图 1.
催化丙二酸二甲酯加氢反应示意图
Figure 1.
Reaction process for the catalytic hydrogenation of dimethyl malonate

1, 3-丙二醇(1, 3-propanediol, 1, 3-PDO)是合成聚酯的重要原料[1], 近年来1, 3-PDO的制备研究备受关注.目前, 1, 3-PDO主要通过石化路线获得, 如丙烯醛水合氢化法[2]和环氧乙烷羰基化法[3].此外微生物发酵法[4]、甘油氢解法[5]等也是很多课题组研究开发的路线.随着近年来煤基合成气路线经草酸二酯、进而加氢制备乙醇酸酯以及乙二醇研究的开展[6], 可比拟的丙二酸酯加氢制备1, 3-PDO的技术路线研究引起化学家们的特别关注[7].丙二酸二甲酯(dimetyl malonate, DMM)在催化加氢过程中主要发生的反应如图 1所示, DMM首先部分加氢得到中间产物3-羟基丙酸甲酯(3-hydroxypropanoic acid methyl ester, 3-HPM), 进一步加氢得到最终产物1, 3-PDO.但是, DMM容易发生脱羧反应生成乙酸甲酯, 同时3-HPM的β位羟基稳定性较低, 易在酸性位点脱水转化生成丙烯酸甲酯、丙酸甲酯(methyl propionate, MP)等副产物.因此, DMM高选择性催化加氢制备3-HPM是这一反应路线的核心步骤.
目前, 催化丙二酸酯加氢多使用负载型多相催化剂, 反应条件苛刻[8].众所周知, 均相催化剂可在较为温和条件下高选择性地获取目标产物, 相比于多相催化剂有明显优势[9].近年来, 国内Zhou[10]、Ding[11]、Zhang[12]等课题组在均相催化加氢领域开展了众多富有意义的工作, 取得了显著的研究成果.最近, 在草酸二甲酯均相催化加氢制乙醇酸甲酯及乙二醇方面进行了系列研究[13], 在此基础上, 进一步将研究工作扩展到丙二酸酯的催化加氢.本文中, 采用系列膦胺配体L1~L8(图 2), 分别与乙酰丙酮钌[Ru(acac)3]组成催化剂体系, 考察了催化丙二酸酯加氢的反应性能.同时选择L1-Ru(acac)3体系, 研究了对其它酯类分子E2~E7(图 2)的催化加氢性能.
自1995年Noyori等[14]报道发现膦配体和胺配体构成的Ru(Ⅱ)催化剂体系可有效催化醛、酮等羰基衍生物的加氢反应以来, 这一催化体系的合成及应用研究取得了显著的进展[9].近年来, 我们课题组合成了多种膦胺配体构成的Ru(Ⅱ)化合物, 并成功应用于草酸酯、脂肪酸酯等多种酯类分子的加氢反应研究中[13a, 13b], 但将此类催化剂应用于DMM催化加氢制3-HPM或1, 3-PDO时结果不甚理想. 1997年, Elsevier等[15]报道采用Ru(acac)3作为金属前驱体, 与膦配体构成催化剂体系.在甲醇溶液中, 这一体系可有效催化草酸二甲酯加氢制乙二醇.紧接着, 该课题组[16]研究发现这一催化剂体系也可以催化芳香酸酯的加氢反应. 2010年以来, Leitner等[17]进一步将这一催化剂体系应用于生物质酸、CO2等加氢难度较高底物分子的加氢反应研究中, 均取得不错的催化反应效果.基于上述研究进展, 我们尝试采用Ru(acac)3和膦胺配体构成催化体系, 应用于DMM均相催化加氢.
类比文献反应条件[15], 首先以甲醇为溶剂, 在393 K、初始H2压力7 MPa、反应时间16 h以及Ru(acac)3和配体用量分别为1 mol%、2 mol%条件下考察了配体对体系催化加氢性能的影响.如表 1所示, L1-Ru(acac)3在反应条件下取得了55%的DMM转化率和45%的3-HPM收率(Entry 1, 表 1).有趣的是, 将L1结构中的胺氢替换为甲基所得的配体L2所构成的催化体系没有催化活性(Entry 2, 表 1), 类似的实验现象文献已有报道[11, 18].衍生于L1配体的L3~L5分别组成的催化体系表现出不同的催化活性(Entries 3~6, 表 1).具体来说, L3-Ru(acac)3催化活性低于L1-Ru(acac)3 (Entry 3, 表 1), L5-Ru(acac)3催化活性与L1-Ru(acac)3基本相当(Entry 5, 表 1), 而L4-Ru(acac)3基本没有催化活性(Entry 4, 表 1).由于L4配体与Ru中心可以采用N, N, P-三齿螯合配位, 过多配位基堵塞活性中心, 因此L4-Ru(acac)3催化活性低.相比之下, 配体L5与L1相似, 采用双齿与金属中心螯合, 所构成体系催化活性相当. L3-Ru(acac)3催化活性低于L1-Ru(acac)3和L5-Ru(acac)3的原因, 可能是由于L3结构中氮原子所连基团的空间位阻较大所致.在相同反应条件下, 较为柔软的双齿膦胺配体L6~L8组成的催化体系性能均较差(Entries 7~9, 表 1).
 下载:
导出CSV
下载:
导出CSV
| Entry | Ligand | Conv./% | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | |||
| 1 | L1 | 55 | 45 | 1 | 3 | 6 |
| 2 | L2 | 0 | — | — | — | — |
| 3 | L3 | 33 | 25 | 0 | 2 | 6 |
| 4 | L4 | 6 | 2 | 0 | 0 | 4 |
| 5 | L5 | 55 | 43 | 0 | 8 | 4 |
| 6 | L6 | 16 | 6 | 0 | 2 | 8 |
| 7 | L7 | 19 | 10 | 0 | 3 | 6 |
| 8 | L8 | 9 | 0 | 0 | 1 | 8 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), ligand (2 mol%), methanol (10 mL), temp.=393 K, p(H2)=7 MPa, time=16 h. b Mainly containing methyl acetate and methyl acrylate. | ||||||
选择最优配体L1, 进一步考察了配体用量对体系催化性能的影响, 结果如表 2所示.可以看出, 当n(L1)/n(Ru(acac)3)比值为2时体系取得了最优的催化反应结果(Entries 2~4, 表 2), 低于或高于这一比值均不利于DMM酯基加氢反应(Entries 2 & 4, 表 2).值得注意的是, 当没有使用配体时, 反应体系无催化活性(Entry 1, 表 2).综合以上结果, 在下文我们选用物质的量比为2:1的L1与Ru(acac)3作为最优的催化体系开展进一步研究.
下载:
导出CSV
| Entry | n(L1)/n(Ru) | Conv./% | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | |||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 1 | 14 | 10 | 0 | 1 | 3 |
| 3 | 2 | 55 | 45 | 1 | 3 | 6 |
| 4 | 3 | 3 | 1 | 0 | 0 | 2 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), methanol (10 mL), temp.=393 K, p(H2)=7 MPa, time=16 h. b Mainly containing methyl acetate and methyl acrylate. | ||||||
在确定配体结构和用量后, 我们对反应条件进行系统的优化.首先考察了温度和时间对L1-Ru(acac)3体系催化性能的影响, 结果如表 3所示.当温度为373 K时, 反应体系活性较低, 难以有效催化DMM加氢(Entry 1, 表 3).将反应温度提高到393 K, 催化活性明显提高, 反应8 h即实现44% DMM转化, 并取得36% 3-HPM收率(Entry 2, 表 3).延长反应时间可进一步提高DMM转化率和3-HPM收率(Entries 3 & 4, 表 3).但是, 对比Entry 3和Entry 4数据可以看出, 393 K反应16 h后体系基本达到平衡; 继续延长反应时间, 底物转化率和目标产物收率无明显提高.提高反应温度至413 K, 虽然8 h内体系即实现50% DMM转化, 但是目标产物3-HPM收率较低, 仅为34% (Entry 5, 表 3).这可能是由于反应温度过高导致发生3-HPM β位羟基脱水等副反应所致.因此, 393 K和16 h是当前体系较为合适的反应温度和时间.
下载:
导出CSV
| Entry | Temp./ K | Time/ h | Conv./ % | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | ||||
| 1 | 373 | 16 | 7 | 5 | 0 | 0 | 2 |
| 2 | 393 | 8 | 44 | 36 | 0 | 2 | 6 |
| 3 | 393 | 16 | 55 | 45 | 1 | 3 | 6 |
| 4 | 393 | 24 | 58 | 47 | 2 | 4 | 5 |
| 5 | 413 | 8 | 50 | 34 | 0 | 5 | 11 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), L1 (2 mol%), methanol (10 mL), p(H2)=7 MPa. b Mainly containing methyl acetate and methyl acrylate. | |||||||
在以上研究中, 采用甲醇作为溶剂.在反应过程中, 甲醇一方面作为溶剂, 另一方面起到还原Ru(acac)3生成Ru2+, Ru2+与配体配位原位生成催化剂的作用[19].众所周知, Ru等过渡金属化合物与伯醇作用将诱导发生醇脱羰基的反应[20].在前期研究中, 我们发现羰基与Ru金属中心配位将占据催化剂活性位点, 导致催化剂中毒和失活[13a].此外, Bergens等[21]报道指出小分子醇易与Ru2+作用生成烷氧基配合物, 阻碍加氢过程的进行.在当前反应体系中, 过多甲醇的存在极有可能导致发生上述毒化过程.因此, 我们采用甲醇和四氢呋喃(THF)混合溶液作为溶剂, 并固定溶剂总体积为10 mL, 考察了甲醇用量对L1-Ru(acac)3催化加氢性能的影响, 结果如图 3所示.
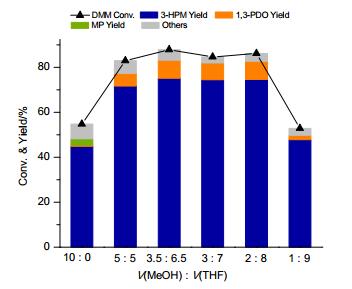
从图 3结果可以看出, 相比于使用甲醇作为溶剂, DMM转化率以及3-HPM收率随着反应体系中THF体积的增加呈现出先增加后降低的近火山型变化趋势.当甲醇和THF体积分别为2和8 mL时, 体系取得了较优的催化反应结果, 此时DMM转化率以及3-HPM收率分别达到86%和75%, 同时还伴有8%收率的二步加氢产物1, 3-PDO生成.此外, 在甲醇和THF体积分别为3和7 mL(或3.5和6.5 mL等)条件下, 也取得了优异的催化反应结果.以上结果说明, 在一定范围内采用甲醇-THF混合溶剂可以有效提升L1-Ru(acac)3催化体系的催化加氢性能.这主要得益于THF的引入, 有效减缓了反应体系中甲醇与Ru2+作用生成烷氧基配合物的可能, 以及甲醇分解产生CO毒化催化剂的进程.
考察了L1-Ru(acac)3催化不同结构酯类分子加氢的反应性能, 结果如表 4所示.表 4中所用到酯类分子结构见图 2.从表 4结果可以看出, L1-Ru(acac)3难以有效催化3-HPM加氢制1, 3-PDO.这一结果与L1-Ru(acac)3催化DMM加氢取得8% 1, 3-PDO收率的结果一致(Entry 1, 表 4), 导致出现这一现象的原因还有待进一步探究.相比于催化稳定性较差的3-HPM加氢, L1-Ru(acac)3在较高温度下可以有效催化2-羟基丙酸甲酯(E3)加氢制1, 2-丙二醇(Entry 3, 表 4). L1-Ru(acac)3在较低温度下也可以有效催化草酸酯分子(E4 & E5)部分加氢制得乙醇酸酯(Entries 4 & 5, 表 4).进一步研究发现, L1-Ru- (acac)3催化γ-戊内酯(E6)加氢制1, 4-戊二醇, 可以取得近乎完全的底物转化率和目标产物收率(Entry 6, 表 4).值得注意的是, L1-Ru(acac)3在催化碳酸乙烯酯(E7)加氢制甲醇和乙二醇的反应中, 也表现出了一定的催化活性(Entry 7, 表 4).作为碳酸酯的一种, 碳酸乙烯酯可以从环氧乙烷和CO2制备得到[22].然而, 由于共轭结构的存在, 碳酸酯稳定性很高, 较难被加氢[23]. Ding等[11]报道指出, 耦合碳酸乙烯酯制备及其加氢的“omega过程”, 在催化CO2和环氧乙烷转化制甲醇和乙二醇之间架起了一座新的桥梁.因此, Ding等认为高效催化碳酸乙烯酯加氢制甲醇是解决全球能源问题的理想方案之一.
下载:
导出CSV
| Entry | Substrate | Temp./K | Conv./% | Hydrogenation yield/% | |
| Product Ab | Product Bc | ||||
| 1 | E1 | 393 | 86 | 75 (3-HPM) | 8 (1, 3-PDO) |
| 2 | E2 | 393 | 7 | 3 (1, 3-PDO) | — |
| 3 | E3 | 423 | 83 | 82 (1, 2-PDO) | — |
| 4 | E4 | 388 | 100 | 98 (MG) | 1 (EG) |
| 5d | E5 | 383 | 98 | 97 (MPEG) | 1 (EG) |
| 6 | E6 | 413 | 97 | 97 (1, 4-PDO) | — |
| 7d | E7 | 413 | 63 | 59 (EG) | 54 (MeOH) |
| a Reaction conditions: Substrate (7.5 mmol), Ru(acac)3 (1 mol%), L1 (2 mol%), 2 mL methanol and 8 mL THF as the solvent, P(H2)=7 MPa, Time=16 h. b Yield of product A. c Yield of product B. d 2 mL ethanol and 8 mL THF as the solvent. MG: methyl glycolate; EG: ethylene glycol; MPEG: ethyl glycolate; 1, 2-PDO: 1, 2-propanediol; 1, 4-PDO: 1, 4-pentanediol. | |||||
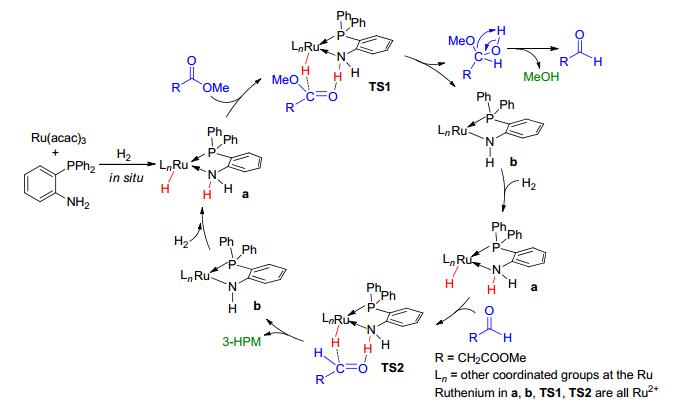
基于文献报道以及我们课题组前期的工作基础[11, 13a, 13b, 24], 推测了如图 4所示的L1-Ru(acac)3催化DMM加氢的反应机理.首先, 在原位条件下反应形成同时具有Ru—H和N—H官能团的催化活性物种a. a通过Ru—H与N—H分别与DMM羰基作用形成过渡态TS1, 进一步转化生成3-氧代丙酸甲酯和甲醇以及催化剂中间体b.中间体b可以通过活化氢气分子返回到活性态a, 实现催化循环.最后, 中间产物3-氧代丙酸甲酯与a通过形成过渡态TS2, 最终转化生成目标产物3-HPM.综合以上反应过程可以看出, a是通过结构中N—H和Ru—H的协同作用实现加氢反应的催化循环.由不含N—H官能团的L2配体与Ru(acac)3所构成的催化体系, 在反应条件下没有催化活性的实验结果验证了这一反应机理(Entry 2, 表 1).
首次以Ru(acac)3和o-二苯基膦苯胺构成均相催化剂体系, 应用于DMM加氢制3-HPM或1, 3-PDO, 系统探究了配体用量、反应温度和时间以及溶剂等反应条件对催化性能的影响.研究发现, Ru(acac)3和o-二苯基膦苯胺物质的量比为1:2较为合适; 此外, 相比于使用甲醇作为溶剂, 采用甲醇-THF混合溶剂可有效提升催化体系的催化加氢性能.在优化的反应条件下, DMM转化率达到86%, 目标产物3-HPM和1, 3-PDO收率分别为75%和8%.这一催化体系在一定反应条件下也可以有效催化草酸酯、γ-戊内酯、碳酸乙烯酯等多种酯类分子的加氢.
催化剂活性评价采用配备有聚四氟乙烯内衬的PARR 5500型高压反应釜, 釜体积为100 mL.反应液采用配备有氢焰离子化检测器(FID)、KB-Wax色谱柱(60 m×0.32 mm×0.33 μm)以及香港Collect公司生产的自动进样器(AS-2920)的福立-9790II型气相色谱进行分析.
甲苯、己烷、THF以及甲醇等有机溶剂购买自国药集团上海试剂公司; Ru(acac)3、配体合成所需原料、DMM等酯类化合物以及相应的加氢产物购买自Aldrich、Alfa Aesar、百灵威以及阿拉丁等化学试剂公司.文中所使用配体L1~L8参考文献方法合成[25].
实验中涉及无氧无水的操作均采用标准Schlenk技术或在Mbraum手套箱(O2和H2O含量低于1.0 ppm)中进行.有机溶剂甲苯、己烷、THF等用钠丝预处理后, 氮气气氛下用钠钾合金回流后取用. DMM等液体酯类用氢化钙或无水硫酸镁室温搅拌2 d后氮气气氛下蒸馏, 储存在手套箱中备用.草酸二甲酯和碳酸乙烯酯直接减压蒸馏后储存在手套箱中备用.
以催化DMM加氢为例说明活性评价过程:首先, 称取一定量Ru(acac)3、配体、DMM以及溶剂于釜体中, 反应釜组装完成后转移出手套箱; 紧接着, 用冰水冷却釜体至约278 K, 用H2洗气三次并充H2压力至7 MPa.然后, 将釜体置于加热装置中加热升温至指定温度并维持一定时间; 反应完成后, 快速将釜体冷却至约278 K并排去釜中残余的氢气.最后, 在反应液中加入一定量对二甲苯作为内标, 搅拌均匀后用气相色谱进行分析.气相色谱分析具体参数如下:采用N2作为载气, 并固定其流速为25 mL/min; 气化室和FID检测器温度分别为533和523 K; 程序升温过程如下: 313 K维持5 min, 后以10 K/min升高温度至473 K并在这一温度下维持20 min.根据所得谱图中产物、原料的峰面积进行转化率和产率的计算.
辅助材料(Supporting Information) 部分活性数据及反应产物的气相色谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Kraus, G. A. Clean 2008, 36, 648.
Arntz, D.; Wiegand, N. US 5015789, 1991.
(a) Slaugh, L. H.; Weider, P. R. US 5256827, 1993.
(b) Powell, J. B.; Mullin, S. B.; Weider, P. R.; Eubanks, D. C.; Arhancet, J. P. US 5770776, 1998.
(a) Kaur, G.; Srivastava, A. K.; Chand, S. Biochem. Eng. J. 2012, 64, 106.
(b) Lee, C. S.; Aroua, M. K.; Daud, W. M. A. W.; Cognet, P.; Pérès-Lucchese, Y.; Fabre, P. L.; Reynes, O.; Latapie, L. Renewable Sustainable Energy Rev. 2015, 42, 963.
(a) Wang, Y.; Zhou, J.; Guo, X. RSC Adv. 2015, 5, 74611.
(b) Sun, D.; Yamada, Y.; Sato, S.; Ueda, W. Appl. Catal. B Environ. 2016, 193, 75.
(a) Chen, L. F.; Guo, P. J.; Qiao, M. H.; Yan, S. R.; Li, H. X.; Shen, W.; Xu, H. L.; Fan, K. N. J. Catal. 2008, 257, 172.
(b) He, Z.; Lin, H.; He, P.; Yuan, Y. J. Catal. 2011, 277, 54.
(c) Peng, S. Y.; Xu, Z. N.; Chen, Q. S.; Chen, Y. M.; Sun, J.; Wang, Z. Q.; Wang, M. S.; Guo, G. C. Chem. Commun. 2013, 49, 5718.
(d) Ma, X. B.; Chi, H. W.; Yue, H. R.; Zhao, Y. J.; Xu, Y.; Lv, J.; Wang, S. P.; Gong, J. L. AIChE J. 2013, 59, 2530.
(a) Ding, T.; Tian, H.; Liu, J.; Wu, W.; Zhao, B. Catal. Commun. 2016, 74, 10.
(b) Ding, T.; Tian, H.; Liu, J.; Wu, W.; Yu, J. Chin. J. Catal. 2016, 37, 484.
(a) He, L.; Gong, X.; Ye, L.; Duan, X.; Yuan, Y. J. Energy Chem. 2016, 25, 1038.
(b) Yu, J.; Cao, J.; Du, L.; Wei, Y.; Wang, T.; Tian, H. Appl. Catal., A 2018, 555, 161.
(a) Zhao, B. G.; Han, Z. B.; Ding, K. L. Angew. Chem., Int. Ed. 2013, 52, 4744.
(b) Werkmeister, S.; Junge, K.; Beller, M. Org. Process Res. Dev. 2014, 18, 289.
(c) Pritchard, J.; Filonenko, G. A.; van Putten, R.; Hensen, E. J. M.; Pidko, E. A. Chem. Soc. Rev. 2015, 44, 3808.
Li, W.; Xie, J. H.; Yuan, M. L.; Zhou, Q. L. Green Chem. 2014, 16, 4081. doi: 10.1039/C4GC00835A
Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2012, 51, 13041. doi: 10.1002/anie.201207781
(a) Tan, X.; Wang, Y.; Liu, Y.; Wang, F.; Shi, L.; Lee, K. H.; Lin, Z.; Lv, H.; Zhang, X. Org. Lett. 2015, 17, 454.
(b) Tan, X.; Wang, Q.; Liu, Y.; Wang, F.; Lv, H.; Zhang, X. Chem. Commun. 2015, 51, 12193.
(a) Fang, X.; Zhang, C.; Chen, J.; Zhu, H.; Yuan, Y. RSC Adv. 2016, 6, 45512.
(b) Fang, X.; Sun, M.; Zheng, J.; Li, B.; Ye, L.; Wang, X.; Cao, Z.; Zhu, H.; Yuan, Y. Sci. Rep. 2017, 7, 3961.
(c) Zhang, Y. W.; Chen, Y. L.; Fang, X. L.; Yuan, Y. Z.; Zhu, H. P. Chin. J. Org. Chem. 2017, 37, 2275 (in Chinese).
(张亦伟, 陈艺林, 方霄龙, 袁友珠, 朱红平, 有机化学, 2017, 37, 2275.)
Ohkuma, T.; Ooka, H.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2675. doi: 10.1021/ja00114a043
Teunissen, H. T.; Elsevier, C. J. Chem. Commun. 1997, 667.
Teunissen, H. T.; Elsevier, C. J. Chem. Commun. 1998, 1367.
(a) Geilen, F. M. A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Angew. Chem., Int. Ed. 2010, 49, 5510.
(b) Wesselbaum, S.; vom Stein, T.; Klankermayer, J.; Leitner, W. Angew. Chem., Int. Ed. 2012, 51, 7499.
Saudan, L. A.; Saudan, C. M.; Debieux, C.; Wyss, P. Angew. Chem., Int. Ed. 2007, 46, 7473. doi: 10.1002/(ISSN)1521-3773
Geilen, F. M. A.; Engendahl, B.; H lscher, M.; Klankermayer, J.; Leitner, W. J. Am. Chem. Soc. 2011, 133, 14349. doi: 10.1021/ja2034377
(a) Van der Sluys, L. S.; Kubas, G. J.; Caulton, K. G. Organometallics 1991, 10, 1033.
(b) Chen, Y. Z.; Chan, W. C.; Lau, C. P.; Chu, H. S.; Lee, H. L.; Jia, G. Organometallics 1997, 16, 1241.
Hamilton, R. J.; Bergens, S. H. J. Am. Chem. Soc. 2006, 128, 13700. doi: 10.1021/ja065460s
Mirza, C.; Christian, B.; Bernhard, R.; Wolfgang, A. H.; Fritz, E. K. Angew. Chem., Int. Ed. 2011, 50, 8510. doi: 10.1002/anie.201102010
Ito, M.; Ootsuka, T.; Watari, R.; Shiibashi, A.; Himizu, A.; Ikariya, T. J. Am. Chem. Soc. 2011, 133, 4240. doi: 10.1021/ja1117254
(a) John, J. M.; Takebayashi, S.; Dabral, N.; Miskolzie, M.; Bergens, S. H. J. Am. Chem. Soc. 2013, 135, 8578.
(b) Tan, X.; Wang, Y.; Liu, Y.; Wang, F.; Shi, L.; Lee, K. H.; Lin, Z.; Lv, H.; Zhang, X. Org. Lett. 2015, 17, 454.
(a) Herd, O.; Heß ler, A.; Hingst, M.; Tepper, M.; Stelzer, O. J. Organomet. Chem. 1996, 522, 69.
(b) Hingst, M.; Tepper, M.; Stelzer, O. Eur. J. Inorg. Chem. 1998, 1998, 73.
(c) Habtemariam, A.; Watchman, B.; Potter, B. S.; Palmer, R.; Parsons, S.; Parkin, A.; Sadler, P. J. J. Chem. Soc., Dalton Trans. 2001, 1306.
(d) Doherty, S.; Knight, J. G.; Scanlan, T. H.; Elsegood, M. R. J.; Clegg, W. J. Organomet. Chem. 2002, 650, 231.
(e) Han, F. B.; Zhang, Y. L.; Sun, X. L.; Li, B. G.; Guo, Y. H.; Tang, Y. Organometallics 2008, 27, 1924.
(f) Richard, V.; Ipouck, M.; Mérel, D. S.; Gaillard, S.; Whitby, R. J.; Witulski, B.; Renaud, J. L. Chem. Commun. 2014, 50, 593.

图 1 催化丙二酸二甲酯加氢反应示意图
Figure 1 Reaction process for the catalytic hydrogenation of dimethyl malonate

图 3 溶剂对L1-Ru(acac)3催化DMM加氢的影响
Figure 3 Influence of solvent on the catalytic hydrogenation of DMM

图 4 DMM催化加氢制3-HPM可能的反应机理图
Figure 4 Proposed reaction mechanism for the reduction of DMM into 3-HPM
表 1 配体对催化DMM加氢反应的影响a
Table 1. Influence of ligands on the catalytic hydrogenation of DMM
| Entry | Ligand | Conv./% | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | |||
| 1 | L1 | 55 | 45 | 1 | 3 | 6 |
| 2 | L2 | 0 | — | — | — | — |
| 3 | L3 | 33 | 25 | 0 | 2 | 6 |
| 4 | L4 | 6 | 2 | 0 | 0 | 4 |
| 5 | L5 | 55 | 43 | 0 | 8 | 4 |
| 6 | L6 | 16 | 6 | 0 | 2 | 8 |
| 7 | L7 | 19 | 10 | 0 | 3 | 6 |
| 8 | L8 | 9 | 0 | 0 | 1 | 8 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), ligand (2 mol%), methanol (10 mL), temp.=393 K, p(H2)=7 MPa, time=16 h. b Mainly containing methyl acetate and methyl acrylate. | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 L1用量对催化DMM加氢反应的影响a
Table 2. The influence of the dosage of L1 on the catalytic hydrogenation of DMM
| Entry | n(L1)/n(Ru) | Conv./% | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | |||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 1 | 14 | 10 | 0 | 1 | 3 |
| 3 | 2 | 55 | 45 | 1 | 3 | 6 |
| 4 | 3 | 3 | 1 | 0 | 0 | 2 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), methanol (10 mL), temp.=393 K, p(H2)=7 MPa, time=16 h. b Mainly containing methyl acetate and methyl acrylate. | ||||||
下载: 导出CSV
表 3 反应温度和时间对催化DMM加氢反应的影响a
Table 3. Influence of reaction temperature and time on the catalytic hydrogenation of DMM
| Entry | Temp./ K | Time/ h | Conv./ % | Yield/% | |||
| 3-HPM | 1, 3-PDO | MP | Othersb | ||||
| 1 | 373 | 16 | 7 | 5 | 0 | 0 | 2 |
| 2 | 393 | 8 | 44 | 36 | 0 | 2 | 6 |
| 3 | 393 | 16 | 55 | 45 | 1 | 3 | 6 |
| 4 | 393 | 24 | 58 | 47 | 2 | 4 | 5 |
| 5 | 413 | 8 | 50 | 34 | 0 | 5 | 11 |
| a Reaction conditions: DMM (7.5 mmol), Ru(acac)3 (1 mol%), L1 (2 mol%), methanol (10 mL), p(H2)=7 MPa. b Mainly containing methyl acetate and methyl acrylate. | |||||||
下载: 导出CSV
表 4 其它酯类化合物的催化加氢反应a
Table 4. Substrate scope for the catalytic system
| Entry | Substrate | Temp./K | Conv./% | Hydrogenation yield/% | |
| Product Ab | Product Bc | ||||
| 1 | E1 | 393 | 86 | 75 (3-HPM) | 8 (1, 3-PDO) |
| 2 | E2 | 393 | 7 | 3 (1, 3-PDO) | — |
| 3 | E3 | 423 | 83 | 82 (1, 2-PDO) | — |
| 4 | E4 | 388 | 100 | 98 (MG) | 1 (EG) |
| 5d | E5 | 383 | 98 | 97 (MPEG) | 1 (EG) |
| 6 | E6 | 413 | 97 | 97 (1, 4-PDO) | — |
| 7d | E7 | 413 | 63 | 59 (EG) | 54 (MeOH) |
| a Reaction conditions: Substrate (7.5 mmol), Ru(acac)3 (1 mol%), L1 (2 mol%), 2 mL methanol and 8 mL THF as the solvent, P(H2)=7 MPa, Time=16 h. b Yield of product A. c Yield of product B. d 2 mL ethanol and 8 mL THF as the solvent. MG: methyl glycolate; EG: ethylene glycol; MPEG: ethyl glycolate; 1, 2-PDO: 1, 2-propanediol; 1, 4-PDO: 1, 4-pentanediol. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们