Scheme 1.
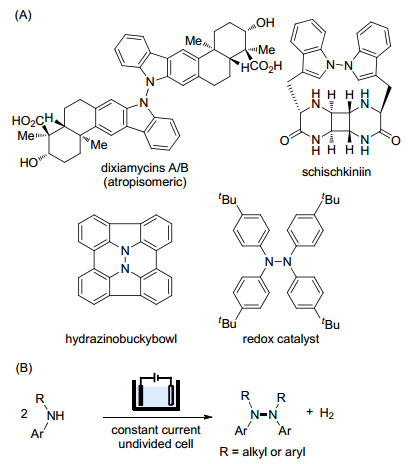
(A) Selected compounds containing the tetrasubstituted hydrazine moiety, and (B) electrochemical dehydrogenative dimerization of secondary amines

Tetrasubstituted hydrazines have been found in natural products and materials (Scheme 1A).[1] We have recently discovered that 1, 1, 2, 2-tetrakis(4-(tert-butyl)phenyl)- hydrazine is an efficient redox catalyst for electrosynthesis.[2] Tetrasubstituted hydrazines can be synthesized through transition metal-catalyzed oxidative coupling of secondary amines in the presence of a terminal oxidant such as oxygen or diaziridinone.[3] While these methods are attractive, the use of transition metal catalysts cause the concern of trace-metal contamination of the product.
Organic electrochemistry, which employs traceless electron as "reagents" to achieve redox reactions, has been attracting increasing interests among organic chemists.[4, 5] In addition to its reagent-free feature, electrochemical methods can achieve transformations difficult for alternative methods that employ chemical oxidants. As an example, Baran and coworkers reported that the electrochemical oxidation outperformed chemical oxidants in promoting the N—N dimerization of carbazoles.[6] In this method, careful control of the electrode potential is essential for success. We have been interested in developing novel redox catalysts for organic electrosynthesis.[7] To further explore the potential of tetrasubstituted hydrazines as redox catalysts, we need an transition metal-free and scalable method to access these compounds. Herein we report the development of an electrochemical synthesis of tetrasubstituted hydrazines through dehydrogenative dimerization of secondary arylamines (Scheme 1B). The reactions are conveniently carried out under constant current conditions in an undivided cell.
The oxidative dimerization of bis(4-(tert-butyl)phenyl)- amine (1a) was selected as a model reation to search for optimal electrolysis conditions (Table 1). Screening experiments revealed that an isolated yield of 89% of tetraarylhydrazine 2a could be obtained when the reaction was carried out in a mixed solvent of tetrahydrofuran (THF)/MeOH (V:V=3:1) in the presence of 1 equiv. pyridine (Entry 1). Conveniently, the electrolysis was conducted using a constant current (janode=0.1 mA•cm−2)in an undivided cell equipped with a reticulated vitreous carbon (RVC) anode and a platinum plate cathode. The basic additive was needed for optimal results as its absence resulted in a lower yield of 64% (Entry 2). However, the base could be reduced to 0.5 equiv. (Entry 3) or replaced with inorganic salts, such as KOAc (Entry 4), K2CO3 (Entry 5) or KHCO3 (Entry 6), without affecting the yield of 2a. The volume ratio of THF/MeOH could be lowered to 1:1 (Entry 7) but not to 1:3 (Entry 8). Importantly, the electrosynthesis was not sensitive to oxygen, as the reaction of 1a under air afforded 2a in 93% yield (Entry 9).
 下载:
导出CSV
下载:
导出CSV
 |
||
| Entry | Deviation from the standard condition | Yieldb/% |
| 1 | None | 96 (89)c |
| 2 | No base | 64c |
| 3 | 0.5 equiv. pyridine as base | 91 |
| 4 | 1 equiv. KOAc as base | 93 |
| 5 | 1 equiv. K2CO3 as base | 94 |
| 6 | 1 equiv. KHCO3 as base | 95 |
| 7 | THF/MeOH (V:V=1:3) as solvent | 38 |
| 8 | THF/MeOH (V:V=1:1) as solvent | 93 |
| 9 | Under air | 93 |
| a Reaction conditions: RVC anode, Pt plate cathode, undivided cell, 1a (0.5 mmol), base (0.5 mmol), solvent (10 mL), Et4NBF4 (0.5 mmol), r.t.. b Yields were determined by 1H NMR analysis using 1, 3, 5-trimethoxybenzene as the internal standard. c Isolated yield. | ||
The substrates scope was next investigated (Table 2). The reaction was compatible with diphenylamines substituted at the para-position of both phenyl rings with Me (2b), Ph (2c), F (2d), Br (2e) or I (2f). Unsymmetrically para-substituted diphenylamines (2g~2i) and 4-methyl-N- phenylaniline (2j) bearing an unsubstituted phenyl ring were also viable substrates. However, the reaction of the parent diphenylamine 1k failed to afford the tetraphenylhydrazine (2k). Diarylamines containing a pyridyl ring also underwent efficient dimerization (2l). Further investigation revealed that 3, 6-disubstituted carbazoles were suitable substrates (2m and 2n). N-Alkylanilines also underwent N—N dimerization but required the use of KOAc as the base and a higher reaction temperature (2o~2u). The electrosynthesis could be scaled up to gram scale as demonstrated for the synthesis of 2b~2e.
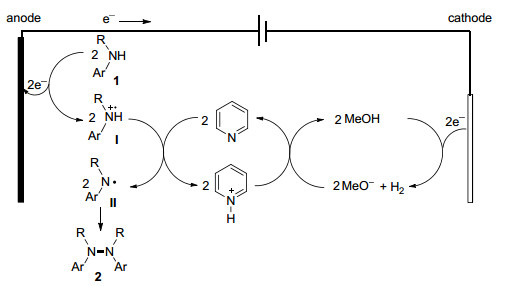
A possible reaction mechanism was proposed (Scheme 2). The secondary amine 1 was oxidized on the anode to generate radical cation I, which loses a proton to the added base to generate aminyl radical Ⅱ. Homodimerization of Ⅱ affords the tetrasubstituted hydrazine product 2.[7] At the cathode, MeOH solvent was reduced to generate hydrogen gas and MeO-. The cathodically generated MeO- reacts with pyridinium to regenerate MeOH and pyridine. Hence, MeOH and pyridine are not consumed during the electrolysis. Since the oxidation potential of the hydrazine product [Ep/2(2a)=0.68 V vs SCE in MeCN] is lower than that of the starting amine [Ep/2(1a)=0.80 V vs SCE in MeCN], the oxidative dimerization reaction is likely self-catalyzed, e.g. the hydrazine product serves as a redox catalyst for the anodic generation of radical cation I.
In summary, an electrochemical method for the synthesis of tetrasubstituted hydrazines with diverse electronic properties through dehydrogenative homodimerization of secondary arylamines has developed. The reactions do not need oxidizing reagents and transition metal catalysts and can be carried out efficiently on gram scale.
Anhydrous tetrahydrofuran was obtained from sodium/ benzophenone by distillation under argon. Flash column chromtography was performed with silica gel (230~400 mesh). NMR spectra were recorded on a Bruker AV-500 instruments. High resolution mass spectra (ESI HRMS) were recorded on a Micromass QTOF2 Quadruple/ Time-of-Flight Tandem mass spectrometer by the instrumentation center of Department of Chemistry, Xiamen University. Cyclic voltammograms were obtained on a CHI 760E potentiostat. Infrared spectra (IR) and melting point (m.p.) were recorded on a Nicolet-Avater 330 spectrometer and BUCHI melting point M-560, respectively.
A 25 mL three-necked round-bottomed flask was charged with substrate (0.5 mmol), Et4NBF4 (0.5 mmol) and pyridine (0.5 mmol). The flask was equipped with a reticulated vitreous carbon (100 PPI, 1.0 cm×1.0 cm×1.2 cm) anode, a platinum plate (1 cm×1 cm) cathode and then flushed with argon. The electrodes were fixed on the flask using thermometer adaptors. THF (7.5 mL) and MeOH (2.5 mL) were added. The electrolysis was carried out at room temperature using a constant current of 7.5 mA until complete consumption of the substrate (monitored by TLC or 1H NMR). For the synthesis of compounds 2o~2u, the reactions were conducted under reflux (oil bath temperature set at 80 ℃) employing KOAc (0.5 mmol) as the base. The reaction mixture was concentrated under reduced pressure, and the residue was chromatographed through silica gel eluting with ethyl acetate/hexanes to give the desired product.
The gram scale synthesis of 2b~2e was conducted in a 250 mL beaker-type cell with a RVC (3.4 cm×3.4 cm×1.2 cm) anode, a Pt plate cathode (3 cm×3 cm), and a constant current of 70 mA. The reaction mixture consisted substrate (6.0 mmol), Et4NBF4 (1.3 g, 6.0 mmol), pyridine (0.47 g, 6.0 mmol), THF (90 mL) and MeOH (30 mL).
1, 1, 2, 2-Tetra([1, 1'-biphenyl]-4-yl)hydrazine (2c): Yield 90%, gram scale yield 85%, yellow solid. m.p. 119.1~120.4 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.54 (d, J=7.7 Hz, 8H), 7.51 (d, J=8.5 Hz, 8H), 7.47 (d, J=8.5 Hz, 8H), 7.39 (t, J=7.7 Hz, 8H), 7.30~7.26 (m, 4H); 13C NMR (126 MHz, CDCl3) δ: 142.8, 140.6, 135.3, 128.9, 128.0, 127.0, 126.7, 118.7; IR (KBr) ν: 1601, 1518, 1485, 1320, 760 cm-1; HRMS (ESI) calcd for C48H36N2Na [M+Na]+663.2771, found 663.2770.
1, 1, 2, 2-Tetrakis(4-iodophenyl)hydrazine (2f): Yield 84%, white solid. m.p. 130.2~132.0 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.49 (d, J=8.4 Hz, 8H), 6.96 (d, J=8.4 Hz, 8H); 13C NMR (126 MHz, CDCl3) δ: 142.4, 138.5, 120.2, 85.6; IR (KBr) ν: 1576, 1484, 1316, 1290, 1180, 809 cm-1; HRMS (ESI) calcd for C24H16I4N2Na [M+Na]+862.7384, found 862.7398.
1, 2-Bis(4-(tert-butyl)phenyl)-1, 2-bis(4-fluorophenyl)hydrazine (2g): Yield 72%, light yellow solid. m.p. 74.1~74.3 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.23~7.21 (m, 4H), 7.21~7.18 (m, 4H), 7.17~7.12 (m, 4H), 6.91~6.84 (m, 4H), 1.26 (s, 18H); 13C NMR (126 MHz, CDCl3) δ: 158.3 (d, JC-F=240.7 Hz), 144.8, 141.4, 140.2 (d, JC–F=2.6 Hz), 126.2, 119.9 (d, JC-F=7.8 Hz), 117.3, 115.9 (d, JC-F=22.6 Hz), 34.3, 31.6; 19F NMR (471 MHz, CDCl3) δ: -121.5; IR (KBr) ν: 1504, 1318, 1226, 1134, 1080, 822 cm-1; HRMS (ESI) calcd for C32H34F2N2Na [M+Na]+507.2582, found 507.2583.
1, 2-Bis(4-(tert-butyl)phenyl)-1, 2-bis(4-chlorophenyl)-hydrazine (2h): Yield 90%, brown solid. m.p. 134.2~136.6 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.24~7.20 (m, 4H), 7.20~7.14 (m, 8H), 7.14~7.10 (m, 4H), 1.26 (s, 18H); 13C NMR (126 MHz, CDCl3) δ: 145.8, 142.7, 140.5, 129.3, 126.3 (2C), 118.8, 118.3, 34.4, 31.5; IR (KBr) ν: 1591, 1487, 1318, 1095, 819 cm-1; HRMS (ESI) calcd for C32H34Cl2N2Na [M+Na]+ 539.1991, found 539.1995.
1, 2-Bis(4-(tert-butyl)phenyl)-1, 2-bis(4-bromophenyl)-hydrazine (2i): Yield 89%, light yellow solid. m.p. 100.0~101.1 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.26 (d, J=6.9 Hz, 4H), 7.23~7.20 (m, 4H), 7.19~7.16 (m, 4H), 7.13~7.09 (m, 4H), 1.27 (s, 18H); 13C NMR (126 MHz, CDCl3) δ: 145.9, 143.2, 140.3, 132.2, 126.3, 119.1, 118.4, 113.7, 34.4, 31.5; IR (KBr) ν: 1584, 1484, 1317, 1134, 1079, 818 cm-1; HRMS (ESI) calcd for C32H34Br2N2Na [M+Na]+629.0960, found 629.0959.
1, 2-Bis(4-(tert-butyl)phenyl)-1, 2-di(pyridin-2-yl)hydra-zine (2l): Yield 63%, light yellow sticky solid. 1H NMR (500 MHz, CDCl3) δ: 8.26 (ddd, J=4.9, 2.0, 1.0 Hz, 2H), 7.46~7.42 (m, 4H), 7.27 (s, 2H), 6.95 (dd, J=8.5, 1.0 Hz, 2H), 6.78 (ddd, J=7.3, 4.9, 1.0 Hz, 2H), 1.27 (s, 18H); 13C NMR (126 MHz, CDCl3) δ: 156.7, 148.0, 146.4, 139.5, 138.3, 125.8, 120.0, 116.7, 110.6, 34.4, 31.6; IR (KBr) ν: 1587, 1512, 1468, 1429, 1331 cm-1; HRMS (ESI) calcd for C30H35N4 [M+H]+ 451.2856, found 451.2861.
3, 3', 6, 6'-Tetra-tert-butyl-9, 9'-bicarbazole (2m): Yield 57%; White solid; m.p. 263.4~266.1 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.18 (d, J=1.7 Hz, 4H), 7.31~7.25 (m, 4H), 6.71 (d, J=8.5 Hz, 4H), 1.43 (s, 36H); 13C NMR (126 MHz, CDCl3) δ: 144.1, 138.6, 124.2, 122.0, 116.6, 108.8, 35.0, 32.2; IR (KBr) ν: 1632, 1486, 1397, 1031, 787 cm-1; HRMS (ESI) calcd for C40H48N2Na [M+Na]+ 579.3710, found 579.3708.
1, 2-Bis(4-fluorophenyl)-1, 2-dimethylhydrazine (2p): Yield 58%, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.00~6.90 (m, 4H), 6.82~6.75 (m, 4H), 2.91 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 156.9 (d, JC–F=237.0 Hz), 145.6 (d, JC–F=1.8 Hz), 115.8 (d, JC–F=22.3 Hz), 113.90 (d, JC–F=7.4 Hz), 33.8; 19F NMR (471 MHz, CDCl3) δ: -126.9; IR (KBr) ν: 2921, 1505, 1225, 1139, 1098, 823 cm-1; HRMS (APCI) calcd for C14H15F2N2 [M+H]+249.1198, found 249.1205.
1, 2-Bis(3-bromophenyl)-1, 2-dimethylhydrazine (2t): Yield 36%, white solid. m.p. 93.1~93.8 ℃; 1H NMR (500 MHz, CDCl3) δ: 7.09 (t, J=8.2 Hz, 2H), 7.01~6.91 (m, 4H), 6.71 (ddd, J=8.2, 2.4, 0.9 Hz, 2H), 2.97 (s, 6H); 13C NMR (126 MHz, CDCl3) δ: 150.1, 130.8, 123.8, 122.0, 115.6, 111.3, 34.2; IR (KBr) ν: 2919, 1587, 1481, 1275, 1029, 750 cm-1; HRMS (APCI) calcd for C14H14Br2N2Na [M+Na]+ 390.9416, found 390.9420.
1, 2-Bis(4-chlorophenyl)-1, 2-di(pent-4-en-1-yl)hydrazine (2u): Yield 61%, yellow oil. 1H NMR (500 MHz, CDCl3) δ: 7.17~7.11 (m, 4H), 6.63~6.59 (m, 4H), 5.78 (ddt, J=16.9, 10.2, 6.7 Hz, 2H), 5.05~4.97 (m, 4H), 3.38 (t, J=8.0 Hz, 4H), 2.08 (qt, J=7.3, 1.4 Hz, 4H), 1.83~1.70 (m, 4H); 13C NMR (126 MHz, CDCl3) δ: 146.9, 137.6, 129.3, 123.4, 115.7, 113.9, 50.5, 31.5, 27.1; IR (KBr) ν: 2921, 1592, 1492, 1384, 1277, 1096, 816 cm-1; HRMS (APCI) calcd for C22H27Cl2N2 [M+H]+ 389.1546, found 389.1548.
Supporting Information Cyclic voltammograms and NMR spectra of new compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Blair, L. M.; Sperry, J. J. Nat. Prod. 2013, 76, 794.
(b) Zhang, Q.; Mándi, A.; Li, S.; Chen, Y.; Zhang, W.; Tian, X.; Zhang, H.; Li, H.; Zhang, W.; Zhang, S.; Ju, J.; Kurtán, T.; Zhang, C. Eur. J. Org. Chem. 2012, 5256.
(c) Shoeb, M.; Celik, S.; Jaspars, M.; Kumarasamy, Y.; MacManus, S. M.; Nahar, L.; Thoo-Lin, P. K.; Sarker, S. D. Tetrahedron 2005, 61, 9001.
(d) Higashibayashi, S.; Pandit, P.; Haruki, R.; Adachi, S.-i.; Kumai, R. Angew. Chem., Int. Ed. 2016, 55, 10830.
Hou, Z. W.; Mao, Z. Y.; Melcamu, Y. Y.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 1636. doi: 10.1002/anie.v57.6
(a) Reddy, C. B. R.; Reddy, S. R.; Naidu, S. Catal. Commun. 2014, 56, 50.
(b) Yan, X.-M.; Chen, Z.-M.; Yang, F.; Huang, Z.-Z. Synlett 2011, 2011, 569.
(c) Ryan, M. C.; Martinelli, J. R.; Stahl, S. S. J. Am. Chem. Soc. 2018, 140, 9074.
(d) Fritsche, R. F.; Theumer, G.; Kataeva, O.; Knolker, H. J. Angew. Chem., Int. Ed. 2017, 56, 549.
(e) Zhu, Y.; Shi, Y. Org. Lett. 2013, 15, 1942.
(a) Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230.
(b) Tang, S.; Liu, Y.; Lei, A. Chem 2018, 4, 27.
(c) Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485.
(d) Yang, Q. L.; Fang, P.; Mei, T. S. Chin. J. Chem. 2018, 36, 338.
(e) Wiebe, A.; Gieshoff, T.; Mö hle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel Siegfried, R.
Angew. Chem., Int. Ed. 2018, 57, 5594.
(f) Moeller, K. D. Chem. Rev. 2018, 118, 4817.
(g) Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265.
(h) Francke, R.; Little, R. D. Chem. Soc. Rev. 2014, 43, 2492.
(a) Hayashi, R.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2016, 138, 8400.
(b) Horn, E. J.; Rosen, B. R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M. D.; Baran, P. S. Nature 2016, 533, 77.
(c) Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem., Int. Ed. 2017, 56, 3009.
(d) Fu, N.; Li, L.; Yang, Q.; Luo, S. Org. Lett. 2017, 19, 2122.
(e) Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346.
(f) Yang, Q. L.; Li, Y. Q.; Ma, C.; Fang, P.; Zhang, X. J.; Mei, T. S. J. Am. Chem. Soc. 2017, 139, 3293.
(g) Zhang, S.; Li, L.; Xue, M.; Zhang, R.; Xu, K.; Zeng, C. Org. Lett. 2018, 20, 3443.
(h) Fu, N.; Sauer, G. S.; Saha, A.; Loo, A.; Lin, S. Science 2017, 357, 575.
(i) Li, J.; Huang, W.; Chen, J.; He, L.; Cheng, X.; Li, G. Angew. Chem., Int. Ed. 2018, 57, 5695.
(j) Yuan, Y.; Cao, Y.; Qiao, J.; Lin, Y.; Jiang, X.; Weng, Y.; Tang, S.; Lei, A. Chin. J. Chem. 2019, 37, 49.
(k) Ye, Z. H.; Ding, M. R.; Wu, Y. Q.; Li, Y.; Hua, W. K.; Zhang, F. Z. Green Chem. 2018, 20, 1732.
(l) Qian, P.; Su, J.-H.; Wang, Y.; Bi, M.; Zha, Z.; Wang, Z. J. Org. Chem. 2017, 82, 6434.
(m) Lin, D. Z.; Huang, J. M. Org. Lett. 2018, 20, 2112.
(n) Zhang, L.; Zhang, Z. X.; Hong, J. T.; Yu, J.; Zhang, J. N.; Mo, F. Y. J. Org. Chem. 2018, 83, 3200.
Rosen, B. R.; Werner, E. W.; O'Brien, A. G.; Baran, P. S. J. Am. Chem. Soc. 2014, 136, 5571. doi: 10.1021/ja5013323
(a) Zhu, L.; Xiong, P.; Mao, Z. Y.; Wang, Y. H.; Yan, X.; Lu, X.; Xu, H.-C. Angew. Chem., Int. Ed. 2016, 55, 2226.
(b) Xiong, P.; Xu, H.-H.; Song, J.; Xu, H.-C. J. Am. Chem. Soc. 2018, 140, 2460.
(c) Qian, X.-Y.; Li, S.-Q.; Song, J.; Xu, H.-C. ACS Catal. 2017, 2730.
(d) Cai, C.-Y.; Xu, H.-C. Nat. Commun. 2018, 9, 3551.
(e) Hou, Z.-W.; Yan, H.; Song, J.-S.; Xu, H.-C. Chin. J. Chem. 2018, 36, 909.
(f) Xiong, P.; Xu, H.-H.; Song, J.; Xu, H.-C. J. Am. Chem. Soc. 2018, 140, 2460.
(g) Zhao, H.-B.; Xu, P.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 15153.
(h) Wu, Z.-J.; Li, S.-R.; Xu, H.-C. Angew. Chem., Int. Ed. 2018, 57, 14070.
(i) Xu, F.; Li, Y.-J.; Huang, C.; Xu, H.-C. ACS Catal. 2018, 3820.
(j) Hou, Z. W.; Mao, Z. Y.; Zhao, H. B.; Melcamu, Y. Y.; Lu, X.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2016, 55, 9168.
(k) Wu, Z.-J.; Li, S.-R.; Long, H.; Xu, H.-C. Chem. Commun. 2018, 54, 4601.
(l) Hou, Z.-W.; Mao, Z.-Y.; Song, J.; Xu, H.-C. ACS Catal. 2017, 5810.
(a) Gieshoff, T.; Kehl, A.; Schollmeyer, D.; Moeller, K. D.; Waldvogel, S. R. J. Am. Chem. Soc. 2017, 139, 12317.
(b) Gieshoff, T.; Schollmeyer, D.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2016, 55, 9437.

Scheme 1 (A) Selected compounds containing the tetrasubstituted hydrazine moiety, and (B) electrochemical dehydrogenative dimerization of secondary amines

Scheme 2 Proposed mechanism for the electrochemical synthesis of tetrasubstituted hydrazines
Table 1. Optimization of reaction conditionsa
| |
||
| Entry | Deviation from the standard condition | Yieldb/% |
| 1 | None | 96 (89)c |
| 2 | No base | 64c |
| 3 | 0.5 equiv. pyridine as base | 91 |
| 4 | 1 equiv. KOAc as base | 93 |
| 5 | 1 equiv. K2CO3 as base | 94 |
| 6 | 1 equiv. KHCO3 as base | 95 |
| 7 | THF/MeOH (V:V=1:3) as solvent | 38 |
| 8 | THF/MeOH (V:V=1:1) as solvent | 93 |
| 9 | Under air | 93 |
| a Reaction conditions: RVC anode, Pt plate cathode, undivided cell, 1a (0.5 mmol), base (0.5 mmol), solvent (10 mL), Et4NBF4 (0.5 mmol), r.t.. b Yields were determined by 1H NMR analysis using 1, 3, 5-trimethoxybenzene as the internal standard. c Isolated yield. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
