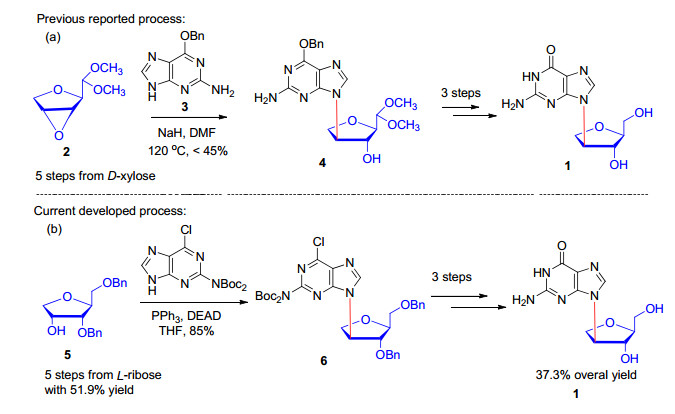
图式 1.
L-鸟嘌呤异核苷的合成策略
Scheme 1.
Synthetic strategies of iso-L-guanosine

核苷是DNA和RNA的基本结构单元, 长期以来对核苷进行结构修饰以期发现新的抗癌和抗病毒药物是新药研发的热点之一[1].迄今为止, 根据美国食品与药品管理局(FDA)统计, 上市的60多个抗肿瘤药物中有9个(15%)是核苷及其类似物, 29个抗病毒药物中有16个(55%)是核苷及其类似物.针对核苷化合物在核苷水解酶作用下易于发生降解的潜在药物代谢问题, Montgomery等[2]在20世纪70年代末首次提出了异核苷的概念(Isomeric nucleoside或者Isonucleoside), 主要是指核苷碱基从核糖呋喃环的1'位移动到糖环的其他位置(例如2'或者3'等).由于碱基位置的移动, 新构建的C—N键在核苷水解酶的作用下非常稳定.在过去的三十年里, 异核苷作为一类新的抗肿瘤和抗病毒药物先导化合物得到了广泛的研究, 药物化学家合成出了大量的异核苷化合物, 主要的研究小组有Nair[3]、Matsuda[4]、张礼和[5]和我们小组[6]等, 可惜的是目前仍未筛选出能进入临床使用的药物分子.
2013年, 杨振军小组首次研究发现异核苷(D-或者L-胸腺嘧啶)嵌杂的寡聚核苷酸在干扰核糖核酸(siRNA)[7]和适配体(Aptamer)[8]中表现出优良的生物学效应; 进一步的研究表明异核苷修饰的血红蛋白结合适配体(TBA)能够增强与血红蛋白的亲和力, 从而增强凝血效果[9], 另一方面异核苷修饰的适配体能够显著增加对杂核核糖蛋白A1 (heterogeneous nuclear ribonucleoprotein A1, hnRNP A1)的亲和力, 从而具有潜在的抑制肿瘤形成作用[10], 为异核苷的研究和应用开辟了新的思路.为了研究L-鸟嘌呤异核苷嵌杂的寡聚核苷酸的生物学性质, 首先需要在克级规模对L-鸟嘌呤异核苷进行合成.在文献中, 张礼和小组报道[11]了以D-木糖为起始原料, 首先经过5步反应构建关键中间体环氧化合物2, 然后通过鸟嘌呤碱基3对环氧化合物2的开环反应构建异核苷C—N键(Scheme 1a).在合成中, 由于鸟嘌呤碱基3在有机溶剂中的溶解度非常差, 同时由于化合物2中环氧结构稳定很高, 在关键步骤中需要使用N, N-二甲基甲酰胺(DMF)作为溶剂, 在120 ℃加热的条件下进行核苷键的构建, 反应条件苛刻, 异核苷中间体4的产率低于45%.该路线不可避免地产生N-9取代和3'取代副产物, 分离困难, 同时未见L-鸟嘌呤异核苷1的详细表征数据报道.本文将报道以L-核糖为起始原料进行L-鸟嘌呤异核苷1的合成工艺研究(Scheme 1b).
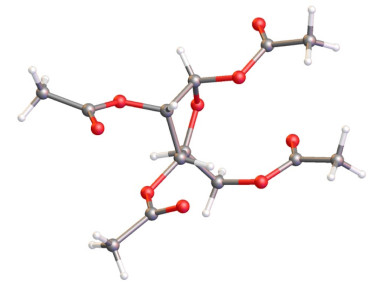
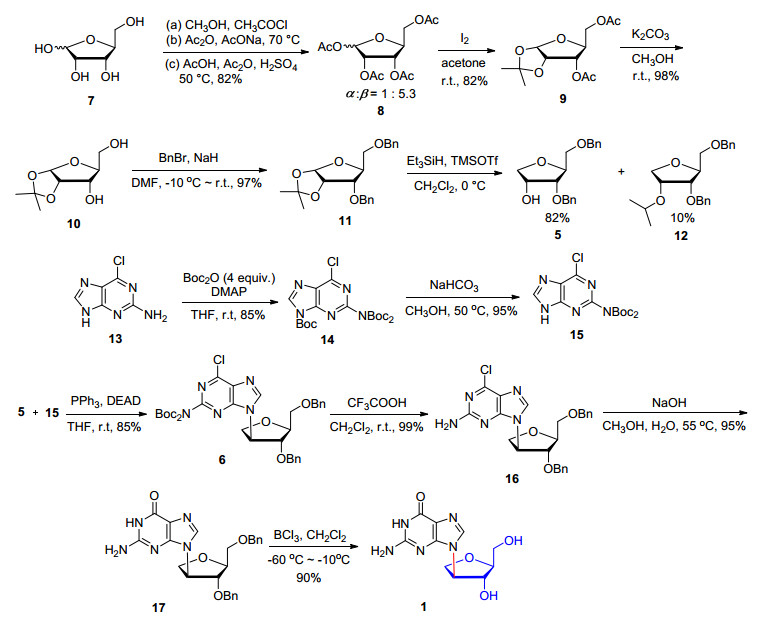
首先在1%的氯化氢甲醇溶液中将L-核糖高产率转化为L-呋喃核糖甲苷, 减压除去溶剂后, 产物不经分离纯化, 直接在醋酸钠和醋酐体系中加热回流, 将剩余羟基进行乙酰化, 然后在醋酐和醋酸混合溶剂中将甲苷在浓硫酸催化下转化为相应的乙酰酯, 经过柱层析分离纯化得到四乙酰化-L-呋喃核糖8[12](α:β=1:5.3), 三步总收率为82.0%, 同时首次得到了β构型的单晶衍射晶体结构(如Scheme 2所示)[13], 该方法步骤操作简便、重现性好, 可以在100克级规模上进行.然后四乙酰呋喃核糖8在干燥的丙酮溶剂中, 以单质碘(I2)为催化剂, 以82%产率得到丙叉保护的核糖9[14], 之后在甲醇溶液中, 以碳酸钾为碱, 几乎定量地脱除乙酰基得到丙叉保护的核糖10.接着以DMF为溶剂, 以氢化钠(NaH)为碱, 用苄溴保护核糖的3, 5位羟基, 同样几乎定量得到糖11.最后用三氟甲磺酸三甲基硅酯(TMSOTf)为路易斯酸, 用三乙基硅氢(Et3SiH)还原原位形成的端头碳正离子, 在得到关键中体5的同时, 不可避免地会产生异丙醚产物12[15].该异丙醚副产物12为热力学产物.通过条件优化, 发现降低反应的浓度(0.09 mmol/mL)和降低反应温度, 可以有效降低副产物的形成, 最后以82%产率得到关键糖中间体5.
在得到关键糖中间体5后, 我们尝试将2位羟基转换为不同的离去基团[16], 例如甲磺酸酯、对甲苯磺酸酯和三氟甲磺酸酯等, 然后与碱基13进行取代反应合成目标异核苷化合物, 经过长时间的尝试, 发现底物容易发生消除反应, 产率很低(<20%), 同时伴随着N-7副产物的形成, 分离非常困难, 因此只能寻求别的合成途径.
近些年来, Mitsunobu反应在核苷化学中得到了广泛的应用[17], 该反应条件温和, 对手性羟基立体专一性发生构型的翻转, 因此尝试着用该反应来解决我们面临的问题.由于6-氯鸟嘌呤在有机溶剂中的溶解度特别差, 同时2-氨基由于带有活泼氢, 可能会和糖中间体5中的2位羟基发生Mitsunobu反应, 因此首先需要将2位氨基进行保护.选择近年来在核苷碱基保护中得到广泛应用的叔丁氧羰基(Boc)来实现目的.通过对文献报道[18]的条件进行摸索和改进, 首先在4 equiv. Boc2O作用下, 对环外氨基和N-9氨基进行全保护, 然后将得到的产物溶解在甲醇中, 用弱碱碳酸氢钠溶液水解去除N-9氨基上的Boc基团, 从而以81%的总收率得到环外氨基双Boc保护的碱基15, 改进后的步骤在100克规模上顺利重复来合成碱基15.
接着尝试用Mitsunobu反应来合成异核苷6, 经过条件优化, 以四氢呋喃(THF)为溶剂, 在三苯基膦(PPh3)和偶氮二甲酸二乙酯(DEAD)作用下, 碱基15和糖中间体5可以顺利发生缩合反应得到异核苷6, 同时HPLC分析未发现碱基N-7位偶联副产物的生成.然后尝试在强碱的作用下, 一步水解异核苷6的6位氯原子和Boc保护基团, 但是反应产物复杂, 很难进行分离.之后, 改变反应策略, 首先在三氟乙酸作用下几乎定量水解脱除Boc保护基团, 再在12.5 mol/L氢氧化钠溶液中回流水解去除6位的氯原子, 顺利以94%产率得到异核苷17.接着, 首先尝试用Pd/C加氢的方法脱除苄基, 但是由于氨基的存在, 反应非常慢, 不能反应完全.最后, 采用二氯甲烷(DCM)为溶剂, 低温下(-60 ℃)用三氯化硼(BCl3)可以顺利脱除苄基[19], 以90%产率得到目标分子L-鸟嘌呤异核苷1, HPLC分析显示纯度大于98.5%.文献报道L-鸟嘌呤异核苷1数据表征不够详细, 本文所有报道合成中间体结构都经严格的光谱数据确证.
以L-核糖为原料, 以Mitsunobu反应进行碱基15和糖中间体5的缩合反应合成异核苷6为关键步骤, 经过9步反应, 以37.3%的总产率完成了L-鸟嘌呤异核苷1的全合成研究.研究表明Mitsunobu反应可以显著提高核苷键生成的产率和避免副反应的发生.该全合成路线反应条件温和、反应中间体容易纯化、总产率高、重现性好, 可以作为鸟嘌呤异核苷的通用合成路线. L-鸟嘌呤异核苷1经过标准的三步反应就可以得到DNA合成单体亚磷酰胺, 嵌入寡聚核苷酸中进行相关的生物学性质研究正在进行中.
Bruker AV400型核磁共振仪(德国Bruker公司); AB SCIEX Triple TOF 4600型高分辨质谱仪(美国AB SCIEX公司); Autopol IV-T A21200-T型全自动数字式旋光仪(美国Rudolph公司); M-560熔点测定仪(BUCHI公司).无水二氯甲烷(CH2Cl2)相应的分析纯试剂加入氢化钙回流3 h, 使用前蒸出; 无水四氢呋喃(THF)为分析纯, 加入四氢铝锂回流3 h, 使用前蒸出; 无水甲醇(CH3OH)为分析纯, 加入镁和碘回流3 h, 蒸出后密封保存.所用常规试剂均购自北京伊诺凯科技有限公司.
冰浴条件下, 向无水甲醇(70 mL)中缓慢加入乙酰氯(5 mL).搅拌30 min后, 分批加入化合物L-核糖7 (5.0 g, 33.3 mmol), 加完后继续反应2 h.薄层色谱(TLC)检测显示反应完全后, 用氢氧化钠甲醇溶液(0.1 g/m)将反应液调至中性, 反应液减压浓缩得到淡黄色油状化合物.氩气保护下, 向该淡黄色油状化合物加入醋酸酐(17.9 g, 175.5 mmol), 无水乙酸钠(14.3 g, 174.4 mmol), 70 ℃反应4 h. TLC检测显示反应完全后, 将反应液倒入冰水(200 mL)中, 用二氯甲烷(200 mL×3)萃取, 有机相分别用水(100 mL×3)、饱和碳酸氢钠溶液(150 mL×3)、饱和食盐水(100 mL×2)萃取和无水硫酸镁干燥.过滤液减压浓缩得到黄色油状物, 将其溶于冰醋酸(100 mL), 加入醋酸酐(5.0 g, 49.0 mmol), 冰浴条件下加入2~3滴浓硫酸, 反应体系升温至50 ℃反应4 h, TLC检测显示反应完全后, 将反应液倒入冰水(200 mL)中, 加入二氯甲烷(200 mL×3)溶解, 有机相分别用水(100 mL×3)、饱和碳酸氢钠溶液(150 mL×3)、饱和食盐水(100 mL×2)洗涤、无水硫酸镁干燥.过滤液减压浓缩得粗产物, 通过硅胶柱层析分离纯化得白色固体8β和8α (10.0 g, α:β=1:5.3), 产率为82%. 8β: m.p. 81~82 ℃ (文献值[12(b)]: 80~83 ℃); [α]25D+13.0 (c 0.10, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 6.02 (s, 1H), 5.24 (d, J=1.9 Hz, 2H), 4.32 (s, 1H), 4.29 (d, J=3.1 Hz, 1H), 4.06 (q, J=5.5 Hz, 1H), 2.09 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.04 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.5, 170.0, 169.8, 169.3, 98.1, 79.5, 74.0, 70.5, 63.5, 21.3, 21.0, 20.8, 20.7; HRMS calcd for C13H18O9Na [M+Na]+ 341.0843, found 341.0840. 8α:油状物, [α]D25-42.6 (c 0.05, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 6.31 (d, J=4.5 Hz, 1H), 5.27~5.15 (m, 1H), 4.38 (q, J=3.5 Hz, 1H), 4.24 (dd, J=12.0, 3.0 Hz, 1H), 4.13 (dd, J=12.0, 3.0 Hz, 1H), 2.08 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.5, 170.2, 169.9, 169.6, 94.1, 81.1, 69.9, 69.7, 63.5, 21.2, 21.0, 20.8, 20.5; HRMS calcd for C13H18O9Na [M+Na]+ 341.0843, found 341.0841.
氩气保护下将化合物8β (5.0 g, 15.7 mmol)溶于丙酮, 加入催化量的碘(0.2 g, 0.8 mmol).反应体系室温下反应3.5 h, TLC检测显示反应完全后, 加入饱和硫代硫酸钠水溶液(25 mL)淬灭反应.反应液减压浓缩蒸去丙酮溶剂, 浓缩物加入水(100 mL)稀释, 用二氯甲烷(100 mL×3)萃取, 分别用水(100 mL×3)、饱和碳酸氢钠溶液(100 mL×3)、饱和食盐水(100 mL×2)洗涤、无水硫酸镁干燥.过滤液减压浓缩得粗产物, 通过进一步硅胶柱层析分离纯化得黄色油状物9 (3.5 g), 产率为82%. [α]D25-125.9 (c 0.11, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 5.81 (d, J=3.6 Hz, 1H), 4.77 (t, J=4.2 Hz, 1H), 4.76~4.61 (m, 1H), 4.24 (d, J=13.0 Hz, 1H), 4.18 (q, J=5.6 Hz, 1H), 4.05 (dd, J=12.2, 5.4 Hz, 1H), 2.06 (s, 3H), 2.03 (s, 3H), 1.45 (s, 3H), 1.27 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 170.6, 170.2, 112.7, 104.4, 77.3, 75.6, 72.4, 62.9, 26.9, 26.8, 21.0, 20.9; HRMS calcd for C12H18O7Na [M+Na]+ 297.0944, found 297.0942.
将化合物9 (5.0 g, 18.2 mmol)溶于甲醇(40 mL)中, 加入无水碳酸钾(3.0 g, 21.7 mmol), 室温下搅拌2 h. TLC检测显示反应完全后, 反应液减压旋干, 粗产物通过柱层析分离纯化得白色固体10 (3.4 g), 产率为98%. m.p. 87~88 ℃(文献值[20]: 88~89 ℃); [α]D25-68.5 (c 0.10, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 5.65 (d, J=3.6 Hz, 1H), 5.00 (d, J=6.5 Hz, 1H), 4.65 (t, J=5.7 Hz, 1H), 4.43 (t, J=3.9 Hz, 1H), 3.73~3.66 (m, 2H), 3.66~3.59 (m, 1H), 3.42~3.35 (m, 1H), 1.43 (s, 3H), 1.26 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 111.6, 103.8, 80.8, 79.6, 71.0, 60.7, 27.1, 26.9; HRMS calcd for C8H14O5Na [M+Na]+ 213.0733, found 213.0739
将化合物10 (5.0 g, 26.3 mmol)溶于无水DMF (50 mL)中, 在-10 ℃低温下, 分批加入60% NaH (4.2 g, 175 mmol).氩气保护下, 反应液搅拌1 h后, 缓慢滴加苄溴(18.0 g, 105.9 mmol).滴加完毕, 继续反应1 h后, 升温至室温反应4 h. TLC检测显示反应完全后, 加入冰水(10 mL)淬灭反应, 将反应液倒入冰水(100 mL)中, 二氯甲烷(200 mL×3)萃取, 有机相分别用水(100 mL×3)、饱和碳酸氢钠溶液(150 mL×3)、饱和食盐水(100 mL×2)萃取, 无水硫酸镁干燥.过滤液减压浓缩得到粗产物, 通过硅胶柱层析分离纯化得淡黄色油状化合物11 9.5 g, 产率为97%. [α]D25-118.5 (c 0.10, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 7.7~7.16 (m, 10H), 5.73 (d, J=3.5 Hz, 1H), 4.71 (t, J=3.8 Hz, 1H), 4.64 (d, J=11.9 Hz, 1H), 4.55~4.39 (m, 3H), 3.99 (dd, J=8.6, 4.7 Hz, 1H), 3.74 (dd, J=9.1, 4.2 Hz, 1H), 3.65 (d, J=11.0 Hz, 1H), 3.48 (dd, J=11.2, 5.0 Hz, 1H), 1.45 (s, 3H), 1.29 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 138.7, 138.4, 128.7, 128.1, 128.0, 127.9, 112.2, 104.3, 77.9, 76.9, 72.8, 71.4, 69.3, 27.1, 27.0; HRMS calcd for C22H26O5Na [M+Na]+ 393.1673, found 393.1673.
冰浴条件下, 将化合物11 (5.0 g, 13.5 mmol)溶于无水二氯甲烷(150 mL)中, 加入三乙基硅烷(7.9 g, 68.0 mmol)后, 缓慢滴加TMSOTf (4.6 g, 20.7 mmol).滴加完毕, 反应体系自然升温至室温反应3 h. TLC检测显示反应完全后, 加入冰水(10 mL)淬灭反应, 将反应液倒入冰水(100 mL)中, 二氯甲烷(100 mL×3)萃取, 有机相分别用水(100 mL×3)、饱和碳酸氢钠溶液(150 mL×3)、饱和食盐水(100 mL×2)萃取, 无水硫酸镁干燥.过滤液减压浓缩得粗产物, 通过硅胶柱层析分离纯化得黄色油状化合物5 3.5 g和淡黄色油状化合物12 0.5 g.
化合物5:产率为82%. [α]D25-92.9 (c 0.08, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 7.42~7.22 (m, 10H), 4.88 (d, J=5.1 Hz, 1H), 4.69 (d, J=12.0 Hz, 1H), 4.57~4.40 (m, 3H), 4.22 (d, J=3.8 Hz, 1H), 3.98~3.82 (m, 2H), 3.80~3.68 (m, 1H), 3.64~3.57 (m, 1H), 3.57~3.50 (m, 1H), 3.49~3.40 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ: 138.9, 138.9, 128.6, 128.6, 128.0, 127.8, 80.0, 79.8, 73.2, 72.7, 71.4, 71.0, 69.3; HRMS calcd for C19H22O4Na [M+Na]+ 337.1410, found 337.1415.
1-脱氧-2-O-异丙基-3, 5-O-二苄基-L-核糖(12):产率为10%. [α]D25-75.0 (c 0.11, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 7.43~7.18 (m, 10H), 4.60 (d, J=11.8 Hz, 1H), 4.47~4.50 (m, 3H), 4.11 (d, J=4.7 Hz, 1H), 3.97~3.91 (m, 1H), 3.91~3.81 (m, 2H), 3.71~3.60 (m, 2H), 3.53 (dd, J=10.7, 3.4 Hz, 1H), 3.45 (dd, J=10.7, 5.0 Hz, 1H), 1.11 (t, J=6.5 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ: 138.9, 138.8, 128.7, 128.6, 128.1, 127.9, 127.8, 80.5, 78.9, 74.9, 72.8, 71.5, 71.1, 70.9, 70.7, 23.0, 22.7; HRMS calcd for C22H28O4Na [M+Na]+ 379.1879, found 379.1873.
氩气保护下, 将6-氯鸟嘌呤13 (8.5 g, 50.0 mmol)和DMAP (0.6 g, 4.9 mmol)溶于无水四氢呋喃(100 mL)中, 冰浴下缓慢滴加Boc2O (43.6 g, 0.2 mol).滴加完毕, 反应体系升温至室温继续反应8 h. TLC检测显示反应完全后, 反应液减压旋干得粗产物, 通过硅胶柱层析分离纯化得白色固体14 (20.0 g), 产率为85%. m.p. 50~52 ℃(文献值[18d]: 50~51 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 9.01 (s, 1H), 1.62 (s, 9H), 1.42 (s, 18H); 13C NMR (101 MHz, DMSO-d6) δ: 152.8, 152.3, 150.7, 150.3, 147.5, 145.5, 130.7, 87.2, 84.0, 27.9, 27.8; HRMS calcd for C20H28ClN5O6Na [M+Na]+ 492.1620, found 492.1623.
将化合物14 (4.0 g, 8.5 mmol)溶于甲醇(100 mL)中, 加入饱和碳酸氢钠溶液(25 mL), 50 ℃反应1.5 h. TLC检测显示完全反应后, 低温旋干甲醇溶剂, 用5 mol/L盐酸调至中性, 析出白色固体, 过滤得化合物15 3.0 g, 产率为95%. m.p. 91~93 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.24 (s, 1H), 1.38 (s, 18H); 13C NMR (101 MHz, DMSO-d6) δ: 162.6, 156.1, 151.6, 148.2, 145.0, 131.1, 82.7, 28.0; HRMS calcd for C15H20ClN5ONa [M+Na]+ 392.1096, found 392.1083.
氩气保护下, 将糖5 (5.0 g, 15.9 mmol)、碱基15 (6.5 g, 17.6 mmol)、三苯基膦(8.3 g, 31.6 mmol)溶于无水四氢呋喃中.冰浴下反应液中缓慢滴加偶氮二甲酸二乙酯(5.54 g, 31.8 mmol).滴加完毕, 反应体系升温至室温, 继续反应10 h. TLC检测显示反应完全后, 减压旋干得到粗产物, 通过硅胶柱层析分离纯化得黄色油状物6 (9.0 g), 产率为85%. [α]D25-69.2 (c 0.1, CH3OH); 1H NMR (400 MHz, CDCl3) δ: 8.37 (s, 1H), 7.31~7.03 (m, 10H), 5.16 (s, 1H), 4.74 (d, J=11.8 Hz, 1H), 4.48~4.29 (m, 3H), 4.23 (d, J=3.0 Hz, 2H), 4.07~3.94 (m, 2H), 3.63 (dd, J=10.7, 2.7 Hz, 1H), 3.44 (dd, J=10.7, 3.6 Hz, 1H), 1.39 (s, 18H); 13C NMR (101 MHz, CDCl3) δ: 152.3, 151.9, 151.3, 151.0, 144.9, 137.3, 137.1, 128.6, 128.6, 128.2, 128.1, 127.9, 85.6, 84.9, 83.9, 73.6, 72.8, 71.7, 68.5, 60.2, 28.1; HRMS calcd for C34H40ClN5O7Na [M+Na]+ 688.2508, found 688.2510.
将化合物6 (5.0 g, 7.5 mmol)溶于二氯甲烷(40 mL)中, 缓慢滴加三氟乙酸(10 mL, 131.6 mmol), 室温下反应3 h. TLC检测显示反应完全后, 加入冰水(10 mL)淬灭反应, 将反应液倒入冰水(100 mL)中, 二氯甲烷(100 mL×3)萃取, 有机相分别用水(100 mL×3)、1 mol/L氢氧化钠溶液(150 mL×3)、饱和食盐水(100 mL×2)萃取, 无水硫酸镁干燥.过滤液减压浓缩得到粗产物, 通过硅胶柱层析分离纯化得淡黄色油状物16 3.5 g, 产率为99%. [α]D25-138.0 (c 0.09, CH3OH); 1H NMR (400 MHz, DMSO-d6) δ: 8.02 (d, J=8.1 Hz, 1H), 7.45~7.21 (m, 8H), 7.16 (d, J=8.0 Hz, 2H), 6.98 (s, 2H), 4.95 (s, 1H), 4.67 (d, J=12.2 Hz, 2H), 4.44 (s, 2H), 4.34 (d, J=12.0 Hz, 1H), 4.18~4.10 (m, 2H), 3.98 (d, J=4.0 Hz, 1H), 3.57 (d, J=4.0 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 160.1, 154.2, 149.9, 141.2, 138.5, 138.2, 128.6, 128.1, 128.0, 127.9, 127.8, 123.8, 84.8, 83.9, 72.8, 71.8, 70.3, 69.8, 59.9; HRMS calcd for C24H24ClN5O3Na [M+Na]+ 488.1460, found 488.1459.
将化合物16 (5.0 g, 10.7 mmol)溶于甲醇(80 mL)中, 加入氢氧化钠水溶液(20 mL, 12.5 mol/L), 55 ℃加热反应3 h. TLC检测显示反应完全后, 减压浓缩除去甲醇, 将反应液倒入冰水(100 mL)中, 加入二氯甲烷(100 mL×3)溶解, 有机相分别用水(100 mL×3)、饱和食盐水(100 mL×2)萃取, 无水硫酸镁干燥.过滤液减压浓缩得到粗产物, 通过硅胶柱层析分离纯化得到白色固体17 4.8 g, 产率为95%. [α]D25-97.6 (c 0.09, CH3OH); m.p. 212~214 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.65 (s, 1H), 7.62 (s, 1H), 7.22~7.21 (m, 10H), 6.48 (s, 2H), 4.83 (s, 1H), 4.65 (d, J=11.4 Hz, 1H), 4.53 (d, J=11.8 Hz, 1H), 4.42 (s, 2H), 4.18 (d, J=7.3 Hz, 1H), 4.09 (d, J=4.9 Hz, 1H), 4.02 (s, 1H), 3.93 (s, 1H), 3.53 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 157.3, 154.1, 151.4, 138.6, 138.2, 135.4, 128.7, 128.2, 128.1, 127.9, 116.9, 85.4, 83.8, 72.8, 71.7, 70.9, 70.0, 59.6; HRMS calcd for C24H25N5O4Na [M+Na]+ 470.1799, found 470.1798.
氩气保护下, 将化合物17 (1.0 g, 2.3 mmol)溶于10 mL无水二氯甲烷中.在-60 ℃低温下, 缓慢加入浓度1 mol/L的三氯化硼的二氯甲烷溶液(8.9 mL, 8.9 mmol).反应3 h后, 将温度升至-10 ℃继续反应6 h. TLC检测显示反应完全后, 向反应液加入2 mol/L氢氧化钠溶液(5 mL), 室温搅拌5 h.反应液减压旋干, 粗产物通过硅胶柱层析分离纯化得0.5 g白色固体1, 产率为90%. [α]D25-12.8 (c 0.03, CH3COOH); m.p. 276~278 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.81 (brs, 1H, NH), 7.76 (s, 1H, H-8), 6.64 (s, 2H, NH2), 5.76 (s, 1H, OH), 4.94 (s, 1H, OH), 4.65 (d, J=4.4 Hz, 1H, H-2'), 4.28 (s, 1H, H-4'), 4.21~4.02 (m, 1H, H-1'), 3.92 (dd, J=9.2, 4.5 Hz, 1H, H-1'), 3.74~3.49 (m, 3H, H-3', H-5'); 13C NMR (101 MHz, DMSO-d6) δ: 157.5 (C-6), 154.2 (C-2), 151.6 (C-4), 135.7 (CH-8), 116.8 (C-5), 86.4 (CH-3'), 76.4 (CH-4'), 70.5 (CH2-1'), 61.9 (CH-2'), 61.3 (CH2-5'); HRMS calcd for C10H13N5O4Na [M+Na]+ 290.0860, found 290.0865.
辅助材料(Supporting Information) 化合物1和7~17的核磁共振谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Yates, M. K.; Seley-Radtke, K. L. Antiviral Res. 2019, 162, 5.
(b) Seley-Radtke, K. L.; Yates, M. K. Antiviral Res. 2018, 154, 66.
(c) Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447.
(d) Zhou, X.-X.; Littler, E. Curr. Top. Med. Chem. 2006, 6, 851.
(e) Fan, X.-S.; Zhang, X.-Y.; Wang, X.; Qu, G.-R. Chin. J. Org. Chem. 2008, 28, 1888 (in Chinese).
(范学森, 张新迎, 王霞, 渠桂荣, 有机化学, 2008, 28, 1888.)
(a) Montgomery, J. A.; Clayton, S. D.; Thomas, H. J. J. Org. Chem. 1975, 40, 1923.
(b) Montgomery, J. A.; Thomas, H. J. J. Org. Chem. 1978, 43, 541.
(a) Nair, V.; Jahnke, T. S. Antimicrob. Agents Chemother. 1995, 39, 1017.
(b) Nair, V.; Piotrowska, D. G.; Okello, M.; Vadakkan, J. Nucleosides Nucleotides Nucleic Acids 2007, 26, 687.
(c) Chun, B. K.; Vadakkan, J. J.; Nair, V. Nucleosides Nucleotides Nucleic Acids 2005, 24, 725.
(a) Ogino, T.; Sato, K.; Matsuda, A. ChemBioChem 2010, 11, 2597.
(b) Kira, T.; Kakefuda, A.; Shuto, S.; Matsuda, A.; Baba, M.; Shigeta, S. Nucleosides Nucleotides Nucleic Acids 1995, 14, 571.
(c)Yoshimura, Y.; Asami, K.; Matsui, H.; Tanaka, H.; Takahata, H. Org. Lett. 2006, 8, 6015.
(a) Yu, H. W.; Zhang, H. Y.; Yang, Z. J.; Min, J. M.; Ma, L. T.; Zhang, L. H. Pure App. Chem. 1998, 70, 435.
(b) Tian, X. B.; Min, J. M.; Zhang, L. H. Tetrahedron: Asymmetry 2000, 11, 1877.
(c) Yu, H. W.; Zhang, L. R.; Zhou, J. C.; Ma, L. T.; Zhang, L. H. Bioorg. Med. Chem. 1996, 4, 609.
(a) Song, Y.; Yang, R.; Ding, H.; Sun, Q.; Xiao, Q.; Ju, Y. Synthesis-Stuttgart 2011, 1213.
(b) Sun, Z. D.; Zhu, Y. L.; Huang, H. Y.; Song, X. R.; Xiao, Q. Chin. J. Org. Chem. 2016, 36, 2729 (in Chinsese).
(孙志东, 朱云龙, 黄海洋, 宋贤荣, 肖强, 有机化学, 2016, 36, 2729.)
Huang, Y.; Chen, Z.; Chen, Y.; Zhang, H.; Zhang, Y.; Zhao, Y.; Yang, Z.; Zhang, L. Bioconjugate Chem. 2013, 24, 951. doi: 10.1021/bc300642u
Cai, B.; Yang, X.; Sun, L.; Fan, X.; Li, L.; Jin, H.; Wu, Y.; Guan, Z.; Zhang, L.; Zhang, L.; Yang, Z. Org. Biomol. Chem. 2014, 12, 8866. doi: 10.1039/C4OB01525H
Fan, X.; Sun, L.; Li, K.; Yang, X.; Cai, B.; Zhang, Y.; Zhu, Y.; Ma, Y.; Guan, Z.; Wu, Y.; Zhang, L.; Yang, Z. Mol. Ther.-Nucleic Acids 2017, 9, 218. doi: 10.1016/j.omtn.2017.09.010
Li, L.; Yang, X.; Li, K.; Zhang, G.; Ma, Y.; Cai, B.; Li, S.; Ding, H.; Deng, J.; Nan, X.; Sun, J.; Wu, Y.; Shao, N.; Zhang, L.; Yang, Z. Org. Biomol. Chem. 2018, 16, 7488. doi: 10.1039/C8OB01454J
(a) Zhang, S.; Cao, M.; Guan, Z.; Wang, Z.; Cao, Y. L.; Guo, Y.; Yang, Z. J.; Zhang, L. H. Chin. J. Med. Chem. 2011, 2, 4.
(b) Zhang, S. M.S. Thesis, Peking University, Beijing, 2011 (in Chinese).
(张烁, 硕士论文, 北京大学, 北京, 2011.)
(c) Zhang, H. Y.; Wu, X. J.; Yu, H. W.; Ma, L. T.; Zhang, L. H. Chin. Chem. Lett. 1996, 7, 1089.
(d) Zhang, H. Y.; Zhang, M. L.; Piao, Z. S.; Ma, L. T.; Zhang, L. H. Acta Pharm. Sin. 1999, 34, 363 (in Chinese).
(张虎翼, 张铭龙, 朴志松, 马灵台, 张礼和, 药学学报, 1999, 34, 363.)
(a) Zhang, P. S.; Dong, E Z. M.; Cleary, T. P. Org. Process Res. Dev. 2005, 9, 583.
(b) Forsman, J. J.; Waerna, J.; Murzin, D. Y.; Leino, R. Eur. J. Org. Chem. 2009, 5666.
CCDC 1892867 (Compound 8) contain the supplementary crystallographic data for this paper.
Houston, T. A.; Koreeda, M. Carbohydr. Res. 2009, 344, 2240. doi: 10.1016/j.carres.2009.08.026
Kakefuda, A.; Shuto, S.; Nagahata, T.; Seki, J.; Sasaki, T.; Matsuda, A. Tetrahedron 1994, 50, 10167. doi: 10.1016/S0040-4020(01)81749-X
Ohrui, H.; Waga, T.; Meguro, H. Biosci. Biotechnol. Biochem. 1993, 57, 1040. doi: 10.1271/bbb.57.1040
(a) Yoshimura, Y. Heterocycles 2017, 94, 1625.
(b) Kitkowska, J. D.; Tabaczynska, Z. A.; Draminski, M. Wiad. Chem. 2013, 67, 843.
(c) Leclerc, E. In Chemical Synthesis of Carbocyclic Analogues of Nucleosides, John Wiley & Sons, Inc., New York, 2013, pp. 535~604.
(a) Jacobsen, M. F.; Knudsen, M. M.; Gothelf, K. V. J. Org. Chem. 2006, 71, 9183.
(b) Mercurio, M. E.; Tomassi, S.; Gaglione, M.; Russo, R.; Chambery, A.; Lama, S.; Stiuso, P.; Cosconati, S.; Novellino, E.; Di Maro, S.; Messere, A. J. Org. Chem. 2016, 81, 11612.
(c) Zhou, J.; Du, X.; Chen, X.; Xu, B. Biochemistry 2018, 57, 4867.
(d) Porcheddu, A.; Giacomelli, G.; Piredda, I.; Carta, M.; Nieddu, G. Eur. J. Org. Chem. 2008, (34), 5786.
(a) Tzeng, C.-C.; Hwang, L.-C.; Chen, C.-C.; Wei, D.-C. Nucleoside Nucleotides 1995, 14, 1425.
Meade, E. A.; Wotring, L. L.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 1997, 40, 794. doi: 10.1021/jm960631x
Lenagh-Snow, G. M. J.; Araujo, N.; Jenkinson, S. F.; Rutherford, C.; Nakagawa, S.; Kato, A.; Yu, C.-Y.; Weymouth-Wilson, A. C.; Fleet, G. W. J. Org. Lett. 2011, 13, 5834. doi: 10.1021/ol2024482

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

