

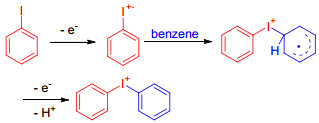
图式 1.
碘苯和苯阳极氧化偶联生成二芳基碘盐的机理
Scheme 1.
Mechanism of the anodic oxidation of iodobenzene and arene coupling

高价态的碘化物被称为高价碘试剂, 其分子结构中没有普通的π键, 而是一个线性的通过碘原子的5p轨道与两个配体的L轨道重叠而形成的三中心四电子的化学键(L—I—L), 称之为“高价键”.由于“高价键”是一种比较弱并且高度极化的化学键, 所以高价碘试剂具有特殊的结构特征以及反应活性, 在有机合成中应用广泛[1], 如烯烃和环丙烷的双官能化[2]、酚类化合物的脱芳构化[3]、氧化偶联[4]、氧化环化[5]、氧化胺基化[6]、卤化[7]、芳基化[8]和重排[9]等; 利用手性高价碘试剂或手性芳基碘化物和氧化剂组成的催化体系, 可高对映选择性地催化不对称反应[10]; 作为单电子氧化剂, 可产生自由基中间体[11].高价碘试剂具有反性能多样、容易制备、反应条件温和、环境友好等优点, 通常代替重金属氧化剂进行反应.然而, 其合成需要使用过量的氧化剂[12], 如m-CPBA, H2O2, Oxone, Selectfluor等, 氧化剂不但价格昂贵, 而且反应后处理棘手.因此, 通过单价碘化物的阳极氧化进行高价碘试剂的电化学合成受到科学家的广泛关注, 这是由于有机电化学合成方法用电流代替了氧化剂和还原剂, 与传统方法相比具有可持续性高、反应条件温和及化学选择性高等优点[13].在过去的30年当中, 出版了很多关于高价碘试剂的书籍和综述[1a~1h], 也见证了高价碘试剂的蓬勃发展, 然而高价碘试剂的电化学合成应用综述尚未报道.本文从二芳基碘盐的电化学合成、含氟高价碘试剂的电化学合成及其介导的氟化反应和双(三氟乙氧基)碘苯的电化学合成及其介导的氧化环化三个方面总结了近年来高价碘试剂的电化学合成方法及其促进的各种化学反应.
二芳基碘盐是一类重要的高价碘试剂, 其分子结构中有两个碳配体和一个抗衡离子, 通常对空气和水稳定.二芳基碘盐既是一类高效的氧化剂, 也是一类高效的亲电芳基化试剂, 能与含氧、硫、氮、碳的亲核试剂发生偶联反应, 还可作为芳炔前体分子参与有机反应[14].其经典的合成方法通常需要两步反应, 首先, 在酸的催化下将芳基碘化物氧化生成三价碘试剂, 随后与芳烃发生偶联反应, 对称和不对称的二芳基碘盐均可通过这一方法来制备[15].近年来, 不对称二芳基碘盐的一锅法和流动化学法的合成也有报道[16]. 1966年, Miller和Hoffmann[17]报道了在芳烃存在下芳基碘的电化学阳极氧化, 一步合成了二芳基碘盐.作者研究了碘苯(1a)的恒电位氧化, 将1a溶解于0.5 mol/L高氯酸锂的乙腈溶液中, 1.6 V (vs. Ag/Ag+)的条件下进行电解, 最终以47%的产率得到4-碘苯苯基高氯酸碘(3a), 利用该方法分别以32%和56%的产率合成了二芳基碘盐3b和3c (Eq. 1).
|
|
(1) |
由于苯在1.9 V以下不能被氧化, 因此, 二芳基碘盐3的形成是来源于芳基碘1的氧化及随后与芳烃2所发生的偶联. 1975年, Wendt小组[18]对这一反应进行了深入地研究, 并提出更合理的反应机理, 他们借助极普法和动力学研究了苯存在下碘苯在金电极表面上的阳极氧化, 结果表明反应生成4-碘苯苯基醋酸碘(5a)和二苯基醋酸碘(5b)的产率几乎相等, 即在金电极上发生的偶联反应没有选择性.然而, 酸催化下的偶联反应, 选择性很高(Eq. 2), 生成碘盐5a和5b的相对反应速率为1:30.两种截然相反的实验结果表明, 电化学生成二芳基碘盐的机理不是经过三价碘中间体的生成及随后与芳烃发生偶联的过程, 而是通过在电极上生成自由基阳离子的中间体, 进而与苯发生偶联, 机理如Scheme 1所示.
|
|
(2) |
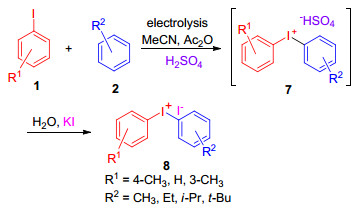
Pletcher小组[19]证明了上述方法的可行性, 并且利用烷基取代的芳基碘和芳烃的阳极氧化/偶联, 合成了系列对称和不对称的二芳基碘盐.方法是以乙酸酐-硫酸为介质, 在以碳毡为阳极、石墨棒为阴极构成的无隔膜电解槽中进行电解, 便以48%~92%的产率得到各种取代的二芳基碘盐化合物.然而, 该方法的局限性在于底物芳基碘和芳烃不能含有易被氧化的官能团.另外, 当芳基碘化物含有吸电子基团时, 增加了其氧化电位, 不利于阳极氧化; 当芳烃中连有吸电子基团时, 降低了芳烃的亲核性, 不利于偶合反应的进行, 因此, 将4-硝基碘苯与甲苯或4-碘甲苯与硝基苯进行反应, 均不能得到二芳基碘盐.当芳基碘和芳烃中连有供电子基团时, 既可以降低芳基碘化合物的氧化电位, 也可提高芳烃的亲核性, 将有利于反应的顺利进行.例如, 将4-甲氧基碘苯(1c)和苯甲醚(2c)在质量分数为2%乙酸酐和5%硫酸的乙酸电解质中电解(Eq. 3), 以42%的产率得到4, 4'-二甲氧基二苯基碘盐6.
|
|
(3) |
Wirth等[20]利用电化学微反应器成功地开发了一种用于合成二芳基碘盐的流动化学方法.微反应器由两个铂电极构成, 电极被一个含有250 μm厚的聚全氟异丙烯箔片隔开.将溶有芳基碘和芳烃底物的乙腈/醋酸酐/硫酸的混合溶液, 用注射泵(80 μL/min)通过反应器的进料口注入, 在恒定电流30 mA下电解, 反应结束后, 将反应混合物收集到碘化钾的溶液中, 产物二芳基碘盐便会沉淀析出, 通过该方法, 以18%~72%的产率合成了系列二芳基碘盐8 (Scheme 2).
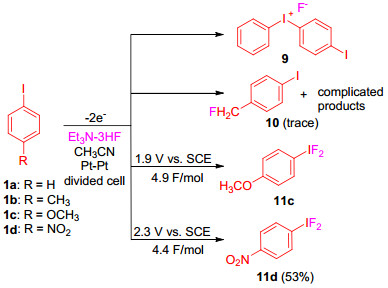
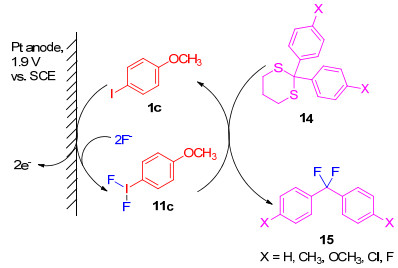
1960年, Schmidt和Meinert[21]首次报道了二氟碘苯的电化学合成, 方法是以氟化银为电解质和氟源, 通过碘苯在乙腈溶液中电解得到. 1976年, Rozhkov[22]指出该实验结果不可重复, 原因是溶液的电导率太低. 1994年, Fuchigami等[23]研究了芳基碘化物的电化学氟化, 结果表明, 碘苯在Et3N-3HF存在下的阳极氧化, 不能生成二氟碘苯, 而是形成了碘盐9.若将碘苯换作4-甲基碘苯时, 同样也没有生成预期的二氟碘苯的产物, 而是形成了甲基氟化产物10.但是在相同条件下, 用4-硝基碘苯作为底物时, 产物4-硝基二氟碘苯(11d)在电解过程中逐渐沉淀析出并通过简单的过滤得以纯化, 产率达到53%, 这是由于硝基官能团吸电子性能强, 致使4-硝基碘苯(1d)在高的电位(2.3 V vs. SCE)下发生阳极氧化; 含有强供电子基团的4-甲氧基碘苯(1c)可以在较低的电位(1.9 V vs.SCE)下发生氧化, 但生成的产物4-甲氧基二氟碘苯(11c)的稳定性较差并且不能分离纯化(Scheme 3).
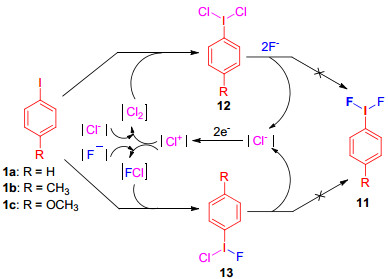
芳基碘化物高的氧化电位, 成为阻碍其发生阳极氧化的主要问题, 理论上, 这可以通过加入介质[24]如氯离子进行间接阳极氧化来解决(Scheme 4).实验表明, 当加入氯离子时, 反应能够顺利进行, 但没有生成二氟碘苯11, 而是生成了氯化氟化的三价碘化合物13[25].高价碘化合物12和13可以促进二硫代缩醛的间接阳极氟化, 氟化既可用槽外法, 也可用槽内法进行, 并且槽内法的氟化反应仅需催化量的芳基碘即可, 例如, 当加入5%的4-甲氧基碘苯时, 就能高产率地生成双氟代产物15 (Scheme 5).
1998年, Hara等[26]发现了在羰基化合物的电化学氧化中, 电解质Et3N-5HF性能要优于Et3N-3HF, 并且以Et3N-5HF为电解质, 合成了高效的氟化试剂4-甲基二氟碘苯(11b), 并用于β-二羰基化合物的间接阳极氟化.方法是将4-碘甲苯(1b)和β-二羰基化合物16按1:1混合, 置于无隔膜电解槽中恒电位(1.5 V vs. Ag/Ag+)电解, 以50%~79%的产率得到α-氟代的β-二羰基化合物17 (Eq. 4).结果表明, α位不含取代基的β-酮酯类化合物的氟化具有较高的选择性, 主要生成单氟化的产物(17a~17d); α位不含取代基的β-二酮类化合物的氟化选择性较差, 除了生成单氟化产物之外, 也能生成双氟化的产物, 例如化合物16f, 除了以50%的产率生成单氟化产物17f外, 也以6%的产率生成α位双氟化的产物.
|
|
(4) |
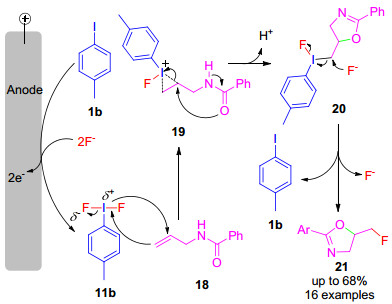
2019年, Waldvogel小组[27]将11b用于烯丙基酰胺(18)的氟化环化反应中, 生成了5-氟甲基-2-噁唑啉类化合物21.反应机理为:首先, 4-碘甲苯(1b)在阳极发生氧化生成11b, 接着受到18双键的亲核进攻生成碘鎓离子中间体19, 随后在羰基的作用下发生开环生成三价碘中间体20, 最后, 在亲核试剂F-的进攻下发生SN2取代生成目标产物21 (Scheme 6).
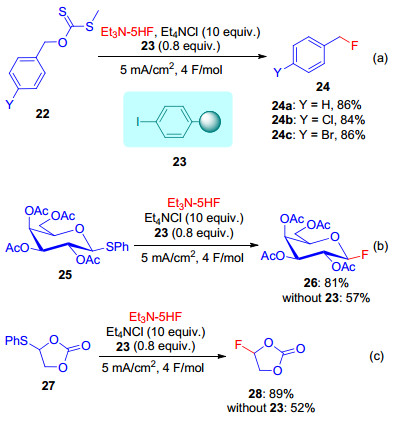
可回收循环利用的聚苯乙烯负载的碘苯和功能化离子液体介导的选择性电催化氟化亦有报道[28]. 2010年, Fuchigami小组[28a]利用聚苯乙烯负载的碘苯23实现了有机硫化合物黄原酸盐22、糖衍生物25、碳酸乙烯酯27的间接阳极氟化, 高产率地生成氟化产物24、26和28.反应是在装有Et3N-5HF和Et4NCl混合液的无隔膜电解槽中恒流(5.0 mA/cm2)条件下电解完成(Scheme 7).聚苯乙烯负载的碘苯23经过滤可从电解液中分离, 并且循环10次之余, 氟化产率仍能高达80%左右.反应机理如Scheme 8所示, 首先, Cl-在阳极发生氧化生成Cl+, 接着与23反应生成PhI+Cl, 随后PhI+Cl捕获F-得到三价碘试剂30, 最后, 30作为有机硫化物的氧化剂进行氧化氟化得到氟化产物.
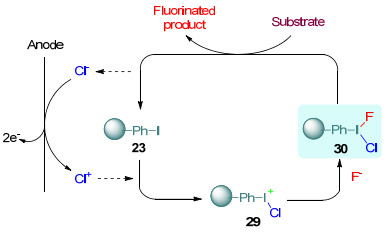
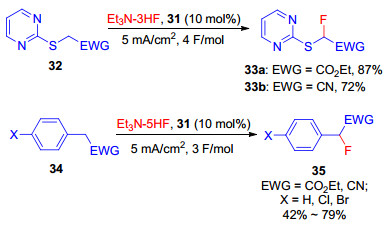
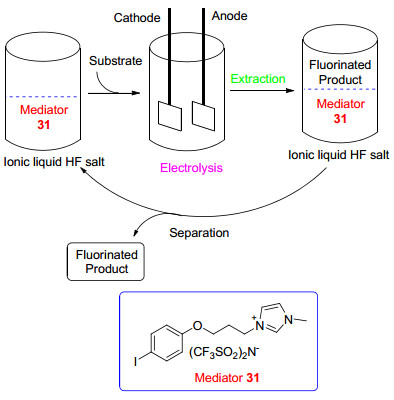
在功能化离子液体31介导的间接阳极氟化中, 反应产物可通过有机溶剂如乙醚等萃取分离, 功能化的离子液体可高效回收循环利用(图 1).伏安法测得31在Et4NF-4HF/MeCN中的氧化电位为E1/2=1.91 V (vs. SCE), 与简单芳基碘化物的氧化电位相当, 因此, 很多底物如2-嘧啶硫化物32以及具有活性苄位的化合物34等都能在装有铂电极无隔膜电解槽中恒电流(5.0 mA/cm2)条件下高效氟化(Scheme 9)[28b].
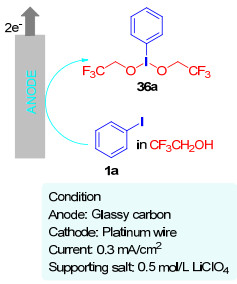
2006年, Nishiyama小组[29a]报道了高价碘试剂双(三氟乙氧基)碘苯36的电化学合成(图 2), 并成功将其用于天然黄原酮衍生物芒果苷的氧化[29b]及各种含杂原子的有机分子和天然产物[29c]的合成之中.
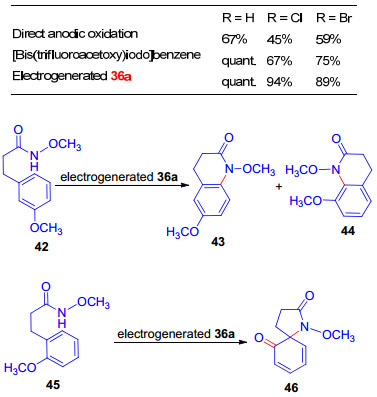
将芳基碘化物阳极氧化生成36, 不经分离, 直接用于酚类化合物的氧化螺环化, 反应是将芳基碘置于由玻璃碳烧杯作为阳极、铂丝作为阴极的无隔膜电解槽中, 恒电流(0.3 mA/cm2)条件下, 用含LiClO4 (0.05 mol/L)的三氟乙醇作为支撑电解质进行电解, 除4-硝基碘苯, 其电解产物能够分离纯化为稳定的固体之外, 其它芳基碘化物电解生成的高价碘试剂只能在溶液中稳定存在.实验表明, 电化学合成的三价碘试剂36, 其氧化性能优于双三氟乙酸碘苯, 在酚类化合物的螺环化反应中, 能以几乎定量的产率(高达97%)生成目标产物38 (Scheme 10)[30a].

此外, 电化学合成的高价碘试剂36还可用于甲氧基酰胺衍生物39的脱芳构化/螺内酰胺化反应, 高产率地得到产物40 (Eq. 5)[29a].实验结果表明, 当甲氧基酰胺衍生物39的取代基R为氢或卤素时, 生成唯一的螺环产物40; 然而, 当取代基R为供电子基团如甲氧基时, 反应没有选择性, 将以大约1:1的比例得到40和41的混合物.另一方面, 苯环中甲氧基的位置也影响反应的结果, 例如当甲氧基和酰胺处于间位时, 生成甲氧基对位亲电进攻的产物喹啉酮衍生物43要多于邻位环化产物44(比例为6.5:1);当甲氧基和酰胺处于邻位时, 将以82%的产率生成唯一的螺环产物46 (Scheme 11).
|
|
(5) |
为提高邻位环化产物的产率, Nishiyama等[30b]将酰胺衍生物苯环中甲氧基的对位用Cl, OAc, Br, CN取代, 结果表明, CN取代的酰胺发生了邻位的环化, 以17%的产率得到预期产物51d, Cl, OAc, Br取代的酰胺则发生对位环化, 高产率地生成喹啉酮类化合物50, 反应机理见Scheme 12.作者又将酰胺衍生物苯环中OCH3换作BnO和AcO, 前者以较高的产率生成50, 而后者生成了复杂的混合物.
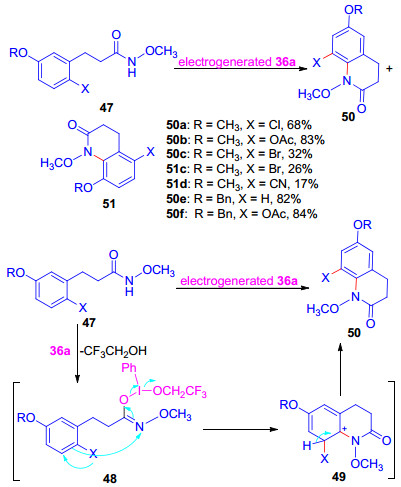
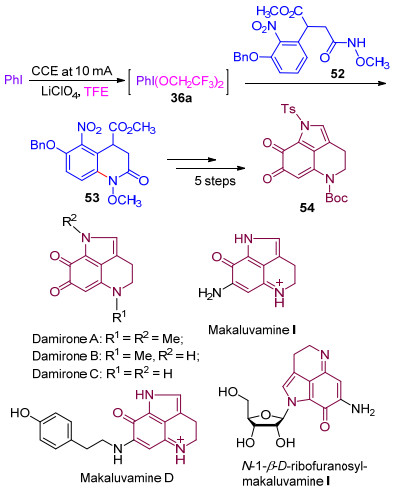
上述喹啉酮类化合物的合成方法曾被成功地用于四氢吡咯亚氨基醌生物碱的合成[30c, 30d].将甲氧基酰胺衍生物52通过电化学合成的三价碘试剂36a的氧化环化, 以62%的产率生成喹啉酮化合物53, 随后, 经过5步反应, 最终以31%的产率得到生物碱Damirone和Makaluvamine的重要中间体α-二酮54 (Scheme 13)[30e].
N-乙酰色胺(55)在电化学合成的36a或醋酸碘苯作用下发生氧化环化, 分别以30%和38%的产率合成了吡咯并吲哚的衍生物56a和56b.以56b为起始原料, 经过三步反应, 将以28%和35%的总收率得到天然产物CPC-1和Debromofrustaminol B (Eq. 6)[30f].
|
|
(6) |
苯乙酰苯胺衍生物57也可在电化学合成的三价碘试剂36a的作用下发生氧化环化反应[30g], 以0~91%的产率生成咔唑类化合物(Eq. 7).然而, 当底物57中的取代基R3为甲氧基时, 仅以42%的产率生成目标产物(58c); 另外, 酰胺官能团对位取代基R4无论是吸电子的硝基还是供电子的甲氧基, 都不能生成目标产物; 其它取代基的底物都能以较好的产率发生环化生成咔唑类化合物58a~58f, 作者还利用该方法合成了抗菌剂甘唑啉.
|
|
(7) |
Möckel等[31]报道了电化学产生的三价碘试剂介导的乙烯基苯甲酸酯60的内酯化反应(Eq. 8), 合成了异色满酮类化合物61, 并将三氟乙氧基引入到产物分子中.反应是在装有0.9 mol/L三氟乙酸的10 mL三氟乙醇溶液和0.8 mol/L高氯酸锂的H-型电解池中, 分别将碘苯和乙烯基苯甲酸酯加入到阳极室, 室温、恒电流密度(7.5 mA/cm2)下电解, 将以较高的产率生成目标产物异色满酮类化合物.实验结果表明, 苯环取代基的电子效应对反应有一定的影响, 例如, 当R1为氢或吸电子基团时, 反应能够顺利进行, 当R1为供电子基团如甲氧基时(61f), 产物稳定性差, 会发生分解; 芳环取代基的位阻效应对反应影响不大.然而双键位取代基的位阻对产率和dr值有一定的影响, 例如, 当取代基为立体位阻大的环己基时(61l), 其dr值高达90:10, 但其产率较低, 仅有45%, 当取代基为立体位阻小的正丙基时(61j), 虽然产率能达到60%, 但是dr值很低, 只有64:36.
|
|
(8) |
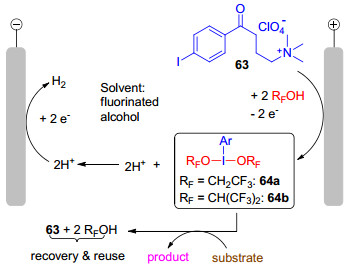
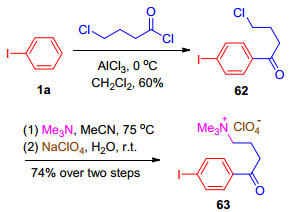
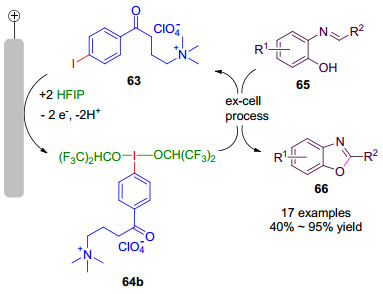
2016年, Francke小组[32a]报道了一种新型可回收循环利用的一价碘/三价碘氧化还原介质, 并成功实现了C—N和C—C键的直接氧化偶联.在一价碘化合物63中, 4-碘苯部分充当了氧化还原活性单元, 烷基铵盐既能提供导电的离子, 又能便于产物和电解质的分离, 还能回收利用.其合成方法是以碘苯1a为原料, 经三步反应, 以74%的总收率得到63 (Scheme 14).一价碘化物铵盐63在氟代醇溶剂如三氟乙醇和1, 1, 1, 3, 3, 3-六氟异丙醇中发生阳极氧化, 生成相应的高价碘试剂64(图 3), 用于各种氧化偶联反应当中, 氟代醇溶剂的选择是基于其具有优良的电化学特性, 如高的导电率和良好的稳定性, 同时还可以稳定产生的三价碘试剂64, 实验表明, 64b在六氟异丙醇中可稳定存在, 然而若将六氟异丙醇移除, 64b便会逐渐分解.
2017年, Francke小组[32b]又将高价碘试剂64b用于2-(亚苄基氨基)苯酚(65)的氧化环化反应之中(Scheme 15), 以40%~95%的产率合成系列苯并噁唑类化合物66, 这种槽外法电化学合成苯并噁唑类化合物的方法具有官能团适应性广泛等优点, 例如含双键、溴、羧酸的底物均适应于该反应.
综述了阳极氧化芳基碘化物制备高价碘试剂的电化学合成方法及其应用.无论是在隔膜还是无隔膜电解槽中, 芳基碘化物的阳极氧化都能在恒电流、恒电位条件下成功实现.利用绿色的电化学技术合成高价碘试剂, 避免了使用化学氧化剂, 减少污染, 节约资源, 并且电化学合成的高价碘试剂, 反应性能与商业化试剂如醋酸碘苯相当, 甚至有时高于醋酸碘苯, 因此, 无论采用槽外法还是槽内法, 都可以促进β-二羰基等化合物的氟化及甲氧基酰胺等化合物的氧化环化, 还可以成功地用于Debromofrustaminol B等天然产物的全合成.另外, 一价碘/三价碘氧化还原介质介导的碳碳键、碳氮等化学键的形成反应已有较广泛的研究, 并证明其性能高效; 同时, 可回收的芳基碘介质也可用于氧化环化反应中.尽管高价碘试剂的电化学合成及应用目前已有深入地研究, 然而仍具有一定的局限性, 这使得该领域的研究将具有更广阔的前景.
(a) "Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis" in Topics in Current Chemistry, Vol. 373, Ed.: Wirth, T., Springer-Verlag, Switzerland, 2016,
(b) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299.
(c) Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650.
(d) Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328.
(e) Duan, Y.-N.; Jiang, S.; Han, Y.-C.; Sun, B.; Zhang, C. Chin. J. Org. Chem. 2016, 36, 1973(in Chinese).
(段亚南, 姜山, 韩永超, 孙博, 张弛, 有机化学, 2016, 36, 1973.)
(f) Zhang, H.; Tang, R.; Wu, J.; Hu, Y. Chemistry 2018, 681(in Chinese).
(张怀远, 唐蓉萍, 伍家卫, 胡雨来, 化学通报, 2018, 681.)
(g) Ma, J.; Chen, L.; Yuan, Z.; Cheng, H. Chin. J. Org. Chem. 2018, 38, 1586(in Chinese).
(马姣丽, 陈立成, 袁中文, 程辉成, 有机化学, 2018, 38, 1586.)
(h) Yan, Y.; Cui, C.; Li, Z. Chin. J. Org. Chem. 2018, 38, 2501(in Chinese).
(闫溢哲, 崔畅, 李政, 有机化学, 2018, 38, 2501.)
(a) Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. J. Am. Chem. Soc. 2017, 139, 4354.
(b) Fujita, M.; Miura, K.; Sugimura, T. Beilstein J. Org. Chem. 2018, 14, 659.
(c) Banik, S. M.; Mennie, K. M.; Jacobsen, E. N. J. Am. Chem. Soc. 2017, 139, 9152.
Smith, D. C.; Vitaku, E.; Njardarson, J. T. Org. Lett. 2017, 19, 3508. doi: 10.1021/acs.orglett.7b01479
Hori, M.; Guo, J.-D.; Yanagi, T.; Nogi, K.; Sasamori, T.; Yorimitsu, H. Angew. Chem., Int. Ed. 2018, 57, 4663. doi: 10.1002/anie.201801132
(a) Zhang, H.; Huang, D.; Wang, K.-H.; Li, J.; Su, Y.; Hu. Y. J. Org. Chem. 2017, 82, 1600.
(b) Chi, Y.; Zhang, W.-X.; Xi, Z. Org. Lett. 2014, 16, 6274.
(c) Chi, Y.; Yan, H.; Zhang, W.-X.; Xi, Z. Chem.-Eur. J. 2017, 23, 757.
(d) Chi, Y.; Yan, H.; Zhang, W.-X.; Xi, Z. Org. Lett. 2017, 19, 2694.
(e) Alazet, S.; Vaillant, F. L.; Nicolai, S.; Courant, T.; Waser, J. Chem.-Eur. J. 2017, 23, 9501.
(f) Colomer, I.; Batchelor-McAuley, C.; Odell, B.; Donohoe, T. J.; Compton, R. G. J. Am. Chem. Soc. 2016, 138, 8855.
(g) Shen, H.; Deng, Q.; Liu, R.; Feng, Y.; Zheng, C.; Xiong, Y. Org. Chem. Front. 2017, 4, 1806.
(h) Wang, Z.; Zhong, J.; Zheng, C.; Fan, R. Org. Chem. Front. 2017, 4, 1005.
Zhang, H.; Huang, D.; Wang, K.-H.; Li, J.; Su, Y.; Hu, Y. Org. Biomol. Chem. 2017, 15, 5337. doi: 10.1039/C7OB00855D
Pluta, R.; Krach, P. E.; Cavallo, L.; Falivene, L.; Rueping, M. ACS Catal. 2018, 8, 2582. doi: 10.1021/acscatal.7b03118
Zhang, H.; Wang, K.-H.; Wang, J.; Su, Y.; Huang, D.; Hu, Y. Org. Biomol. Chem. 2019, 17, 2940. doi: 10.1039/C9OB00236G
Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T. Chem.-Eur. J. 2016, 22, 4030. doi: 10.1002/chem.201504844
Haubenreisser, S.; Wöste, T. H.; Martínez, C.; Ishihara, K.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 413. doi: 10.1002/anie.201507180
Martínez, C.; Bosnidou, A. E.; Allmendinger, S.; Muñiz, K. Chem.- Eur. J. 2016, 22, 9929. doi: 10.1002/chem.201602138
Zhdankin, V. V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, Chichester, UK, 2013, pp. 21~143.
(a) Yang, Q.-L.; Wang, X.-Y.; Lu, J.-Y.; Zhang, L.-P.; Fang, P.; Mei, T.-S. J. Am. Chem. Soc. 2018, 140, 11487.
(b) Xiong, P.; Xu, H.-H.; Song, J.; Xu, H.-C. J. Am. Chem. Soc. 2018, 140, 2460.
(c) Yan, M.; Kawamata, Y.; Baran, P. S. Angew. Chem., Int. Ed. 2018, 57, 4149.
(d) Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 6018.
(e) Elsherbini, M.; Wirth, T. Chem.-Eur. J. 2018, 24, 13399.
(f) Zhang, Z.; Zhang, L.; Cao, Y.; Li, F.; Bai, G.; Liu, G.; Yang, Y.; Mo, F. Org. Lett. 2019, 21, 762.
(g) Chang, X.; Zhang, Q.; Guo, C. Org. Lett. 2019, 21, 10.
(h) Lian, F.; Sun, C.; Xu, K.; Zeng, C. Org. Lett. 2019, 21, 156.
Stuart, D. R. Synlett 2017, 28, 275.
(a) Bielawski, M.; Olofsson, B. Chem. Commun. 2007, 2521.
(b) Bielawski, M.; Zhu, M.; Olofsson, B. Adv. Synth. Catal. 2007, 349, 2610.
(c) Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602.
(d) Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052.
(e) Jalalian, N.; Olofsson, B. Tetrahedron 2010, 66, 5793.
(f) Bouma, M. J.; Olofsson, B. Chem.-Eur. J. 2012, 18, 14242.
(a) Lindstedt, E.; Reitti, M.; Olofsson, B. J. Org. Chem. 2017, 82, 11909.
(b) Laudadio, G.; Gemoets, H. P. L.; Hessel, V.; Noël, T. J. Org. Chem. 2017, 82, 11735.
Miller, L. L.; Hoffmann, A. K. J. Am. Chem. Soc. 1967, 89, 593. doi: 10.1021/ja00979a022
Hoffelner, H.; Lorch, H. W.; Wendt, H. J. Electroanal. Chem. 1975, 66, 183. doi: 10.1016/S0022-0728(75)80002-7
(a) Peacock, M. J.; Pletcher, D. Tetrahedron Lett. 2000, 41, 8995.
(b) Peacock, M. J.; Pletcher, D. J. Electrochem. Soc. 2001, 148, D37.
(a) Folgueiras-Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446.
(b) Folgueiras-Amador, A. A.; Qian, X.-Y.; Xu, H.-C.; Wirth, T. Chem.-Eur. J. 2018, 24, 487.
(c) Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573.
(d) Folgueiras-Amador, A. A.; Wirth, T. J. Flow Chem. 2017, 7, 94.
(e) Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108.
Schmidt, H.; Meinert, H. Angew. Chem. 1960, 72, 109.
Rozhkov, I. N. Russ. Chem. Rev. 1976, 45, 615. doi: 10.1070/RC1976v045n07ABEH002697
Fuchigami, T.; Fujita, T. J. Org. Chem. 1994, 59, 7190. doi: 10.1021/jo00103a003
Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492. doi: 10.1039/c3cs60464k
Fujita, T.; Fuchigami, T. Tetrahedron Lett. 1996, 37, 4725. doi: 10.1016/0040-4039(96)00951-3
Hara, S.; Hatakeyama, T.; Chen, S.-Q.; Ishi-i, K.; Yoshida, M.; Sawaguchi, M.; Fukuhara, T.; Yoneda, N. J. Fluorine Chem. 1998, 87, 189. doi: 10.1016/S0022-1139(97)00144-9
Haupt, J. D.; Berger, M.; Waldvogel, S. R. Org. Lett. 2019, 21, 242. doi: 10.1021/acs.orglett.8b03682
(a) Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Adv. Synth. Catal. 2010, 352, 2757.
(b) Sawamura, T.; Kuribayashi S.; Inagi, S.; Fuchigami, T. Org. Lett. 2010, 12, 644.
(a) Amano, Y.; Nishiyama, S. Tetrahedron Lett. 2006, 47, 6505.
(b) Nishihama, Y.; Amano, Y.; Ogamino, T.; Nishiyama, S. Electrochemistry 2006, 74, 609.
(c) Kajiyama, D.; Saitoh, T.; Nishiyama, S. Electrochemistry 2013, 81, 319.
(a) Amano, Y.; Nishiyama, S. Heterocycles 2008, 75, 1997.
(b) Amano, Y.; Inoue, K.; Nishiyama, S. Synlett 2008, 134.
(c) Izawa, T.; Nishiyama, S.; Yamamura, S. Tetrahedron 1994, 50, 13593.
(d) Faulkner, D. J. Nat. Prod. Rep. 2001, 18, 1.
(e) Inoue, K.; Ishikawa, Y.; Nishiyama, S. Org. Lett. 2010, 12, 436.
(f) Kajiyama, D.; Saitoh, T.; Yamaguchi, S.; Nishiyama, S. Synthesis 2012, 44, 1667.
(g) Kajiyama, D.; Inoue, K.; Ishikawa, Y.; Nishiyama, S. Tetrahedron 2010, 66, 9779.
Möckel, R.; Babaoglu, E.; Hilt, G. Chem.-Eur. J. 2018, 24, 15781. doi: 10.1002/chem.201804152
(a) Broese, T.; Francke, R. Org. Lett. 2016, 18, 5896.
(b) Koleda, O.; Broese, T.; Noetzel, J.; Roemelt, M.; Suna, E.; Francke, R. J. Org. Chem. 2017, 82, 11669.

图式 1 碘苯和苯阳极氧化偶联生成二芳基碘盐的机理
Scheme 1 Mechanism of the anodic oxidation of iodobenzene and arene coupling

图式 3 碘苯衍生物在Et3N-3HF存在下的间接阳极氧化
Scheme 3 Direct anodic oxidation of iodobenzene derivatives in Et3N-3HF

图式 4 氯离子参与的芳基碘的间接阳极氧化
Scheme 4 Indirect anodic oxidation of aryl iodine mediated by chloride ions

图式 6 N-烯丙基酰胺的氟化环化反应机理
Scheme 6 Mechanism for the fluorocyclization of N-allyl- carboxamides

图式 8 聚苯乙烯负载的碘苯介导的间接阳极氟化机理
Scheme 8 Mechanism of indirect anodic fluorination mediated by polystyrene-supported iodobenzene

图式 9 2-嘧啶硫化物和苄位的间接阳极氟化
Scheme 9 Indirect anodic fluorination of 2-pyrimidylsulfides and benzylic positions

图 2 电化学合成高价碘试剂36
Figure 2 Electrochemical generation of the hypervalent iodine reagent 36

图式 10 碘苯衍生物1的阳极氧化及利用36进行酚的氧化
Scheme 10 Anodic oxidation of iodobenzenes 1 and use of 36 in phenolic oxidation

图式 12 化合物47环化重排机理
Scheme 12 Mechanism of the cyclization and rearrangement of compound 47

图式 13 α-二酮54的合成及四氢吡咯亚氨基醌生物碱的结构
Scheme 13 Synthesis of the α-diketone 54 and the stucture of tetrahydropyrroloiminoquinone alkaloids

图 3 一价碘/三价碘氧化还原电对作为电合成的介质体系
Figure 3 Iodine(I)/iodine(III) redox couple as a mediatory system for electrosynthesis

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:






 下载:
下载:






