Res.2018, 42, 476. doi: 10.3184/174751918X15359929315492
(a) Yang, Z.-H.; Tan, H.-R.; An, Y.-L.; Zhao, Y.-W.; Lin, H.-P.; Zhao, S.-Y. Adv.Synth.Catal.2018, 360, 173.
(b) Yang, Z.-H.; Zhu, J.-N.; Jin, Z.-H.; Zheng, J.; Zhao, S.-Y. Synthesis 2018, 50, 4627.
Yang, Z.-H.; Tan, H.-R.; Zhu, J.-N.; Zheng, J.; Zhao, S.-Y. Adv.Synth.Catal. 2018, 360, 1523. doi: 10.1002/adsc.201701431
(a) Maruoka, H.; Okabe, F.; Koutake, Y.; Fujioka, T.; Yamagata, K. Heterocycles 2009, 77, 617.
(b) Bai, J.-F.; Guo, Y.-L.; Peng, L.; Jia, L.-N.; Xu, X.-Y.; Wang, L.-X. Tetrahedron 2013, 69, 1229.
(b) Petrelli, A.; Samain, E.; Pradeau, S.; Halila, S.; Fort, S. ChemBioChem 2017, 18, 206.
Baker, J.-R.; Tedaldi, L.-M.; Aliev, A.-E. Chem.Commun. 2012, 48, 4725. doi: 10.1039/c2cc31673k
Lin, C.; Zhen, L.; Cheng, Y.; Du, H.-J.; Zhao, H.; Wen, X.; Kong, L.-Y.; Xu, Q.-L.; Sun, H. Org.Lett.2015, 17, 2684. doi: 10.1021/acs.orglett.5b01078
Ding, G.; Wu, X.; Jiang, L.; Zhang, Z.; Xie, X. Org.Lett.2017, 19, 6048. doi: 10.1021/acs.orglett.7b02739
Qiu, S.; Lee, R.; Zhu, B.; Coote, M.-L.; Zhao, X.; Jiang, Z. J.Org.Chem.2016, 81, 8061. doi: 10.1021/acs.joc.6b01451
Nishikawa, Y.; Nakano, S.; Tahira, Y.; Terazawa, K.; Yamazaki, K.; Kitamura, C.; Hara, O. Org.Lett.2016, 18, 2004. doi: 10.1021/acs.orglett.6b00608
Kumar, G.-V.; Govindaraju, M.; Renuka, N.; Khatoon, B.-B.-A.; Mylarappa, B.-N.; Kumar, K.-A. Rasayan J.Chem.2012, 5, 338.
Liu, G.-N.; Luo, R.-H.; Zhou, Y.; Zhang, X.-J.; Li, J.; Yang, L.-M.; Zheng, Y.-T.; Liu, H. Molecules 2016, 21, 1198/1. doi: 10.3390/molecules21091198
Zhang, X.-N.; Chen, G.-Q.; Tang, X.-Y.; Wei, Y.; Shi, M. Angew.Chem., Int.Ed.2014, 53, 10768. doi: 10.1002/anie.201406100
Sharma, S.; Han, S.H.; Oh, Y.; Mishra, N.-K.; Lee, S.-H.; Oh, J.-S.; Kim, I.-S. Org.Lett.2016, 18, 2568. doi: 10.1021/acs.orglett.6b00909
Morita, T.; Akita, M.; Satoh, T.; Kakiuchi, F.; Miura, M. Org.Lett.2016, 18, 4598. doi: 10.1021/acs.orglett.6b02244
Yu, W.; Zhang, W.; Liu, Y.; Liu, Z.; Zhang, Y. Org.Chem.Front.2017, 4, 77. doi: 10.1039/C6QO00479B
Zhu, C.; Falck, J.-R. Chem.Commun.2012, 48, 1674. doi: 10.1039/C2CC16963K
Miura, W.; Hirano, K.; Miura, M. Org.Lett.2015, 17, 4034. doi: 10.1021/acs.orglett.5b01940

图 1 具有重要生物活性的天然产物或生物活性分子的化学结构
Figure 1 Selected examples of biologically relevant maleimide derivatives

图式 1 Lewis酸催化吲哚和吡咯与马来酰亚胺的Michael加成
Scheme 1 Lewis acid catalyzed Michael addition of indoles and pyrroles to maleimides

图式 3 芳基重氮四氟硼酸盐与马来酰亚胺的加成反应
Scheme 3 Addition of arenediazonium tetrafluoroborates and maleimides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



































































 下载:
下载:



