Table 1.
Optimization of the reaction conditionsa
Citation:
Wang Yunlong, Zhang Linbao, Niu Junlong, Song Maoping. Copper-Promoted Direct Nitration of Arenes Assisted by an N, O-Bidentate Directing System[J]. Chinese Journal of Organic Chemistry,
2019, 39(6): 1761-1766.
doi:
10.6023/cjoc201901015

N, O-双齿螯合作用下铜促进的C-H键直接硝基化反应
English
Copper-Promoted Direct Nitration of Arenes Assisted by an N, O-Bidentate Directing System
Abstract:
Cu(Ⅱ)-promoted C-H nitration of arenes has been disclosed with the aid of N, O-bidentate directing group. The protocol was operationally simple by using NaNO2as the nitration source. Various amide substrates were tolerated in the reaction system, which establishes opportunities for developing simple and facile methods, and enriches the strategies to access aromatic nitro derivatives.
-
Key words:
- C-H activation
- / bidentate direction
- / Cu-catalyzed
- / nitration
-
1. Introduction
Aromatic nitro compounds are widely applied in preparation of dyes, nonlinear optics, pharmaceuticals, and functional materials.[1] Considerable attention has been paid to develop a practical and facile method for the nitration of arenes. Up to now, the most common approachs to nitroarenes have been focused on electrophilic aromatic substitution with nitrating agents.[2] However, the nitration of benzoic acid needs a harsh reaction system (conc. H2SO4/ HNO3) and always suffers from regioselectivity and functional group tolerance problems that remain difficult to overcome.[3] Alternatively, nitroarenes could be synthesized via ipso-nitration of aryl halides, arylmetals, or aryl carboxylic acids, [4] while the use of prefunctionalized starting materials restricted the practical application in organic synthesis. As a consequence, it would be beneficial if nitroarenes could be accessed by chelation-assisted ortho-C—H activation with high chemoselectivity, broad substrate tolerance, and obviating prefunctionalization of the starting materials.[5] Following this strategy, several groups have made great development in chelation-assisted aromatic C—H nitration.[6] For example, Liu and co- workers reported Pd-catalyzed direct ortho-nitration of N-heterocycles.[6a~6b] Rh(Ⅲ)-catalyzed C—H nitration of arenes has been developed by Li and co-workers.[6e] Moreover, Cu catalyzed nitration of anilines using HNO3 as the nitration agent has also been reported by Carretero and co-workers.[6f] A silver-satalyzed chemo- and regio- selective nitration of anilides using sodium nitrite as a nitrating source was demonstrated by Nasab and co-workers in 2018.[6g] However, most of the reported protocols suffered several drawbacks such as the use of expensive metal catalysis, as well as a narrow substrate scope.
Chelation-assisted C—H bond functionalizations using bidentate-directing groups have emerged as a powerful tool for the synthesis of value-added organic molecules due to the pioneering work of Daugulis and co-workers for the Pd-catalyzed arylation of Csp2—H bond.[7] Then diverse C—H bond functionalization methods employing various directing groups have demonstrated the success of this chelation-assisted strategy utilizing relatively cheaper first-row metal catalysts such as iron[8], cobalt[9], nickel[10] and copper[11]. As for Cu salts, copper-catalyzed or copper-medi- ated C—H bond functionalization including sulfenylation, amination, fluorination, etherification, hydroxylation, alkynylation and trifluoromethylation have been reported by Daugulis, Yu and other groups.[7m~7w] On the other hand, copper-mediated ortho-nitration of (hetero)arenecarboxy- lates was first reported by Gooβen and co-workers on the use of 8-aminoquinoline as a bidentate directing group recently.[6h] The costly AgNO2 as nitration source and the external oxidant N-methylmorpholine N-oxide (NMO) implies that there exists plenty of room to improve the method. Our laboratory has reported copper-mediated direct aryloxylation and alkoxylation of arenes using an N, O-bidentate directing group.[12] As part of our interest in the field, [9e~9f, 12] we herein present the copper-promoted direct nitration of arenes using NaNO2 as the nitration source under external-oxidant-free conditions.
2. Results and discussion
Initially, 2-benzamidopydidine 1-oxide (1a) was subjected to the reaction conditions (0.5 equiv. CuF2•2H2O, 2.0 equiv. NaNO2, 130 ℃, 0.5 mL DMSO, 12 h, Table 1, Entry 1) and the desired nitration product was achieved in 17% yield. After various Cu salts were investigated (Table 1, Entries 2~4), CuCl2•2H2O exhibited the best efficiency in 31% yield (Table 1, Entry 4). The addition of additives failed to give a satisfactory result (see the Supporting Information). Changing from DMSO to other solvents (see the Supporting Information), N-methyl-2-pyrrolidone (NMP) provided a higher nitration conversion efficiency (Table 1, Entry 5). Decreasing the reaction temperature and increasing the amount of NaNO2 were beneficial for the reaction (Table 1, Entries 6~7), as the formation of by-product 2-(2-hydroxybenzamido)pyridine 1-oxide was diminished. To our delight, the further increase (2 equiv of CuCl2•2H2O) led to improved yields of the nitration product to 62% (Table 1, Entries 8~9). No product was obtained in the absence of copper salts (Table 1, Entry 10). Thus, the optimized reaction conditions was presented as follows: 1a (0.2 mmol), CuCl2•2H2O (0.4 mmol, 2 equiv.), NaNO2 (1.6 mmol, 8.0 equiv.), NMP (0.5 mL) under an air atmosphere at 110 ℃ for 12 h.
Table 1

 下载:
导出CSV
下载:
导出CSV

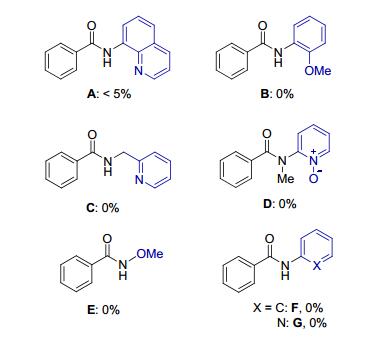
Entry Cu salt Yield/% 1 CuF2•2H2O 17 2 Cu(NO3)2•3H2O < 5 3 Cu(OAc)2•H2O 0 4 CuCl2•2H2O 31 5b CuCl2•2H2O 34 6b, c CuCl2•2H2O 36 7b, c, d CuCl2•2H2O 40 8b, c, d, e CuCl2•2H2O 45 9b, c, d, f CuCl2•2H2O 62 10 0 0 a Reaction conditions: [Cu] (0.1 mmol), NaNO2 (0.4 mmol), 1a (0.2 mmol), DMSO (0.5 mL), air atmosphere, 130 ℃, 12 h, isolated yields. b NMP (0.5 mL) as solvent. c Run at 110 ℃. d NaNO2 (1.6 mmol). e CuCl2•2H2O (0.2 mmol). f CuCl2•2H2O (0.4 mmol). NMP=N-methyl-2-pyrrolidone. Other bidentate directing groups (A~C) including 8-aminoquinoline were also examined to verify the paralleled property of N, O-auxiliary, and the result showed that no reaction occurred under the optimized reaction conditions. In addition, no products were found in structurally similar monodentate groups (D~G) by TLC analysis. These experiments indicate that 2-aminopyridine 1-oxide auxiliary is essential under our reaction system (Figure 1).
Figure 1
We next examined the reaction scope with the respect to the aromatic amides, and the results were listed in Table 2. The nitration of Csp2—H bond exclusively took place at the ortho-position of aromatic ring. Substrates possessing electron-withdrawing or electron-donating groups proceeded nitration smoothly to afford corresponding products (2a~2q) in moderate to good yields. In the case of meta-substituted substrate (1c), nitration occurred at the less hindered position of arenes. Aromatic amide derivatives bearing methyl, methoxy, tert-butyl, halogen groups on the para-position could successfully yield the corresponding nitration products as well, indicating that the reaction system exhibits a broad functional group compatibility. For ortho-substituted substrates (1b, 1h, and 1i), the reaction underwent well to afford the products in good yields. Moreover, the polycyclic substrate (1n) furnished the nitration compound in 54% yield. In all cases, the formation of the dinitrated product was not detected under the optimized reaction conditions.
Table 2
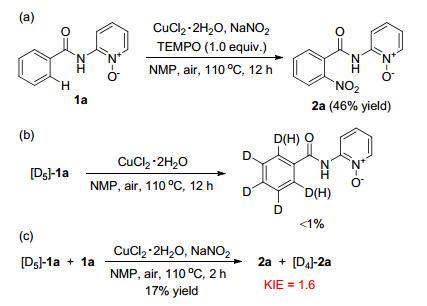
The mechanism was investigated by several control experiments. The addition of the radical quenchers, 2, 2, 6, 6- tetramethyl-1-piperidinyloxy (TEMPO, 1.0 equiv.) to the reaction system did not suppress product formation (Scheme 1a).[13] When [D5]-1a was treated with CuCl2•2H2O and NMP in the absence of the NaNO2 for 12 h, no obvious hydrogen incorporation was detected (Scheme 1b). These results suggested that the C—H cleavage step was largely irreversible. A 1:1 mixture of [D5]-1a and 1a, performed under the optimized reaction conditions, resulted in a kinetic isotope effect (KIE) of 1.6 (see the Supporting Information), which confirmed that C—H bond activation might be the rate limiting step (Scheme 1c). Whereas these control experiments and the mechanistic findings reported by Stahl[14] and metallacycle intermediated heteroarylation proposed by Miura, [11i] a plausible mechanism involving Cu(Ⅲ) species was proposed for the ortho-nitration of arenes.
Scheme 1
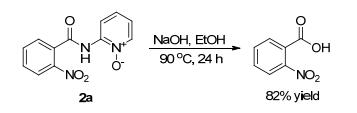
Finally, the N, O-bidentate directing group could be easily removed in one step. As shown in Scheme 2, the nitration product 2a was treated with NaOH in EtOH at 90 ℃ for 24 h, affording 2-nitrobenzoic acid in high yield.
Scheme 2
3. Conclusions
In conclusion, a copper-promoted chelation-assisted C—H nitration of aromatic amides was developed using NaNO2 as the coupling partner. The nitration selectively took place at the ortho-position of aromatic ring with the help of N, O-bidentate directing group. Various functional groups were tolerated in the reaction system, and the protocol is operationally simple. Moreover, the directing group could be removed easily, providing a new way for the synthesis of ortho- nitro benzoic acids.
4. Experimental section
4.1 General information
1H NMR and 13C NMR spectra were recorded at 600 MHz and 150 MHz, respectively on a Bruker DPX 600 instrument using tetramethylsilane as an internal standard. Compounds for HRMS were analyzed on a Waters Q-Tof Micro MS/MS System ESI spectrometer. Melting points were measured on a WC-1 instrument and uncorrected. Unless otherwise noted, materials were obtained from commercial suppliers without further purification and all procedures were performed under ambient air.
4.2 General procedure for nitration of sp2-C—H bonds
A 15 mL screw cap tube was equipped with a magnetic stir bar and charged with aromatic amides 1 (0.2 mmol), CuCl2•2H2O (67.6 mg, 0.4 mmol, 2 equiv), NaNO2 (110 mg, 1.6 mmol, 8 equiv.), and NMP (0.5 mL). The reaction system was stirred evenly at room temperature and heated at 110 ℃ for 12 h, then cooled to room temperature. The reaction system was diluted with 50 mL of ethylacetate, and filtered through a celite pad. The filtrate was quenched with aqueous 1 mol•L-1 HCl solution (10 mL), and then washed with 50 mL saturated brines. The organic layer was collected and dried over Na2SO4. Product 2 was purified by preparative TLC on silica gel (ethylacetate/ MeOH, V:V=10:1).
2-(2-Nitrobenzamido)pyridine 1-oxide (2a): Yellow solid, yield 62%. m.p. 191~192 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.41 (s, 1H), 8.58 (d, J=8.4 Hz, 1H), 8.27 (d, J=6.4 Hz, 1H), 8.16 (d, J=8.1 Hz, 1H), 7.77 (t, J=7.4 Hz, 1H), 7.70 (t, J=7.9 Hz, 2H), 7.43 (t, J=8.0 Hz, 1H), 7.09 (t, J=7.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 164.5, 146.5, 144.0 137.2, 134.0, 131.6, 131.5, 128.4, 128.3, 124.9, 119.5, 115.2. HRMS (positive, ESI) calcd for C12H10N3O4 [M+H]+ 260.0666, found 260.0667.
2-(2-methyl-6-nitrobenzamido)pyridine 1-oxide (2b): Yellow solid, yield 71%. m.p. 208~209 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.25 (s, 1H), 8.63 (dd, J=8.5, 1.6 Hz, 1H), 8.24 (dd, J=6.5, 0.9 Hz, 1H), 8.10 (d, J=8.1 Hz, 1H), 7.62 (d, J=7.6 Hz, 1H), 7.55 (t, J=7.9 Hz, 1H), 7.43 (td, J=9.0, 4.5 Hz, 1H), 7.11~7.04 (m, 1H), 2.50 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 165.0, 145.7, 143.9 137.7, 137.2, 136.6, 131.4, 130.3, 128.3, 122.3, 119.4, 115.2, 19.1. HRMS (positive, ESI) calcd for C13H12N3O4 [M+ H]+ 274.0823, found 274.0824.
2-(5-Methyl-2-nitrobenzamido)pyridine 1-oxide (2c): Yellow solid, yield 26%. m.p. 156~157 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.33 (s, 1H), 8.58 (d, J=8.2 Hz, 1H), 8.27 (d, J=6.2 Hz, 1H), 8.08 (d, J=8.2 Hz, 1H), 7.44 (dd, J=22.2, 8.2 Hz, 3H), 7.08 (t, J=6.5 Hz, 1H), 2.51 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 164.9, 145.8, 144.0 137.2, 137.1, 131.7, 131.7, 128.9, 128.5, 124.9, 119.4, 115.3, 21.5. HRMS (positive, ESI) calcd for C13H12N3O4 [M+H]+ 274.0823, found 274.0825.
2-(4-Methyl-2-nitrobenzamido)pyridine 1-oxide (2d): Yellow solid, yield 50%. m.p. 189~190 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.37 (s, 1H), 8.56 (d, J=8.3 Hz, 1H), 8.27 (d, J=6.1 Hz, 1H), 7.93 (s, 1H), 7.56 (dd, J=18.0, 7.4 Hz, 2H), 7.42 (t, J=8.0 Hz, 1H), 7.07 (t, J=6.9 Hz, 1H), 2.53 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 170.4, 151.4, 148.6 147.2, 143.0, 139.8, 134.1, 134.0, 132.5, 129.5, 125.5, 120.7, 25.8. HRMS (positive, ESI) calcd for C13H12N3O4 [M+H]+ 274.0823, found 274.0824.
2-(3, 5-Dimethoxy-2-nitrobenzamido)pyridine 1-oxide (2e): Yellow solid, yield 20%. m.p. 219~220 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.60 (s, 1H), 8.50 (d, J=7.6 Hz, 1H), 8.27 (s, 1H), 7.38 (s, 1H), 7.26 (s, 1H), 7.07 (s, 1H), 6.78 (s, 1H), 6.67 (d, J=21.8 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 162.89, 162.14, 153.82, 143.93, 137.12, 133.62, 131.43, 128.30, 119.44, 115.15, 103.69, 102.19, 56.83, 56.21. HRMS (positive ESI) calcd for C14H14N3O6 [M+H]+ 320.0877, found 320.0879.
2-(5-Methoxy-2-nitrobenzamido)pyridine 1-oxide (2f): Yellow solid, yield 54%. m.p. 162~163 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.35 (s, 1H), 8.58 (dd, J=8.4, 1.5Hz, 1H), 8.26~8.18 (m, 2H), 7.46~7.39 (m, 1H), 7.11~7.02 (m, 3H), 3.94 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 164.68, 164.00, 143.96, 138.63, 137.19, 134.14, 128.30, 127.45, 119.40, 115.78, 115.24, 113.49, 56.37. HRMS (positive, ESI) calcd for C13H12N3O5 [M+H]+ 290.0772, found 290.0772.
2-(4-Methoxy-2-nitrobenzamido)pyridine 1-oxide (2g): Yellow solid, yield 13%. m.p. 187~188 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.40 (s, 1H), 8.55 (d, J=8.3 Hz, 1H), 8.27 (d, J=6.3 Hz, 1H), 7.63 (d, J=8.4 Hz, 1H), 7.56 (s, 1H), 7.41 (t, J=7.9 Hz, 1H), 7.21 (d, J=8.4 Hz, 1H), 7.07 (t, J=6.9 Hz, 1H), 3.95 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 164.1, 161.8, 148.5, 144.1, 137.1, 129.7, 128.3, 123.3, 119.3, 118.9, 115.1, 110.2, 56.2. HRMS (positive, ESI) calcd for C13H12N3O5 [M+H]+ 290.0772, found 290.0774.
2-(2, 4-Dimethyl-6-nitrobenzamido)pyridine 1-oxide (2h): Yellow solid, yield 47%. m.p. 219~220 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.17 (s, 1H), 8.62 (d, J=8.3 Hz, 1H), 8.26 (d, J=6.1 Hz, 1H), 7.90 (s, 1H), 7.43 (m, 2H), 7.08~7.06 (t, J=6.72 Hz, 1H), 2.47 (s, 3H), 2.45 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 165.2, 145.8, 144.0 141.1, 137.3, 137.2, 137.1, 128.8, 128.3, 122.7, 119.3, 115.1, 21.1, 19.0. HRMS (positive ESI) calcd for C14H14N3O4 [M+H]+ 288.0979, found 288.0981.
2-(3, 6-Dimethyl-2-nitrobenzamido)pyridine 1-oxide (2i): Yellow solid, yield 39%. m.p. 209~210 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.30 (s, 1H), 8.61 (dd, J=8.3, 1.0 Hz, 1H), 8.17 (d, J=6.2 Hz, 1H), 7.87 (s, 1H), 7.41 (t, J=8.0 Hz, 2H), 7.05 (t, J=6.3 Hz, 1H), 2.46 (s, 3H), 2.44 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 165.3, 145.7, 144.0 141.0, 137.3, 137.1, 128.8, 128.4, 122.6, 119.2, 115.2, 21.1, 19.1. HRMS (positive ESI) calcd for C14H14N3O4 [M+H]+ 288.0979, found 288.0980.
2-(3, 5-Dimethoxy-2-nitrobenzamido)pyridine 1-oxide (2j): Beige solid, yield 30%. m.p. 175~176 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.70 (d, J=67.9 Hz, 1H), 8.52 (d, J=8.0 Hz, 1H), 8.29 (d, J=4.7 Hz, 1H), 7.43 (s, 1H), 7.38 (s, 1H), 7.31 (s, 1H), 7.07 (s, 1H), 2.46 (d, J=11.6 Hz, 3H), 2.40 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.4, 147.5, 141.6, 137.1, 135.5, 132.1, 128.7, 128.3, 126.1, 119.3, 115.1, 21.2, 17.8. HRMS (positive ESI) calcd for C14H14N3O4 [M+H]+ 288.0979, found 288.0981.
2-(4, 5-Dimethoxy-2-nitrobenzamido)pyridine 1-oxide (2k): Yellow solid, yield 63%. m.p. 216~217 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.38 (s, 1H), 8.58 (dd, J=8.4, 1.5 Hz, 1H), 8.25~8.17 (m, 1H), 7.68 (s, 1H), 7.48~7.39 (m, 1H), 7.10~7.03 (m, 1H), 7.01 (s, 1H), 4.01 (s, 3H), 4.00 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 164.9, 153.6, 150.0, 144.0, 138.8, 137.2, 128.3, 126.0, 119.3, 115.2, 109.7, 107.3, 56.8, 56.7. HRMS (positive, ESI) calcd for C14H14N3O6 [M+H]+ 320.0877, found 320.0878.
2-(4-(tert-Butyl)-2-nitrobenzamido)pyridine 1-oxide (2l): White solid, yield 46%. m.p. 220~221 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.46 (s, 1H), 8.56 (dd, J=8.4, 1.6 Hz, 1H), 8.23 (d, J=6.2 Hz, 1H), 8.13 (d, J=1.8 Hz, 1H), 7.75 (dd, J=8.0, 1.8 Hz, 1H), 7.61 (d, J=8.0 Hz, 1H), 7.43~7.39 (m, 1H), 7.07~7.04 (m, 1H), 1.40 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 164.6, 156.1, 146.7, 144.1 137.2, 130.8, 128.7, 128.3, 128.2, 121.9, 119.3, 115.2, 35.4, 30.9. HRMS (positive, ESI) calcd for C16H18N3O4 [M+H]+ 316.1292, found 316.1293.
2-(3-Nitro-[1, 1'-biphenyl]-4-ylcarboxamido)pyridine 1- oxide (2m): White solid, yield 35%. m.p. 186~188 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.48 (s, 1H), 8.58 (dd, J=8.5, 1.6 Hz, 1H), 8.33 (d, J=1.7 Hz, 1H), 8.21 (d, J=5.9 Hz, 1H), 7.93 (dd, J=7.9, 1.7 Hz, 1H), 7.75 (d, J=7.9 Hz, 1H), 7.65~7.63 (m, 2H), 7.55~7.46 (m, 3H), 7.43~7.38 (m, 1H), 7.05~7.01 (m, 1H); 13C NMR (150 MHz, CDCl3) δ: 164.4, 147.2, 145.0 144.0, 137.5, 131.9, 129.7, 129.4, 129.3, 128.9, 128.4, 127.2, 123.1, 119.4, 115.3. HRMS (positive, ESI) calcd for C18H14N3O4 [M+H]+ 336.0979, found 336.0981.
2-(2-Nitro-1-naphthamido)pyridine 1-oxide (2n): Yellow solid, yield 54%. m.p. 224~225 ℃ (decompose). 1H NMR (600 MHz, CDCl3) δ 10.43 (s, 1H), 8.75 (d, J=8.2 Hz, 1H), 8.27 (d, J=9.0 Hz, 1H), 8.21 (d, J=6.3 Hz, 1H), 8.10 (d, J=9.14 Hz, 1H), 8.06 (d, J=8.5 Hz, 1H), 8.00 (d, J=8.2 Hz, 1H), 7.74 (t, J=7.39 Hz, 1H), 7.69 (t, J=7.6 Hz, 1H), 7.48 (t, J=7.9 Hz, 1H), 7.09 (t, J=6.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ: 164.6, 144.0, 142.3, 137.3, 135.7, 131.4, 130.5, 130.2, 129.34, 129.31, 128.5, 128.3, 126.9, 119.6, 119.5, 115.3. HRMS (positive ESI) calcd for C16H12N3O4 [M+H]+ 310.0823, found 310.0824.
2-(4-Fluoro-2-nitrobenzamido)pyridine 1-oxide (2o): Yellow solid, yield 24%. m.p. 192~193 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.43 (s, 1H), 8.55 (d, J=8.3 Hz, 1H), 8.27 (d, J=6.2 Hz, 1H), 7.86 (d, J=7.7 Hz, 1H), 7.71 (dd, J=7.8, 5.4 Hz, 1H), 7.48 (t, J=7.6 Hz, 1H), 7.43 (t, J=8.0 Hz, 1H), 7.09 (t, J=6.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 163.4, 143.8, 137.2, 130.33 (JC—F=8.01 Hz), 128.3, 127.8 (JC—F=4.67 Hz), 121.1, 121.0 (JC—F=22.6 Hz), 119.6, 115.2, 113.1, 112.9 (JC—F=26.7 Hz). HRMS (positive ESI) calcd for C12H9FN3O4 [M+H]+ 278.0572, found 278.0575.
2-(4-Chloro-2-nitrobenzamido)pyridine 1-oxide (2p): Yellow solid, yield 22%. m.p. 163~164 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.54 (s, 1H), 8.52 (d, J=8.2 Hz, 1H), 8.22 (d, J=5.6 Hz, 1H), 8.12 (s, 1H), 7.71 (d, J=7.5 Hz, 1H), 7.63 (d, J=8.1 Hz, 1H), 7.42 (t, J=7.8 Hz, 1H), 7.07 (d, J=6.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 163.5, 147.1, 143.8, 137.7, 137.2, 133.9, 129.7, 129.5, 128.4, 125.2, 119.6, 115.3. HRMS (positive ESI) calcd for C12H9ClN3O4 [M+H]+ 294.0276, found 294.0278.
2-(4-Bromo-2-nitrobenzamido)pyridine 1-oxide (2q): Yellow solid, yield 28%. m.p. 184~185 ℃; 1H NMR (600 MHz, CDCl3) δ: 10.56 (s, 1H), 8.53 (d, J=8.3 Hz, 1H), 8.27 (s, 1H), 8.21 (d, J=5.8 Hz, 1H), 7.87 (d, J=7.9 Hz, 1H), 7.56 (d, J=8.0 Hz, 1H), 7.42 (t, J=8.0 Hz, 1H), 7.07 (t, J=6.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 163.6, 147.0, 143.8, 137.2 136.9, 130.1, 129.6, 128.4, 127.9, 125.1, 119.6, 115.3. HRMS (positive ESI) calcd for C12H9BrN3O4 [M+H]+ 337.9771, found 337.9773.
Supporting Information 1H NMR and 13C NMR spectra of compounds 2a~2q. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
-
-
[1]
(a) Olah, G. A.; Malhotra, R.; Narang, S. C. Nitration: Methods and Mechanisms, Wiley-VCH, Weinheim, 1989.
(b) Feuer, H.; Nielsen, A. T. Nitro Compounds: Recent Advances in Synthesis and Chemistry, Wiley-VCH, Weinheim, 1990. -
[2]
(a) Schofield, K. Aromatic Nitrations, Cambridge University Press, Cambridge, 1980.
(b) Ono, N. The Nitro Group in Organic Synthesis, Wiley-VCH, Weinheim, 2001. -
[3]
Hearn, R.; Russell, M. K. R. G. Ann. Rheum. Dis. 1983, 42(Suppl.), 39.
-
[4]
(a) Tani, K.; Lukin, K.; Eaton, P. E. J. Am. Chem. Soc. 1997, 119, 6.
(b) Salzbrunn, S.; Simon, J.; Prakash, G. K. S.; Petasis, N. A.; Olah, G. A. Synlett 2000, 1485.
(c) Prakash, G. K. S.; Panja, C.; Mathew, T.; Surampudi, V.; Petasis, N. A.; Olah, G. A. Org. Lett. 2004, 6, 2205.
(d) Yan, G.; Yang, M. Org. Biomol. Chem. 2013, 11, 2554.
(e) Das, J. P.; Sinha, P.; Roy, S. Org. Lett. 2002, 4, 3055. -
[5]
(a) Kakiuchi, F.; Chatani, N. Adv. Synth. Catal. 2003, 345, 1077.
(b) Ackermann, L.; Vicente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792.
(c) Satoh, T.; Miura, M. Chem.-Eur. J. 2010, 16, 11212.
(d) Wencel-Delord, J.; Drö ge, T.; Liu, F.; Glorius, F. Chem. Soc. Rev. 2011, 40, 4740.
(e) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem. Int. Ed. 2009, 48, 5094.
(f) Ackermann, L. Acc. Chem. Res. 2014, 47, 281.
(g) Misal Castro, L. C.; Chatani, N. Chem. Lett. 2015, 44, 410.
(h) Satoh, T.; Miura, M. Chem. Lett. 2006, 36, 200. -
[6]
(a) Liu, Y.-K.; Lou, S.-J.; Xu, D.-Q.; Xu, Z.-Y. Chem.-Eur. J. 2010, 13590.
(b) Zhang, W.; Lou, S.; Liu, Y.; Xu, Z. J. Org. Chem. 2013, 78, 5932.
(c) Zhang, L.; Liu, Z.; Li, H.; Fang, G.; Barry, B. D.; Belay, T. A.; Bi, X.; Liu, Q. Org. Lett. 2011, 13, 6536.
(d) Zhang, H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2013, 15, 3836.
(e) Xie, F.; Qi, Z.; Li, X. Angew. Chem., Int. Ed. 2013, 52, 11862.
(f) Hernando, E.; Castillo, R. R.; Rodrĭguez, N.; Arrayás, R. G.; Carretero, J. C. Chem. Eur. J. 2014, 20, 13854.
(g) Kianmehr, E.; Nasab, S. B. Eur. J. Org. Chem. 2018, 6447.
(h) Katayev, D.; Pfister, K. F.; Wendling, T.; Gooβen, L. J. Chem.-Eur. J. 2014, 20, 9902.
(i) Liu, J.; Zhuang, S.; Gui, Q.; Chen, X.; Yang, Z.; Tan, Z. Adv. Synth. Catal. 2015, 357, 732. -
[7]
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 13154.
-
[8]
Selected examples.
(a) Asako, S.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 17755.
(b) Shang, R.; Ilies, L.; Matsumoto, A.; Nakamura, E. J. Am. Chem. Soc. 2013, 135, 6030.
(c) Fruchey, E. R.; Monks, B. M.; Cook, S. P. J. Am. Chem. Soc. 2014, 136, 13130. -
[9]
Selected examples.
(a) Grigorjeva, L.; Daugulis, O. Angew. Chem., Int. Ed. 2014, 53, 10209.
(b) Grigorjeva, L.; Daugulis, O. Org. Lett. 2014, 16, 4684.
(c) Grigorjeva, L.; Daugulis, O. Org. Lett. 2014, 16, 4688.
(d) Ma, W.; Ackermann, L. ACS Catal.2015, 5, 2822.
(e) Zhang, L.-B.; Hao, X.-Q.; Zhang, S.-K.; Liu, Z.-J.; Zheng, X.-X.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. Angew. Chem., Int. Ed. 2015, 54, 272.
(f) Zhang, L.-B.; Hao, X.-Q.; Zhang, S.-K.; Zheng, X.-X.; Liu, Z.-J.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. Angew. Chem., Int. Ed. 2015, 54, 10012.
(g) Saxena, P.; Kapur, M. Chem. Asian J. 2018, 13. 861. -
[10]
Selected examples:
(a) Aihara, Y.; Chatani, N. J. Am. Chem. Soc. 2014, 136, 898.
(b) Song, W.; Lackner, S.; Ackermann, L. Angew. Chem., Int. Ed. 2014, 53, 2477.
(c) Shiota, H.; Ano, Y.; Aihara, Y.; Fukumoto, Y.; Chatani, N. J. Am. Chem. Soc. 2011, 133, 14952. -
[11]
Selected examples.
(a) Tran, L. D.; Popov, I.; Daugulis, O. J. Am. Chem. Soc. 2012, 134, 18237.
(b) Truong, T.; Klimovica, K.; Daugulis, O. J. Am. Chem. Soc. 2013, 135, 9342.
(c) Tran, L. D.; Roane, J.; Daugulis, O. Angew. Chem., Int. Ed. 2013, 52, 6043.
(d) Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842.
(e) Shang, M.; Sun, S.-Z.; Wang, H.-L.; Laforteza, B. N.; Dai, H.-X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2014, 53, 10439.
(f) Shang, M.; Wang, H.-L.; Sun, S.-Z.; Dai, H.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 11590.
(g) Li, X.; Liu, Y.-H.; Gu, W.-J.; Li, B.; Chen, F.-J.; Shi, B.-F. Org. Lett. 2014, 16, 3904.
(h) Dong, J.; Wang, F.; You, J. Org. Lett. 2014, 16, 2884.
(i) Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 4457.
(j) Chen, F.-J.; Liao, G.; Li, X.; Wu, J.; Shi, B.-F. Org. Lett. 2014, 16, 5644.
(k) Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726.
(l) Zhang, Q.; Chen, K.; Shi, B.-F. Synlett 2014, 25, 1941.
(m) Daugulis, O.; Roane, J.; Tran, L. D. Acc. Chem. Res. 2015, 48, 1053.
(n) Dou, Y.-D.; Yin, B.; Zhang, P.-F.; Zhu, Q. Eur. J. Org. Chem. 2018, 4571.
(o) Wang, C.-M.; Tang, K.-X.; Gao, T.-H.; Chen, L.; Sun, L.-P. J. Org. Chem. 2018, 83, 8315.
(p) Tu, D.-Q.; Luo, J.; Jiang, C. Chem. Commun. 2018, 54, 2514.
(q) Vinayak, B.; Chandrasekharam, M. Org. Lett. 2017, 19, 3528. -
[12]
Selected our recent reports: (a) Hao, X.-Q.; Chen, L.-J.; Ren, B.; Li, L.-Y.; Yang, X.-Y.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. Org. Lett. 2014, 16, 1104.
(b) Zhang, L.-B.; Hao, X.-Q.; Zhang, S.-K.; Liu, K.; Ren, B.; Gong, J.-F.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2014, 79, 10399. -
[13]
Selected examples.
(a) Wang, D.-W.; Zhao, K.-Y.; Xu, C.-Y.; Miao, H.-Y.; Ding, Y.-Q. ACS Catal. 2014, 4, 3910.
(b) Wang, D.-W.; Ge, B.-Y.; Li, L.; Shan, J.; Ding, Y.-Q. J. Org. Chem. 2014, 79, 8607.
(c) Guo, X-K.; Zhang, L.-B.; Wei, D.-H.; Niu, J.-L. Chem. Sci. 2015, 6, 7059.
(d) Wang, D.-W.; Yu, X.; Yao, W.; Hu, W.-K.; Ge, C.-Y.; Shi, X.-D. Chem.-Eur. J. 2016, 22, 55433.
(e) Yu, X.-L.; Wang, D.-S.; Xu, Z.-J.; Yang, B.-B.; Wang, D.-W. Org. Chem. Front. 2017, 4, 1011.
(f) Wu, Q.; Pan, L.; Du, G.-M.; Zhang, C.; Wang, D.-W. Org. Chem. Front. 2018, 5, 2668.
(g) Xu, Z.-J.; Yu, X.-L.; Sang, X.-X.; Wang, D.-W. Green Chem. 2018, 20, 2571.
(h) Wang, Y.; Du, C.; Wang, Y.-Y.; Guo, X.-K.; Fang, Lei.; Song, M.-P.; Niu, J.-L.; Wei, D.-H. Adv. Synth. Catal. 2018, 360, 2668.
(i) Wang, D.-W.; Yu, X.-L.; Ge, B.-Y.; Miao, H.-Y.; Ding, Y.-Q. Chin. J. Org. Chem. 2015, 35, 676.
(j) Hu, X.-Y.; Yang, B.-B.; Yao, W.; Wang, D.-W. Chin. J. Org. Chem. 2018, 38, 3296. -
[14]
Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797. doi: 10.1021/ja4026424
-
[1]
-
Table 1. Optimization of the reaction conditionsa
Entry Cu salt Yield/% 1 CuF2•2H2O 17 2 Cu(NO3)2•3H2O < 5 3 Cu(OAc)2•H2O 0 4 CuCl2•2H2O 31 5b CuCl2•2H2O 34 6b, c CuCl2•2H2O 36 7b, c, d CuCl2•2H2O 40 8b, c, d, e CuCl2•2H2O 45 9b, c, d, f CuCl2•2H2O 62 10 0 0 a Reaction conditions: [Cu] (0.1 mmol), NaNO2 (0.4 mmol), 1a (0.2 mmol), DMSO (0.5 mL), air atmosphere, 130 ℃, 12 h, isolated yields. b NMP (0.5 mL) as solvent. c Run at 110 ℃. d NaNO2 (1.6 mmol). e CuCl2•2H2O (0.2 mmol). f CuCl2•2H2O (0.4 mmol). NMP=N-methyl-2-pyrrolidone.  下载: 导出CSV
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1374
- HTML全文浏览量: 108
