图式 1.
卤代芳烃与三氟乙酸钠的三氟甲基化反应
Scheme 1.
Trifluoromethylation of aromatic halides with sodium trifluoroacetate

含氟基团具有独特的性质, 这类基团广泛存在于医药[1]、农药[2]和材料[3]中.市场销售的大约20%的医药和30%~40%的农药中含有氟原子, 销量排名前10的4种药物中含有氟原子[4].与不含氟药物相比, 含氟药物通常具有代谢稳定、药物代谢动力学高等特性[4a, 5].在含氟基团中, 三氟甲基基团是一种重要的结构单元, 不仅可以用于染料、高分子材料的改性, 也可以调节药物的代谢活性, 提高脂溶性、生物渗透性和靶向选择性等, 在改善药效和药物动力学方面具有重要作用[6].大量已经上市或处于临床试验中的药物含有三氟甲基, 表现出了相较于其他药物更好的疗效.例如, 用于治疗重度抑郁症、神经性贪食、强迫性神经失调的药物Fluoxetine与传统抗忧郁药物相比副作用较小[6a]; 新一代非甾体抗炎镇痛药celecoxib用于治疗多种临床常见的急性疼痛、术后疼痛等, 也用于治疗慢性疼痛, 如骨关节炎、类风湿关节炎、强直性脊柱炎等[7]; enzalutamide被认为是最好的一个治疗转移性前列腺癌的药物[6f].此外, 还有其他一些含三氟甲基的上市的畅销药, 包括dutasteride、bicalutamide、leflunomide、nilutamide、dexiansoprazole等[6e, 6f].不仅许多医药中含有三氟甲基基团, 很多农药中也含有三氟甲基基团.例如, 除草剂氟乐灵(Trifluralin)、吡氟禾草灵(Fluazifop-butyl)、乙氧氟草醚(Oxyfluorfen)和杀虫剂氟虫腈(Fipronil), 氟啶脲(Chlorfluazuron)以及杀菌剂啶氧菌酯(Picoxystrobin)等.这些含三氟甲基农药在农业生产中发挥了非常重要的作用.
在有机分子中引入三氟甲基基团, 能够使其表现出独特的物理和化学性质, 具有重要的意义[8].因此, 三氟甲基的引入受到了科学工作者的广泛关注, 并且发展出了一系列引入三氟甲基的方法, 包括钯[9]、铜[10]及其他过渡金属催化的三氟甲基化[11~14]或者自由基三氟甲基化等方法[15, 16].新的三氟甲基化试剂的发现加速了三氟甲基化反应的发展[17].三氟甲基化试剂一般可以分为亲电、亲核、自由基三种类型[18].尽管这些三氟甲基化试剂可以用于有机分子的三氟甲基化反应, 但在使用过程中存在许多缺点.例如, 有些三氟甲基化试剂价格昂贵、毒性强、使用不方便, 有些试剂现在还没有商业化或者使用过程中会产生大量的化学废弃物.因此, 为了解决上述三氟甲基化试剂的诸多缺点, 迫切需要开发出廉价且易于处理的新的三氟甲基化试剂.
三氟乙酸及其衍生物作为一种具有广阔前景的三氟甲基化试剂, 具有廉价、易得、后处理方便等优点, 并且反应的副产物是二氧化碳, 符合绿色化学理念[19].本文将系统总结利用三氟乙酸及其衍生物为三氟甲基源来实现三氟甲基化反应的研究进展.
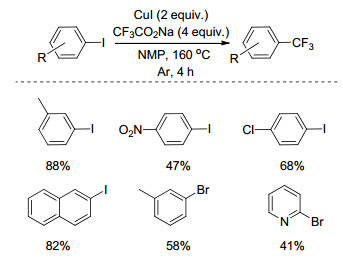
含三氟甲基的芳烃是一类重要的结构单元[20], 因此, 在芳环的特定位置上引入三氟甲基基团具有重要的意义.早在1981年, Kondo课题组[21]以CF3CO2Na为三氟甲基源, CuI为催化剂, N-甲基吡咯烷酮(NMP)为溶剂, 在氩气氛围下实现了碘苯C—X (Br, I)键的三氟甲基化反应(Scheme 1).与之前报道的方法相比, 该方法操作简单、三氟甲基源来源丰富, 产率中等至良好.
1998年, Hünig小组[22]以CF3CO2Na为三氟甲基源, CuI为催化剂, N, N-二甲基乙酰胺(DMA)与PhMe的混合物为溶剂, 实现了多取代碘代芳烃的三氟甲基化反应(Scheme 2).
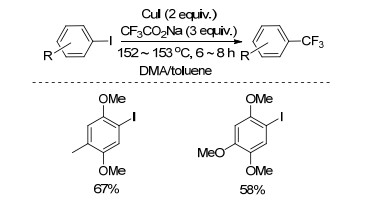
2001年, Branch小组[23]以CF3CO2K为三氟甲基源, CuI为催化剂, N, N-二甲基甲酰胺(DMF)与PhMe的混合物为溶剂, 110 ℃下反应1.5 h, 实现了5-溴代异吲哚碳溴键的三氟甲基化反应(Scheme 3), 该反应产率较高.
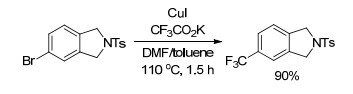
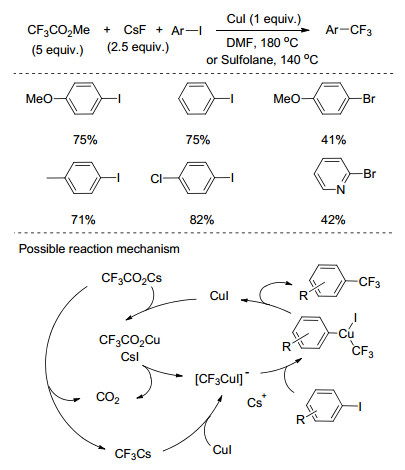
2007年, Langlois课题组[24]报道了芳基卤代物与三氟乙酸甲酯(MTFA)的亲核三氟甲基化反应(Scheme 4).该反应中以三氟乙酸甲酯为三氟甲基源, 芳基碘代物或芳基溴代物都能发生反应, 而芳基氯代物几乎不能发生反应.以DMF为溶剂时, 反应温度需要加热到180 ℃; 而以环丁砜为溶剂时, 反应在140 ℃时就能很好地进行, 这可能是由于环丁砜对三氟甲磺酸亚铜中间体具有配位作用.无论以DMF为溶剂还是以环丁砜为溶剂, 都需要加入过量的碘化亚铜, 这是由于氧化加成的速率远小于脱羧的速率, 脱羧产生的不稳定的三氟甲基负离子需要铜来稳定.
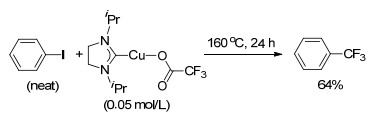
2010年, Vicic小组[25]首次以合成的三氟乙酸N-杂环卡宾(NHC)铜配合物作为催化剂和三氟甲基源, 实现了碘苯或溴苯的碳卤键三氟甲基化反应(Scheme 5).当以(SIiPr)Cu(TFA)为催化剂及三氟甲基源, 碘苯既作溶剂又作反应物, 在160 ℃时反应得到中等收率的三氟甲基化产物; 而以CuI为催化剂, CF3CO2Na为三氟甲基源时, 以碘苯既作溶剂又作反应物, 得不到三氟甲基化的产物.对比试验表明, 在芳基卤溶剂中, 铜配合物的效果要好于无配体的CuI.当把卤苯溶剂与N, N-二甲基乙酰胺按体积比配成1:1时, 上述含三氟甲基铜配合物发生三氟甲基化的效果要差于CuI/CF3CO2Na体系, 该现象说明了基于胺的溶剂可能对CuI有配位作用, 加速了脱羧三氟甲基化反应的进程.
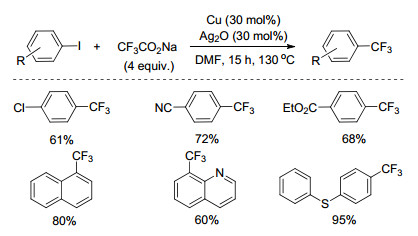
2011年, Duan和Li课题组[26]报道了铜催化的碘苯与CF3CO2Na的三氟甲基化反应(Scheme 6).该反应以对氯碘苯为原料, Cu粉为催化剂, CF3CO2Na为三氟甲基源, Ag2O为添加剂, DMF为溶剂, 130 ℃下反应15 h, 得到61%的产率.该反应具有较好的官能团兼容性, 含有卤素、氰基、酯基、硝基等基团的底物都能很好地发生反应, 产率中等至优秀.反应条件与之前报道的以CF3CO2Na为三氟甲基源的反应相比, 条件较温和.
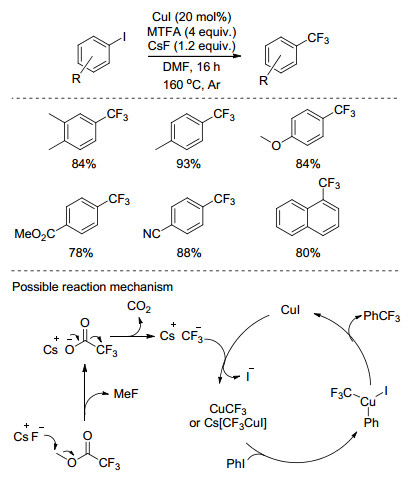
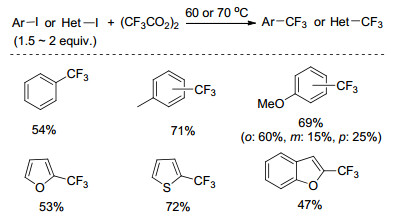
2012年, Wu小组[27]以碘苯或溴苯为底物, CuI为催化剂, 1, 10-菲啰啉为配体, 铯盐为碱, 廉价且易得的三氟乙酸甲酯(MTFA)为三氟甲基源, DMF为溶剂, 首次实现了卤苯与MTFA的三氟甲基化反应(Scheme 7).该反应中, 铯盐对于MTFA的脱羧产生三氟甲基负离子起了至关重要的作用.
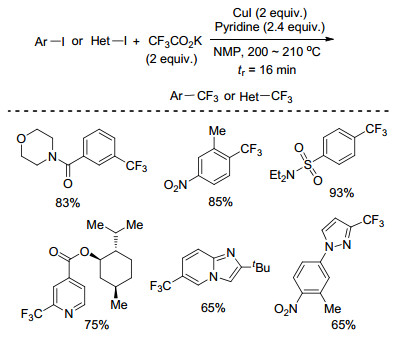
2013年, Buchwald课题组[28]首次实现了在流动体系中CuI促进的碘代芳烃或杂芳烃的碳碘键的三氟甲基化反应(Scheme 8).该反应是在微型反应器中进行的, 以CF3CO2K为三氟甲基源, 吡啶为配体, NMP为溶剂, 200 ℃下反应16 min, 产率较高且可以放大到克级规模.该反应时间短, 官能团兼容性好, 含有硝基、磺酰胺基、吡啶、吲哚、吡唑等单元的底物也能很好地发生反应.
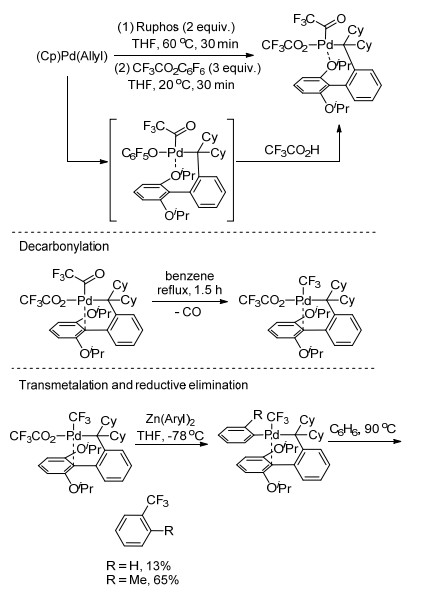
2014年, Sanford课题组[29]以三氟乙酸苯酯为三氟甲基源, 详细研究了钯催化脱羧三氟甲基化反应的催化循环(Scheme 9).作者研究了催化循环的四个基本步骤:首先, (Cp)Pd(allyl)与RuPhos在60 ℃时反应30 min, 生成的(RuPhos)nPd0中间体与三氟乙酸苯酯发生氧化加成, 再与三氟乙酸发生配体交换生成(RuPhos)Pd- (COCF3)(CO2CF3); 然后将(RuPhos)Pd(COCF3)(CO2CF3)在苯中80 ℃回流1.5 h脱去CO生成中间体(RuPhos)- Pd(CF3)(CO2CF3); 接着, (RuPhos)Pd(CF3)(CO2CF3)与Zn(Aryl)2发生转金属化生成(RuPhos)Pd(CF3)(aryl); 最后, (RuPhos)Pd(CF3)(aryl)在苯中90 ℃加热12 h发生还原消除生成三氟甲基化的芳基产物.与其他两个步骤相比, 脱去CO和还原消除需要相对较高的温度.这一反应说明, 将三氟乙酸酯用于交叉偶联反应中具有潜在的可能.
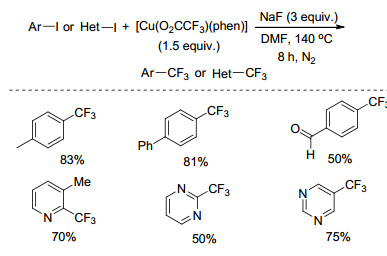
2016年, Weng等[30]以新合成的廉价的[Cu(O2C-CF3)(phen)]络合物为三氟甲基源, NaF为添加剂, DMF为溶剂, 140 ℃下反应, 实现了卤代芳烃或杂芳烃的三氟甲基化反应(Scheme 10).该反应具有良好的官能团容忍性, 含有醛基、乙酰基、硝基、吡啶等基团的底物反应得到中等至良好的收率, 该方法也可以用于雌二醇衍生物的三氟甲基化反应.机理研究表明, 该反应经历了亲核反应的过程而不是自由基的反应过程.
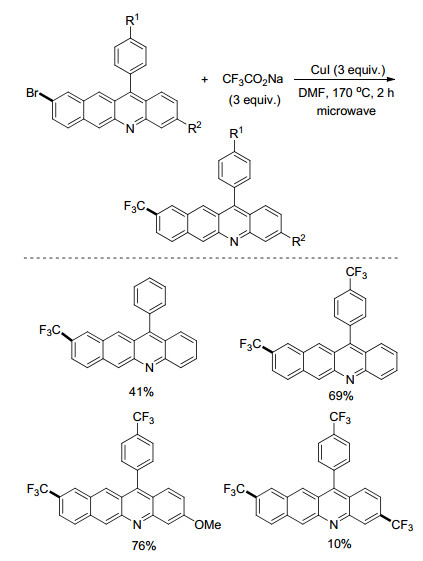
2017年, Torikai小组[31]以CF3CO2Na为三氟甲基源, CuI为催化剂, DMF为溶剂, 在微波辐射下实现了溴代苯并吖啶的三氟甲基化反应(Scheme 11).该反应无需加入配体, 即使两个溴取代的底物也能发生双三氟甲基化反应.而在油浴条件下, 在NMP中回流10.5 h, 该三氟甲基化反应不能发生.
C—H键官能团化反应具有较高的原子经济性和步骤经济性的特点, 符合绿色化学发展理念[32].因此, 直接的C—H键三氟甲基化反应是合成含三氟甲基化合物最简便的方法之一.早在1978年, Vassiliev等[33]通过电解CF3CO2H与CH3CN混合溶液的方法, 在阳极脱羧产生三氟甲基自由基, 实现了苯环的C—H键三氟甲基化反应(Scheme 12).在该反应中, CH3CN对三氟甲基自由基的产生至关重要, 在体系中不加入CH3CN, 生成的是三氟乙酰氧基取代的产物.
1990年, Yoshida等[34]以二(三氟乙酰基)过氧化物为三氟甲基源, 实现了苯环、呋喃环、噻吩环的C—H键三氟甲基化反应(Scheme 13).该反应无需加入金属催化剂, 二(三氟乙酰基)过氧化物在加热条件下就可以分解产生三氟甲基自由基.机理研究表明, 该反应是通过从底物到过氧化物的电子转移进行的, 所以电子转移过程是该反应的决速步骤.含苯环底物的存在加速了过氧化物分解产生三氟甲基自由基的速率.
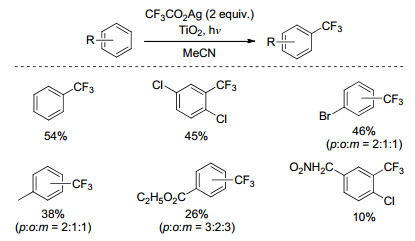
1993年, Mallouk课题组[35]以粉末状的TiO2为光催化剂, CF3CO2Ag为三氟甲基源, MeCN为溶剂, 实现了苯环C—H键三氟甲基化反应(Scheme 14).在该反应中, 生成的银单质附着在光催化剂TiO2表面, 需要加入大大过量的TiO2, 产物的产率较低, 区域选择性较差.反应同时生成了由三氟甲基自由基二聚产生的六氟乙烷, 并且往体系中加入自由基捕捉剂, 三氟甲基化反应被完全抑制, 说明该反应经历了三氟甲基自由基中间体的过程.
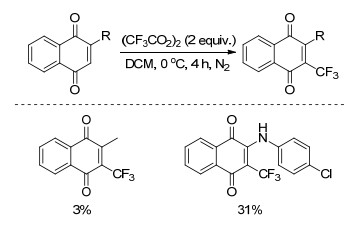
1995年, Matsui小组[36]以三氟乙酸过氧化物((CF3CO2)2)为三氟甲基源, 二氯甲烷(DCM)为溶剂, 0 ℃下反应4 h, 实现了1, 4-萘醌的碳氢键三氟甲基化反应(Scheme 15).该反应经历了自由基的反应历程, 产率较低.
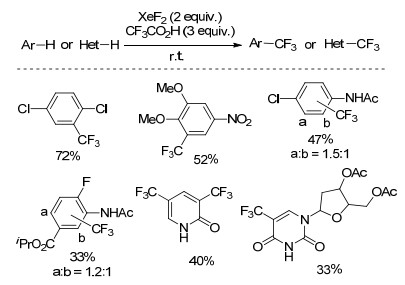
1998年, Matsuo等[37]基于已报道的XeF2与CF3CO2H反应生成六氟乙烷的反应, 以二氯甲烷为溶剂, 往体系中加入含吸电子基团的芳烃, 室温反应得到中等收率的三氟甲基化的产物(Scheme 16).该反应中, 三氟甲基化的区域选择性较差, 富电子的苯甲醚和甲苯主要发生单氟取代的反应.
1999年, Trevin小组[38]通过Kolbe电解氧化CF3CO2H产生三氟甲基自由基, 实现了PhCN的C—H键三氟甲基化反应(Scheme 17).该反应以CH3CN为溶剂, 现场生成的三氟乙酸钠或三氟乙酸吡啶盐为三氟甲基源, 在小试和中试规模下进行了实验, 产率中等, 产物的选择性分别为(o:m:p=46:18:36), 与非电解反应的区域选择性一致.在该试验中, 有机碱吡啶的效果要好于无机碱氢氧化钠, 体系中吡啶的浓度对三氟甲基化的产物产率影响较大, 可能是由于碱浓度的增加促进了三氟乙酸在阳极表面的吸收.较低的电流强度能避免产物进一步发生三氟甲基化, PhCN浓度越低, 产物产率越高.在最优条件下, 带吸电子基团的苯甲醛和苯乙酮反应得到35%的产率, 硝基苯反应得到的产率较低, 而带供电子基团的甲苯不能发生三氟甲基化反应, 生成的是三氟乙酰氧基取代的甲苯.
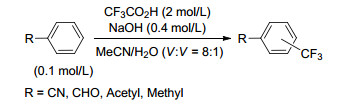
2014年, 我们课题组[18]以CF3CO2H为三氟甲基源, Ag2CO3为催化剂, Na2CO3为碱, MeCN或DCM为溶剂, 120 ℃下反应10 h, 实现了缺电子苯环的C—H键三氟甲基化反应(Scheme 18).该反应体系能够容忍三氟甲基、氰基、酯基等基团, 产率中等至良好.在该反应中, 往体系中加入自由基捕捉剂2, 2, 6, 6-四甲基哌啶氧化物(TEMPO), 三氟甲基化反应被完全抑制, 说明了该反应经历了自由基的历程.
2015年, Stephenson小组[39]以廉价的三氟乙酸酐为三氟甲基源, Ru(bpy)3Cl2•6H2O为光催化剂, 吡啶氮氧盐为氧化剂, 生成的吡啶作为碱, 实现了苯环和杂环的C—H键三氟甲基化反应(Scheme 19).该反应中, 三氟甲基化的区域选择性较差, 反应操作简单, 且可以放大到克级规模, 产率中等.
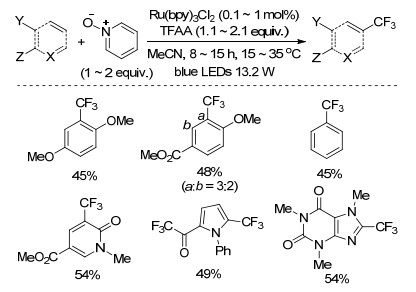
同年, Brase课题组[40]也报道了以三氟乙酸酐为三氟甲基源的芳烃或杂环的C—H键三氟甲基化反应(Scheme 20).该反应无需加入金属催化剂, 以室温下易于储存的过氧化碳酰胺(UHP)为氧化剂, 实现了富电子的呋喃环、噻吩环、吡咯环、苯环的C—H键三氟甲基化反应, 产物产率较低.
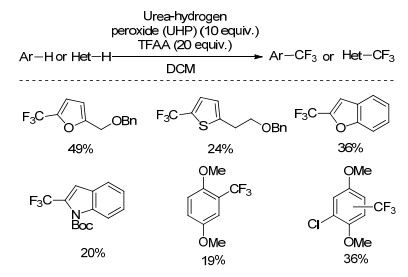
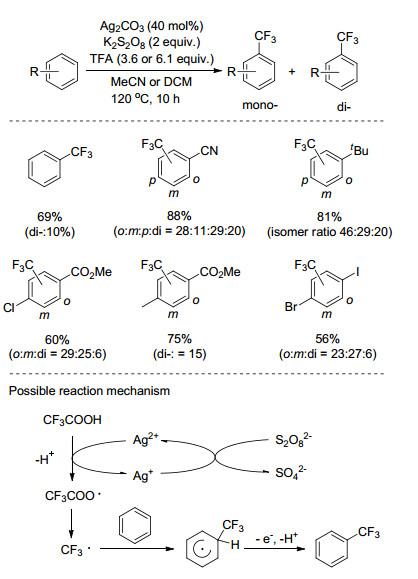
2017年, Su课题组[41]以CF3CO2H为三氟甲基源, Rh修饰的TiO2为光催化剂, 催化量的Na2S2O8为氧化剂, 室温反应下实现苯的C—H键三氟甲基化反应(Scheme 21).该反应条件温和, 底物适用性好, 卤素、乙酰基、酯基、氰基、羧基取代的苯环以及各种氮杂环都能很好的发生反应, 产率中等至良好.机理研究表明, 三氟乙酸分解释放氢气的过程是该反应进行的驱动力, 该反应经历了自由基反应的历程, 往反应体系中加入自由基捕捉剂TEMPO, 成功地捕捉到了三氟甲基自由基.
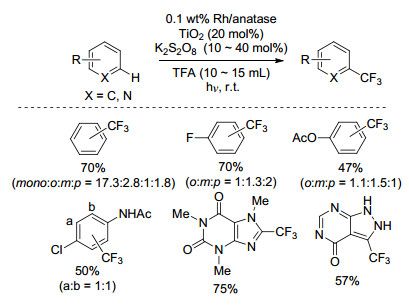
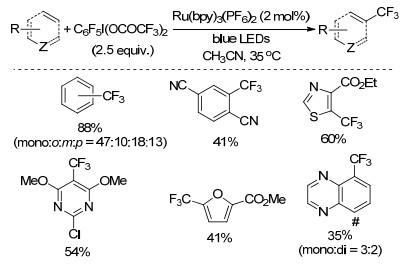
2018年, Qing课题组[42]以二(三氟乙酸)五氟碘苯(C6F5I(OCOCF3)2)为三氟甲基源, Ru(bpy)3(PF6)2为光催化剂, CH3CN为溶剂, 35 ℃下反应12 h, 实现了含吸电子基团苯环的C—H键三氟甲基化反应(Scheme 22).该反应条件温和, 官能团兼容性好, 产率中等至良好, 生成的五氟碘苯可以回收重新制备二(三氟乙酸)五氟碘苯, 提高了反应原子经济性, 但三氟甲基化的区域选择性不高.
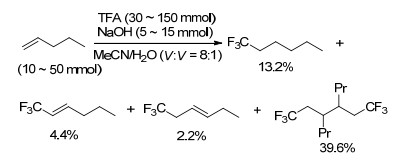
通过双键的官能团化反应引入三氟甲基是构建sp3-C—CF3键的重要方法之一[15a, 43], 其中包括碳碳双键和碳氧双键的三氟甲基化反应. 1974年, Coe等[44]以现场生成的CF3CO2Na为三氟甲基源, CH3CN和H2O的混合液为溶剂, NaOH为碱, 利用Kolbe电解法实现了烯烃双键的三氟甲基化反应(Scheme 23).该反应不能得到单一的产物, 得到的是多种三氟甲基化的混合产物.双键上取代基的电子性质对混合产物的产率影响较大, 吸电子基团取代的双键反应得到的产率较高, 而给电子基团取代的双键反应得到的产率较低.
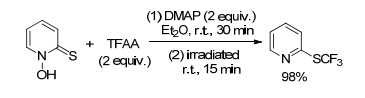
1986年, Barton小组[45]以三氟乙酸酐为三氟甲基源, 4-二甲氨基吡啶(DMAP)为催化剂, 乙醚为溶剂, 实现了1-羟基吡啶-2-硫酮的三氟甲基化反应, 生成了2-三氟甲硫基吡啶(Scheme 24).该反应首先在室温下反应30 min生成相应的酯, 没有被分离出来的酯在钨丝灯下照射15 min, 发生脱羧三氟甲基化反应生成了2-三氟甲硫基吡啶.该反应条件温和, 产率较高.
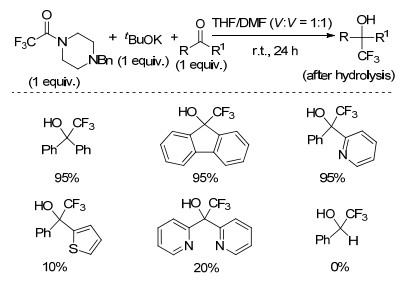
2003年, Langlois课题组[46]以三氟乙酸酯和二取代三氟乙酰胺为亲核三氟甲基化试剂, DMF和THF的混合物为溶剂, 在叔丁醇钾作用下室温反应24 h, 实现了二苯甲酮羰基的三氟甲基化反应(Scheme 25).该反应条件温和, 只需加入1 equiv.的三氟甲基化试剂, 产率中等至良好.该体系中, 双取代的羰基才能发生亲核三氟甲基化反应, 而单取代的羰基则不能发生三氟甲基化反应.
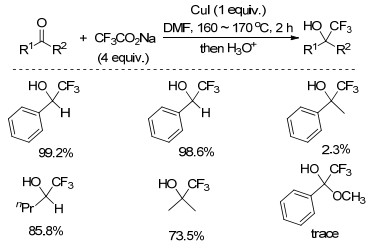
2005年, Chang小组[47]以CF3CO2Na为三氟甲基源, CuI为促进剂, DMF为溶剂, 170 ℃下反应2 h, 实现了苯甲醛羰基的三氟甲基化反应(Scheme 26), 产率中等至优秀.该反应中, DMF加速了三氟甲基阴离子的生成或者促进了三氟甲基阴离子对羰基的亲核进攻.
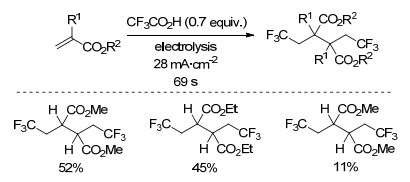
2014年, Wirth小组[48]同样基于Kolbe电解法, 在电化学微反应器中实现了缺电子烯烃的双三氟甲基化反应(Scheme 27).该反应以CF3CO2H为三氟甲基源, 三乙胺为碱, CH3CN和H2O的混合液为溶剂, 在室温下只需反应69 s, 产率中等.与之前报道的反应相比, 该反应条件温和、高效.
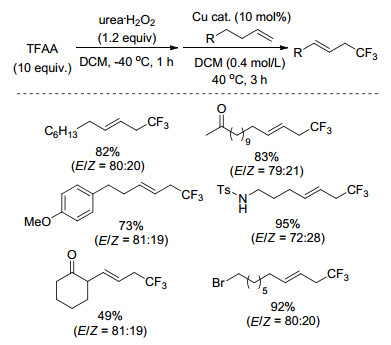
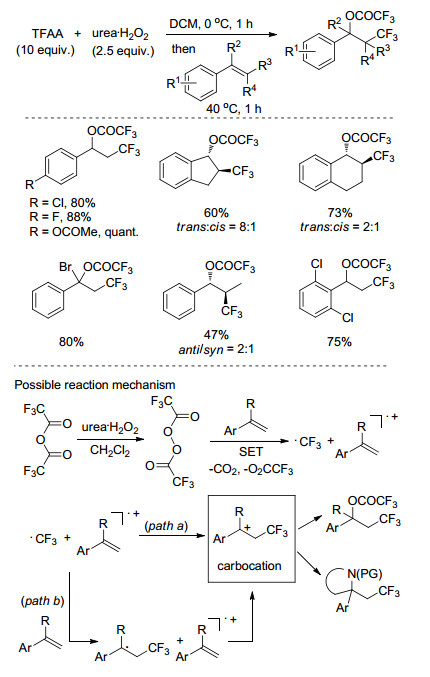
2016年, Sodeoka课题组[49]以三氟乙酸酐(TFAA)为三氟甲基源, 实现了非活化烯丙基的三氟甲基化反应(Scheme 28).该反应中, 三氟乙酸酐与过氧化碳酰胺(UHP)在无金属条件下反应现场生成过氧化物, 然后往体系中加入[Cu(CH3CN)4]PF6催化剂和底物.该反应体系能够容忍羟基、乙酰基等活性基团, 底物适用性广, 产物产率较高.
2018年, Sodeoka课题组[50]同样以三氟乙酸酐(TFAA)为三氟甲基源, 实现了无金属条件下烯烃的三氟甲基化反应(Scheme 29).该反应中, 三氟乙酸酐与过氧化碳酰胺(UHP)在0 ℃下反应1 h生成过氧化物, 然后往体系中加入烯烃, 40 ℃下反应1 h, 生成三氟乙酰氧化三氟甲基化或胺化三氟甲基化的双官能化产物.机理研究表明, 烯烃作为单电子转移的给电子体加速了三氟甲基自由基的产生过程.
此外, 还可以通过三氟乙酸衍生物羰基的缩合反应引入三氟甲基. 2018年, Wang小组[51]通过三氟乙酸酐与胺基的缩合反应合成了含三氟甲基的2-氨基噻二唑(Scheme 30).
本文论述了以三氟乙酸及其衍生物为三氟甲基源的三氟甲基化反应的研究进展, 对其三种反应类型进行了归纳和总结, 并就其相关机理予以阐述.三氟乙酸及其衍生物具有廉价、易得、稳定及副产物是二氧化碳等优点, 将其作为三氟甲基源在有机分子中引入三氟甲基具有广阔的发展前景.然而, 现已报道的以三氟乙酸及其衍生物为三氟甲基源的反应存在着一些有待解决的问题.首先, C—X (X=Br, I)键的三氟甲基化反应虽然具有产物产率较高、区域选择性较好等优点, 但是底物需要预官能化, 原子经济性不高, 脱羧过程需要较高的反应温度.其次, C—H键三氟甲基化反应和碳碳(碳氧)双键的三氟甲基化反应虽然存在着原子经济性和步骤经济性等优点, 但是这类反应普遍存在着产率不高、区域选择性不好等缺点, 这些也给随后的分离纯化带来了困难.对于以三氟乙酸及其衍生物为三氟甲基源的三氟甲基化反应, 如何降低催化剂的使用量和脱羧反应的温度, 如何提高三氟乙酸及其衍生物转化的效率、产物的产率及区域选择性, 将是今后颇具有挑战性的研究方向.
(a) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(b) Li, Y.; Wu, Y.; Li, G.-S.; Wang, X.-S. Adv. Synth. Catal. 2014, 356, 1412.
Jeschke, P. ChemBioChem 2004, 5, 570. doi: 10.1002/cbic.v5:5
(a) O'Hagan, D.; Harper, D. B. J. Fluorine Chem. 1999, 100, 127.
(b) Vaillancourt, F. H.; Yeh, E.; Vosburg, D. A.; GarneauTsodikova, S.; Walsh, C. T. Chem. Rev. 2006, 106, 3364.
(a) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881.
(b) Schlosser, M. Angew. Chem., Int. Ed. 2006, 45, 5432.
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320. doi: 10.1039/B610213C
(a) Ismail, F. M. D. J. Fluorine Chem. 2002, 118, 27.
(b) Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992.
(c) Petrov, V. A. Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2009.
(d) Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, Ltd, Chichester, UK., 2009.
(e) Zhu, W.; Wang, J.; Wang, S. J. Fluorine Chem. 2014, 167, 37.
(f) Zhang, J.; Jin, C.; Zhang, Y. Chin. J. Org. Chem. 2014, 34, 662 (in Chinese). (张霁, 金传飞, 张英俊, 有机化学, 2014, 34, 662.)
(a) Penning, T. D.; Talley, J. J.; Bertenshaw, S.R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K. A.; Veenhuizen, W. Y.; Zhang, Y.; Isakson, P.C. J. Med. Chem. 1997, 40, 1347.
(b) Chakraborti, A. K.; Garg, S. K.; Kumar, R.; Motiwala, H. F.; Jadhavar, P. S. Curr. Med. Chem. 2010, 17, 1563.
(a) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(b) Prakash, S. G. K.; Yudin, A. K. Chem. Rev. 1997, 97, 757.
(a) Ye, Y.; Ball, N. D.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2010, 132, 14682.
(b) Wang, X.; Truesdale, L.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 3648.
(c) Zhang, X.; Dai, H.; Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 11948.
(d) Mu, X.; Chen, S.; Zhen, X.; Liu, G. Chem.-Eur. J. 2011, 17, 6039.
(e) Zhang, L.-S.; Chen, K.; Chen, G.; Li, B.-J.; Luo, S.; Guo, Q.-Y.; Wei, J.-B.; Shi, Z.-J. Org. Lett. 2013, 15, 10.
(f) Miura, M.; Feng, C.-G.; Ma, S.; Yu, J.-Q. Org. Lett. 2013, 15, 5258.
(g) Culkin, D. A.; Hartwig, J. F. Organometallics 2004, 23, 3398.
(h) Grushin, V. V. Acc. Chem. Res. 2010, 43, 160.
(i) Hughes, R. P.; Meyer, M. A.; Tawa, M. D.; Ward, A. J.; Williamson, A.; Rheingold, A. L.; Zakharov, L. N. Inorg. Chem. 2004, 43, 747.
(j) Ball, N. D.; Gary, J. B.; Ye, Y.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 7577.
(k) Ball, N. D.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2010, 132, 2878.
(l) Cho, E. J.; Senecal, T. D.; Kinzel, T.; Zhang, Y.; Watson, D. A.; Buchwald, S. L. Science 2010, 328, 1679.
(a) Shimizu, R.; Egami, H.; Nagi, T.; Chae, J.; Hamashima, Y.; Sodeoka, M. Tetrahedron Lett. 2011, 51, 5947.
(b) Zhang, C.-P.; Wang, Z.-L.; Chen, Q.-Y.; Zhang, C.-T.; Gu, Y.-C.; Xiao, J.-C. Angew. Chem., Int. Ed. 2011, 50, 1896.
(c) Liu, T.; Shen, Q. Org. Lett. 2016, 13, 2342.
(d) Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2012, 134, 1298.
(e) Cai, S.; Chen, C.; Sun, Z.; Xi, C. Chem. Commun. 2013, 49, 4552.
(f) He, Z.; Tan, P.; Hu, J. Org. Lett. 2016, 18, 72.
(g) Li, X.; Zhao, J.; Zhang, L.; Hu, M.; Wang, L.; Hu J. Org. Lett. 2015, 17, 298.
(h) Gao, X.; Xiao, Y.-L.; Wan, X.; Zhang X. Angew. Chem., Int. Ed. 2018, 57, 3187.
(i) Xu, X.; Chen, H.; He, J.; Xu, H. Chin. J. Chem. 2017, 35, 1665.
(a) Hafner, A.; Stefan, B. Angew. Chem., Int. Ed. 2012, 51, 3713.
(b) Ye, Y.; Lee, S. H.; Sanford, M. S. Org. Lett. 2011, 13, 5464.
(c) Loy, R. N.; Sanford, M. S. Org. Lett. 2011, 13, 2548.
(d) Seo, S.; Taylor, J. B.; Greaney, M. F. Chem. Commun. 2013, 49, 6385.
(e) Liu, Y.-R.; Tu, H.-Y.; Zhang, X.-G. Synthesis 2015, 47, 3460.
Kino, T.; Nagase, Y.; Ohtsuka, Y.; Yamamoto, K.; Uraguchi, D.; Tokuhisa, K.; Yamakawa, T. J. Fluorine Chem. 2010, 131, 98. doi: 10.1016/j.jfluchem.2009.09.007
(a) Nagib, D. A.; MacMillan, D. W. C. Nature 2011, 480, 224.
(b) Iqbal, N.; Choi, S.; Ko, E.; Cho, E. J. Tetrahedron Lett. 2012, 53, 2005.
(c) Kamigata, N.; Fukushima, T.; Yoshida, M. Chem. Lett. 1990, 649.
(d) Kamigata, N.; Ohtsuka, T.; Fukushima, T.; Yoshida, M.; Shimizu, T. J. Chem. Soc. Perkin Trans. 1 1994, 1339.
(e) Xie, J.; Yuan, X.; Abdukader, A.; Zhu, C.; Ma, J. Org. Lett. 2014, 16, 1768.
Mejía, E.; Togni, A. ACS Catal. 2012, 2, 521. doi: 10.1021/cs300089y
(a) Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950.
(b) Parsons, A. T.; Buchwald, S. L. Nature 2011, 480, 184.
(a) Ji, Y.; Brueckl, T.; Baxter, R. D.; Fujiwara, Y.; Seiple, I. B.; Su, S.; Blackmond, D. G.; Baran, P. S. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411.
(b) Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herle, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95.
(c) Wu, X.; Chu,L.; Qing, F.-L. Tetrahedron Lett. 2013, 54, 249.
(d) Yang, Y.-D.; Iwamoto, K.; Tokunaga, E.; Shibata, N. Chem. Commun. 2013, 49, 5510.
(e) Fennewald, J. C.; Lipshutz, B. H. Green Chem. 2014, 16, 1097. (f) Cui, L.; Matusaki, Y.; Tada, N.; Miura, T.; Uno, B.; Itoh, A. Adv. Synth. Catal. 2013, 355, 2203.
(a) Umemoto, T. Chem. Rev. 1996, 96, 1757.
(b) Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921.
(c) Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757.
(d) Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, 65.
(e) Chu, L.; Qing, F.-L. Acc. Chem. Res. 2014, 47, 1513.
Shi, G.; Shao, C.; Pan, S.; Yu, J.; Zhang, Y. Org. Lett. 2015, 17, 38. doi: 10.1021/ol503189j
(a) Lopez, S. E.; Salazar, J. J. Fluorine Chem. 2013, 156, 73.
(b) Rui, S.; Lei, L. Sci. China Chem. 2011, 54, 1670.
(c) Rodríguez, N.; Goossen, L. J. Chem. Soc. Rev. 2011, 40, 5030.
Wang, J.; Sanchez-Rosello, M.; Acena, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432. doi: 10.1021/cr4002879
Matsui, K.; Tobita, E.; Ando, M.; Kondo, K. Chem. Lett. 1981, 12, 1719. http://www.researchgate.net/publication/304405568_ChemInform_Abstract_A_CONVENIENT_TRIFLUOROMETHYLATION_OF_AROMATIC_HALIDES_WITH_SODIUM_TRIFLUOROACETATE
Hünig, S.; Bau, R.; Kemmer, M.; Meixner, H.; Metzenthin, T.; Peters, K.; Sinzger, K.; Gulbis, J. Eur. J. Org. Chem. 1998, 2, 335. http://www.researchgate.net/publication/243883451_25-DisubstitutedN_N-Dicyanoquinone_Diimines_(DCNQIs)__Syntheses_and_Redox_Properties
Austin, N. E.; Avenell, K. Y.; Boyfield, I.; Branch, C. L.; Hadley, M. S.; Jeffrey, P.; Johnson, C. N.; Macdonald, G. J.; Nash, D. J.; Riley, G. J.; Smith, A. B.; Stemp, G.; Thewlis, K. M.; Vong, A. K. K.; Wood, M. D. Bioorg. Med. Chem. Lett. 2001, 11, 685. doi: 10.1016/S0960-894X(01)00037-3
Langlois, B. R.; Roques, N. J. Fluorine Chem. 2007, 128. 1318. doi: 10.1016/j.jfluchem.2007.08.001
McReynolds, K. A.; Lewis, R. S.; Ackerman, L. K. G.; Dubinina, G. G.; Brennessel, W. W.; Vicic, D. A. J. Fluorine Chem. 2010, 131, 1108. doi: 10.1016/j.jfluchem.2010.04.005
Li, Y.; Chen, T.; Wang, H.; Zhang, R.; Jin, K.; Wang, X.; Duan, C. Synlett 2011, 1713. http://www.researchgate.net/publication/264703393_ChemInform_Abstract_A_Ligand-Free_Copper-Catalyzed_Decarboxylative_Trifluoromethylation_of_Aryl_Iodides_with_Sodium_Trifluoroacetate_Using_Ag2O_as_a_Promoter
Schareina, T.; Wu, X.-F.; Zapf, A.; Cotte, A.; Gotta, M.; Beller, M. Top Catal. 2012, 55, 426. doi: 10.1007/s11244-012-9824-0
Chen, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2013, 52, 11628. doi: 10.1002/anie.201306094
Maleckis, A.; Sanford, M. S. Organometallics 2014, 33, 2653. doi: 10.1021/om500398z
Lin, X.; Hou, C.; Li, H.; Weng, Z. Chem.-Eur. J. 2016, 22, 2075. doi: 10.1002/chem.201504306
Torikai, K.; Koga, R.; Liu, X.; Umehara, K.; Kitano, T.; Watanabe, K.; Oishi, T.; Noguchi, H.; Shimohigashi, Y. Bioorg. Med. Chem. 2017, 25, 5216. doi: 10.1016/j.bmc.2017.07.067
(a) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242.
(b) Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293.
(c) Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Chem. Rev. 2015, 115, 12138.
(d) Song, G.; Li, X. Acc. Chem. Res. 2015, 48, 1007.
(e) Guo, X.-X.; Gu, D.-W.; Wu, Z.; Zhang, W. Chem. Rev. 2015, 115, 1622.
(f) Zheng, C.; You, S.-L. RSC Adv. 2014, 4, 6173.
(g) Ackermann, L. Chem. Rev. 2011, 111, 1315.
(h) Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936.
(i) Zhang, Y.; Shi, G.; Yu, J.-Q. Carbon-Carbon σ-Bond Formation via C—H Bond Functionalization in Comprehensive Organic Synthesis,2nd ed., Vol. 3, Eds.: Molander, G.; Knochel, P.,Elsevier, Oxford, 2014, pp. 1101~1209.
Grinberg, V. A.; Polishchuk, V. R.; German, L. S.; Kanevskii, L. S.; Vassiliev, Y. B. Izv. Akad. Nauk SSSR, Ser. Khim. 1978, 3, 673.
Sawada, H.; Nakayama, M.; Yoshida, M.; Yoshida, T.; Kamigata, N. J. Fluorine Chem. 1990, 46, 423. doi: 10.1016/S0022-1139(00)82927-9
Lai, C.; Mallouk, T. E. J. Chem. Soc., Chem. Commun. 1993, 17, 1359.
Matsui, M.; Kondoh, S.; Shibata, K.; Muramatsu, H. Bull. Chem. Soc. Jpn. 1995, 68, 1042. doi: 10.1246/bcsj.68.1042
Tanabe, Y.; Matsuo, N.; Ohno, N. J. Org. Chem. 1988, 53, 4583.
Depecker, C.; Marzouk, H.; Trevin, S.; Devynck, J. New J. Chem. 1999, 23, 739. doi: 10.1039/a901305i
Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi: 10.1038/ncomms8919
Zhong, S.; Hafner, A.; Hussal, C.; Nieger, M.; Brase, S. RSC Adv. 2015, 5, 6255. doi: 10.1039/C4RA13430C
Lin, J.; Li, Z.; Kan, J.; Huang, S.; Su, W.; Li, Y. Nat. Commun. 2017, 8, 1. doi: 10.1038/s41467-016-0009-6
Yang, B.; Yu, D.; Xu, X.-H.; Qing, F.-L. ACS Catal. 2018, 8, 2839. doi: 10.1021/acscatal.7b03990
(a) Parsons, A. T.; Buchwald, S. L. Angew. Chem., Int. Ed. 2011, 50, 9120.
(b) Xu, J.; Fu, Y.; Luo, D.-F.; Jiang, Y.-Y.; Xiao, B.; Liu, Z.-J.; Gong, T.-J.; Liu, L. J. Am. Chem. Soc. 2011, 133, 15300.
(c) Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410.
(d) Liu, X.; Xiong, F.; Huang, X.; Xu, L.; Li, P.; Wu, X. Angew. Chem., Int. Ed. 2013, 52, 6962.
Brookes, C. J.; Coe, P. L.; Owen, D. M.; Pedler, A. E.; Tatlow, J. C. J. Chem. Soc., Chem. Commun. 1974, 9, 323.
Barton, D. H. R.; Lacher, B.; Zard, S. Z. Tetrahedron 1986, 42, 2325. doi: 10.1016/S0040-4020(01)90613-1
Jablonski, L.; Joubert, J.; Billard, T.; Langlois, B. R. Synlett 2003, 230.
Chang, Y.; Cai, C. J. Fluorine Chem. 2005, 126, 937. doi: 10.1016/j.jfluchem.2005.04.012
Arai, K.; Watts, K.; Wirth, T. ChemistryOpen 2014, 3, 23. doi: 10.1002/open.201300039
Kawamura, S.; Sodeoka, M. Angew. Chem., Int. Ed. 2016, 55, 8740. doi: 10.1002/anie.201604127
Valverde, E.; Kawamura, S.; Sekine, D.; Sodeoka, M. Chem. Sci. 2018, 9, 7115. doi: 10.1039/C8SC02547A
Zhang, Y.; Li, Z.; Song, H.; Wang, B. Chin. J. Chem. 2018, 36, 635. doi: 10.1002/cjoc.v36.7

图式 1 卤代芳烃与三氟乙酸钠的三氟甲基化反应
Scheme 1 Trifluoromethylation of aromatic halides with sodium trifluoroacetate

图式 2 多取代卤代芳烃与三氟乙酸钠的三氟甲基化反应
Scheme 2 Trifluoromethylation of multisubstituted aromatic halides with sodium trifluoroacetate

图式 3 5-溴代异吲哚与三氟乙酸钠的三氟甲基化反应
Scheme 3 Trifluoromethylation of 5-bromoisoindole with sodium trifluoroacetate

图式 4 卤代芳烃与三氟乙酸甲酯的三氟甲基化反应
Scheme 4 Trifluoromethylation of aromatic halides with methyl trifluoroacetate

图式 6 铜催化的卤代芳烃与三氟乙酸钠的三氟甲基化反应
Scheme 6 Copper-catalyzed trifluoromethylation of aryliodides with sodium trifluoroacetate

图式 7 铜催化的芳基卤与三氟乙酸甲酯的三氟甲基化反应
Scheme 7 Copper-catalyzed trifluoromethylation of aryl halides with methyl trifluoroacetate

图式 8 铜催化的碘代芳烃与三氟乙酸钾的三氟甲基化反应
Scheme 8 Copper-catalyzed trifluoromethylation of aryliodides with potassium trifluoroacetate

图式 9 钯催化的脱羧三氟甲基化反应的催化循环
Scheme 9 Catalytic cycle for Palladium-catalyzed decarbonylative trifluoromethylation

图式 10 以[Cu(O2CCF3)(phen)]为三氟甲基化试剂的反应
Scheme 10 Trifluoromethylation using [Cu(O2CCF3)(phen)] as trifluoromethylating reagent

图式 11 微波辐射下苯并吖啶衍生物的三氟甲基化反应
Scheme 11 Trifluoromethylation of phenyl benzoacridine derivative under microwave irradiation

图式 13 芳烃与三氟乙酸过氧化物的三氟甲基化反应
Scheme 13 Trifluoromethylation of aromatic compounds with bis(trifluoroacetyl)peroxide

图式 14 芳烃的光化学三氟甲基化反应
Scheme 14 Photochemical trifluoromethylation of aromatic compounds

图式 16 二氟化氙促进的芳烃的三氟甲基化反应
Scheme 16 Trifluoromethylation of Aromatics with trifluoroacetic acid mediated by xenon difluoride

图式 17 通过Kolbe电解法进行的芳烃的三氟甲基化反应
Scheme 17 Trifluoromethylation of aromatic compounds via Kolbe electrolysis

图式 18 以三氟乙酸为三氟甲基化试剂的反应
Scheme 18 Trifluoromethylation of arenes using trifluoroacetic acid as the trifluoromethylating reagent

图式 19 规模化且操作简单的自由基三氟甲基化反应
Scheme 19 A scalable and operationally simple radical trifluoromethylation

图式 20 无金属参与的(杂)芳烃的自由基三氟甲基化反应
Scheme 20 Metal-free radical trifluoromethylation of (hetero)arenes

图式 21 光驱动的(杂)芳烃与三氟乙酸的三氟甲基化反应
Scheme 21 Photo-driven trifluoromethylation of (hetero)arenes with trifluoroacetic acid

图式 22 (杂)芳烃与三氟乙酸五氟碘苯的脱羧反应
Scheme 22 Decarboxylation of perfluoroarene Iodine(Ⅲ) trifluoroacetates for trifluoromethylation of (Hetero)arenes

图式 23 烯烃双键与三氟乙酸的三氟甲基化反应
Scheme 23 Trifluoromethylation of olefinic double bonds with trifluoroacetic acid

图式 24 1-羟基吡啶-2-硫酮的三氟甲基化反应
Scheme 24 Trifluoromethylation of 1-hydroxypyridine-2(1H)- thione

图式 25 三氟乙酸衍生物作为亲核三氟甲基化试剂
Scheme 25 Trifluoroacetic acid derivatives as nucleophilic trifluoromethylating reagents

图式 26 碘化亚铜催化的羰基化合物与三氟乙酸钠的三氟甲基化反应
Scheme 26 CuI-catalyzed trifluoromethylation of carbonyl compounds with sodium trifluoroacetate

图式 27 烯烃在电化学微反应器中的三氟甲基化反应
Scheme 27 Trifluoromethylation of alkenes in an electrochemical microreactor

图式 28 惰性烯烃与酸酐的三氟甲基化反应
Scheme 28 Trifluoromethylation of unactivated alkenes with acid anhydrides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
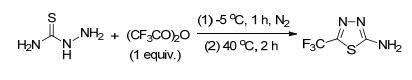
 下载:
下载:




