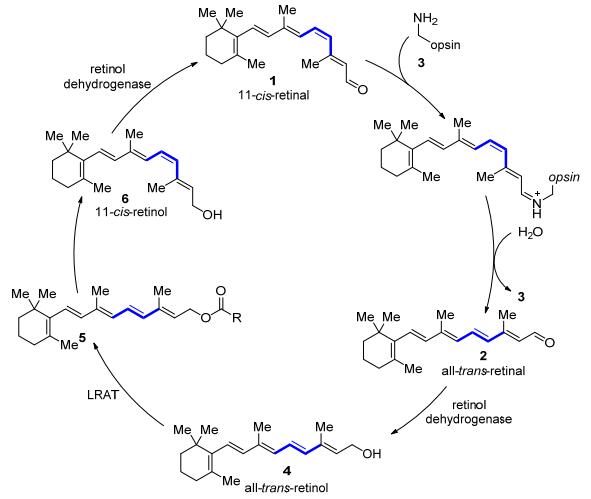
图式 1.
视觉循环
Scheme 1.
Visual cycle

烯烃作为一种重要的结构单元, 广泛地存在于生物活性分子和材料分子中.在有机合成中, 烯烃也是最常用的基团之一, 它们作为起始原料可以很容易地实现官能团化, 来合成多种化学中间体和功能性骨架[1].由于烯烃的立体化学(Z/E构型)极大地影响着其化学、物理和生理相关的性质及功能, 因此高度立体选择性地构建烯烃一直都是有机合成重点的研究内容之一.目前已经有大量合成烯烃方法的报道, 比如, Wittig反应及其改良和变体[2]、Julia反应及其改良和变体[3]、Peterson烯基化反应[4]、烯烃复分解反应[5]、炔烃的选择性还原[6]等反应.但这些方法往往得到的是烯烃立体异构体的混合物.通常来说, 选择性合成热力学稳定的E-式烯烃的方法已经比较成熟, 但是对于热力学不稳定的Z-式烯烃来说, 还是面临着很大的挑战.目前合成Z-构型烯烃的反应往往依赖于反应的动力学控制, 通常都需要使用高能量的试剂来促进Z-式烯烃的合成[7].然而在反应与分离的过程中, 也极易造成热力学不稳定的Z-式烯烃向热力学更加稳定的E-式烯烃的异构化, 所以多数情况下不能得到令人满意的立体选择性结果.此外, 这些策略所需要的反应条件也比较苛刻(如强碱和极低温度等), 并且同时还需要使用昂贵的过渡金属催化剂, 原子经济性也比较低[8].虽然烯烃的合成有了巨大的发展, 但仍然迫切地需要开发出简便高效绿色和立体选择性合成烯烃的方法, 特别是Z-式烯烃的合成.
近年来, 有机光化学和光催化技术, 特别是可见光促进的化学反应, 得到了很大的发展, 越来越多的科学家将其作为解决合成中长期存在的挑战的一种尝试.本文就近年来可见光促进的烯烃异构化的相关研究进展进行总结, 并且对相应的反应机理进行了讨论.
自然界中, 烯烃异构化最典型的例子就是在脊椎动物光感受器中发生的11-顺式视黄醛1异构化为全反式视黄醛2的过程(Scheme 1).该过程依赖于11-顺式-视黄醛1与视蛋白3以席夫碱的形式形成相应的视紫红质.吸收可见光后导致了11-顺式双键的几何异构化并形成具有11-反式双键的变视紫红质II, 视紫红质构型的改变, 启动了对大脑的神经脉冲, 从而产生视觉响应.视黄醛部分也会与视蛋白分离得到游离的11-反式视黄醛2, 在光感受器中经过酶促还原后, 全反式视黄醇4转移至视网膜色素上皮细胞中(RPE), 在卵磷脂视黄醇酰基转移酶(LRAT)的作用下形成视黄酯5.视黄酯5水解产生的能量驱动了最后一步同分异构酶催化的E→Z的异构化过程.最后, 11-顺式视黄醇6被氧化成相应的11-顺式视黄醛1以完成视觉循环[9].
该过程表明可见光促进的烯烃的异构化是可行的.受到此循环过程的启发, 同时在化学家们的不懈努力之下, 烯烃光敏化异构化的机理在理论和实验两个方面都取得了较大的进展, 使得人们对光化学反应有了较为深入的了解.通常在基态反应中, C(sp2)-C(sp2)双键的旋转往往受到高旋转能垒的阻碍, 不易发生异构化.普通的烯烃也不能直接吸收可见光, 不能直接被可见光激发.添加合适的光敏剂则可以克服这一困难[10].可见光激发光敏剂后, 通过能量转移的方式敏化烯烃化合物, 使其跃迁至单线态, 经系间窜越过程得到双自由基的三线态中间体, 此中间体可以进行旋转来实现烯烃的异构化.由于不同构型的烯烃淬灭激发态光敏剂的速率是不同的, 通常共轭的E-式烯烃可以有效地淬灭光敏剂, 而Z-式烯烃由于位阻作用, 是非共轭状态, 通常不能有效地淬灭光敏剂, 从而实现烯烃的E→Z异构化(Scheme 2)[11].高能量的紫外光直接照射也可以实现烯烃的激发, 顺式烯烃激发所需要的能量要比反式烯烃激发所需要的能量要高, 因此, 使用介于顺式与反式烯烃激发波长之间的特定波长的光进行照射, 即可实现选择性地将反式烯烃激发至三线态, 经双自由基中间体的旋转后, 接着向顺式和反式两个方向发生衰减, 而异构化得到的顺式产物不会被再次活化, 所以其衰减系数的大小就决定了最终的平衡体系中顺式产物与反式产物的比例, 即可选择性地得到烯烃定向异构化反应的顺式产物.
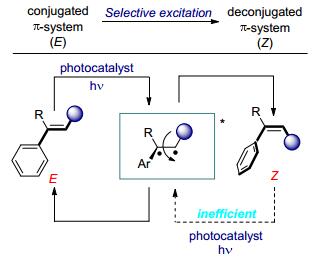
烯烃光化学异构化的基本策略主要有两种, 一种是在光敏剂的催化下, 对既定构型的烯烃化合物直接实现异构化; 另一种则是在通过化学转化构建双键的过程中, 对其进行立体选择性地控制.
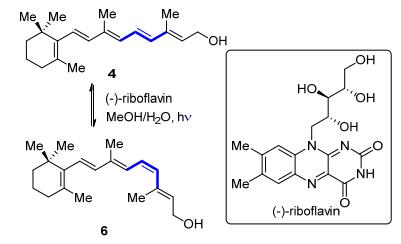
1967年, Walker和Radda[12]发现在紫外光的照射下, 全反式视黄醇4在(-)-核黄素[(-)-riboflavin]溶液中的吸收显著降低, 薄层色谱(TLC)显示有11-顺式视黄醇6的生成(Scheme 3).有趣的是, 在相同的条件下, 使用11-顺式视黄醇6同样可以得到逆向的异构化产物4.尽管他们对核黄素催化的全反式视黄醇4反热力学的E→Z异构化过程只是局限于定性的研究, 但是这项研究为烯烃选择性异构化指明了方向.
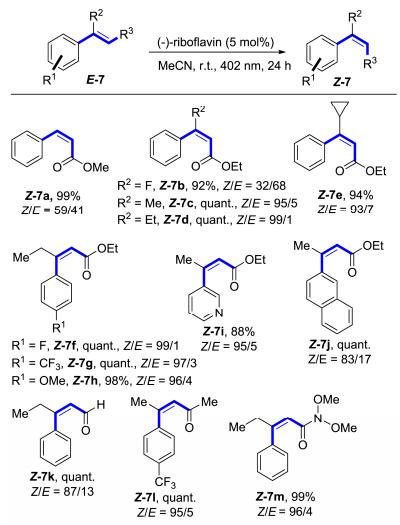
2015年, Gilmour等[13]使用Walker所报道的廉价易得的天然产物核黄素实现了活化烯烃的E→Z异构化(Scheme 4).该反应只需要添加5 mol%的(-)-核黄素, 而不需要其他任何添加剂, 在乙腈溶液中, 用402 nm的可见光照射24 h, 即可实现转换, 反应操作简便, 条件也比较温和.研究表明, R2基团的引入与修饰可以显著地提高异构化的选择性(Z-7c~Z-7e).但是F原子的引入, 异构化效果并不是很理想, 可能是因为卤素的位阻不足以使得顺式异构体的激发能垒增大很多.对于芳环底物来说, 无论是含有缺电子还是富电子基团, 其选择性和反应收率都非常好(Z-7f~Z-7h), 杂环化合物也同样适用(Z-7i).除了肉桂酸酯类的底物, 他们进一步将底物的适用性范围拓展到了醛(Z-7k)、酮(Z-7l)以及酰胺(Z-7m)等.该反应也有其不足之处, 反应底物受R2基团的影响非常大, 同时烯烃也必须是缺电子的烯烃.
在上述异构化的最优条件下, Gilmour等[13]又对肉桂酸衍生物E-8进行了尝试.有趣的是, 该底物成功地以高选择性得到了预期的Z-构型产物, 但是产率却很低, 只有24%, 大部分反应产物却转化成了4-位取代的香豆素9.当该异构化反应在氧气环境中进行时, 分子内环化产物9的收率可以高达96%.这一发现为高效合成香豆素这一重要的药物分子骨架提供了方法.为了验证上述反应广泛的适用性, Gilmour等[14]再次对一锅法合成香豆素类化合物的反应条件进行了筛选和优化.研究表明, (-)-核黄素作为光催化剂, 体积比为1:1的MeCN/MeOH的混合溶液是最佳的反应溶剂, 清洁高效的氧气是最理想的氧化剂, 在波长为402 nm光的照射下, 通过能量转移(ET)实现反式肉桂酸双键的异构化, 随后经过单电子转移(SET)历程可以成功实现分子内环化(Scheme 5).考虑到(-)-核黄素经长时间光照会发生降解, 于是他们每隔12 h补加一次5 mol%的(-)-核黄素以确保第二步的分子内环化反应不会受到前一步异构化反应的影响.此反应整体底物的适用性较好, 无论芳环上是给电子或者是拉电子取代基, 产率都是可以接受的.
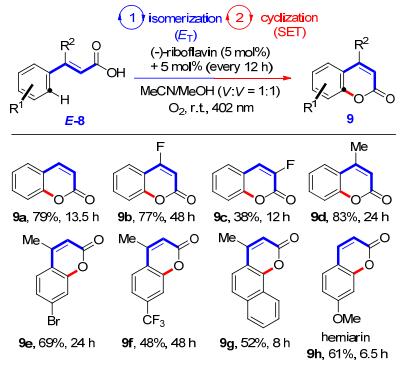
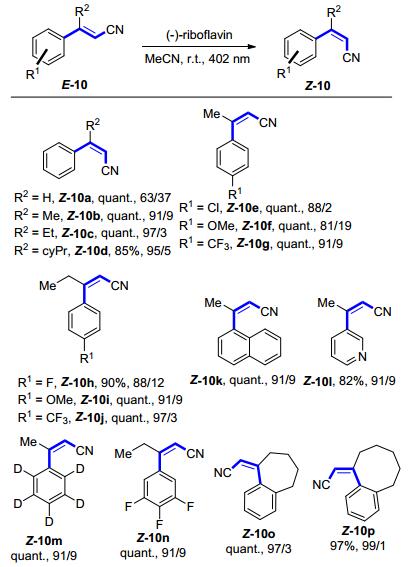
随后, Gilmour等[15]将同样的催化体系应用于肉桂腈的光化学异构化中(Scheme 6).该研究中, 作者从底物取代基的电子效应、位阻因素以及反应中温度和氧气的影响等诸多方面进行了比较详尽的研究.研究结果表明, 在芳环上取代基不变的情况下, 在β位引入取代基, 可以显著地提高Z-异构体的选择性, 并且引入基团的位阻越大, 选择性越好.对芳环邻位取代基的研究, 也得到了同样的结论, 这一结论与前期α, β-不饱和酯的光化学异构化反应所观察到的现象一致.值得一提的是, 通过原子转移自由基聚合反应将(-)-核黄素负载于官能团化的硅纳米粒子上, 并不会影响催化剂的光化学异构化性能[16].
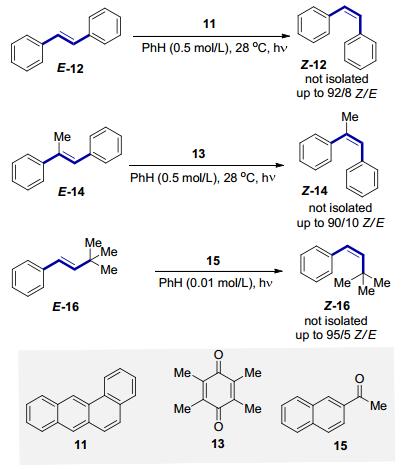
羰基类化合物和多种稠环化合物作为三线态光敏剂, 可以用于烯烃的异构化反应, 已经有很多报道(Scheme 4).早在20世纪60年代, Hammond等[17]就使用苯并[a]蒽11作为光催化剂, 实现了反式二苯乙烯的异构化E-12 (Scheme 7).随后他们又对二苯甲酮、苯乙酮芴酮等一系列酮类化合物的三线态能量进行了测定, 并以450 W的中压汞灯作为光源, 四甲基对苯醌13为光敏剂, 反式1, 2-二苯基丙烯E-14 (1, 2-Diphenylpro- penes)作为研究对象, 用实验证实了顺反异构化反应确实可以通过烯烃的三线态有效地发生[18]. 20世纪80年代, Arai等[19]使用2-萘乙酮15作为光敏剂, 在1 kW的高压汞灯的照射下, 成功实现烷基取代的苯乙烯E-16的E→Z异构化.这些研究的共同特征是使用了化学计量的光敏剂, 尽管没有对最终产物进行分离, 但是这也已经很有力地证明了直接光照和光敏化都是很好的实现富电子E构型烯烃转化为相应的Z构型烯烃的有效途径.
2009年, Liu等[20]借助三线态能量的理论, 尝试了不同的酮类化合物作为光敏剂对多种烯烃进行光异构化.对不同骨架烯烃的尝试, 将底物范围从二苯乙烯类化合物扩展到共轭二烯、三烯等.研究结果给出相同的结论, 烯烃骨架上的取代基位阻的增大, 可以很大程度上增大顺式烯烃的三线态能量, 有助于反式烯烃向顺式烯烃的转换.尽管此次报道的底物范围得到了一定拓展, 但是所尝试的反应大多数需要使用紫外光作为光源照射, 顺式选择性也不理想, 并没能发展成为具有应用价值的通用方法.
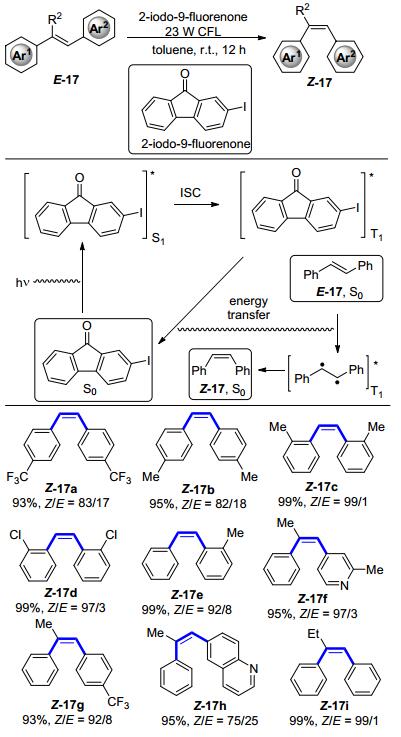
2017年, 王卫等[21]对酮类光敏剂进行了进一步的筛选和结构修饰, 发展出了一种新型的酮类光敏剂2-碘-9-芴酮, 并应用于可见光促进的烯烃异构化反应中, 实现了顺式二芳基乙烯类化合物的绿色高效合成.其反应机理如Scheme 8所示:基态的2-碘-9-芴酮作为光敏剂, 在可见光的驱动下, 被激发至第一激发单线态, 接着发生系间窜越变成激发的三线态, 三线态中间体随后与基态的反式烯烃发生能量交换, 使得双键断裂形成双自由的中间体, 经过C—C键的旋转, 最后得到相应的顺式烯烃.通过对二苯乙烯类衍生物在该反应条件下底物适用性的研究发现, 对于对位取代的二苯乙烯类底物, 无论是吸电子基团或者是给电子基团, 都可以得到高收率并且顺反选择性较好的产物(Z-17a、Z-17b).而当取代基在邻位时, 反应收率与选择性都有显著的提升(Z-17c~Z-17e), 这很可能是因为邻位取代基的引入, 使得顺式构型产物的结构在空间上变得更加拥挤, 进而增大了顺势产物三线态的能量, 而对于反式底物来说, 空间上的结构仍然相对比较平坦, 三线态的能量变化并不大, 最终导致了异构化的发生.
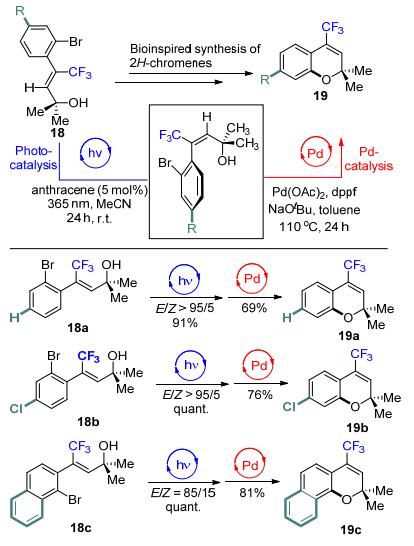
2017年, Gilmour等[22]报道了光诱导的蒽催化的烯丙醇18的Z→E异构化反应, 并通过钯催化的氧化环化反应构建4-三氟甲基取代的2H-色烯类化合物19 (Scheme 9).研究发现, 当使用Z-构型的烯丙醇作为起始原料时, 即使升高温度也不能使得环化反应发生, 而通过光催化的方式, 即可成功地将Z-式的烯丙醇异构化为E-式产物, 在醋酸钯的催化下, 通过环化得到相应的三氟甲基取代的2H-色烯类化合物.
多联吡啶配合物如Ru(Ⅱ), Ir(Ⅲ)等, 基于其电子到配体的电子转移(metal-to-ligand charge transfer, MLCT)激发态具有优良的光化学与光物理性质, 而被广泛地用作可见光激发的光敏剂.其光敏作用主要是通过分子内的能量转移及(或)电子转移来对底物进行活化.
Osawa等[23]在2001年报道了在多联吡啶Ru(Ⅱ)配合物上通过共价键引入叔膦配体来实现反式-4-氰基-二苯乙烯(E-20)的光异构化反应(Scheme 10).此反应使用2 mol%的含膦多联吡啶Ru(Ⅱ)配合物作为光催化剂, 在可见光的照射下(400 W lamp, λ>450 nm), 室温条件下反应2 h即可得到顺式二苯乙烯Z-20为主的混合物(Z/E=85/15), 尽管他们当时没有尝试更多的底物, 但是这个方法的提出为使用金属配合物作为光敏剂, 实现可见光诱导的烯烃异构化反应提供了一个新的思路.
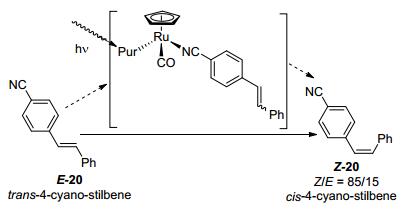
2014年, Weaver等[7]报道了铱配合物作为光催化剂介导的反式芳基烯丙基胺的异构化反应.此反应使用0.7 mol%的fac-Ir(ppy)3作为光催化剂, 在蓝色LED灯的照射下, 可以以最高96%的收率得到反式芳基烯丙基胺E-21的异构化产物Z-21, 分离前的选择性高达98/2.该反应底物整体官能团的容忍性较好, 原子经济性高, 有比较好的应用前景. Weaver就异构化反应提出了两种可能的Ir(ppy)3*催化剂光淬灭的机理, 如Scheme 11所示.第一种是还原淬灭机理:底物胺E-22通过单电子转移过程(single electron transfer, SET)过程被激发态的Ir(Ⅲ)*氧化成氮正离子自由基E-22a.随着胺被氧化, 使得氮原子α-位H的酸性得到增强, 可以被外加碱可逆性地去质子化, 得到烯丙基自由基E-22b.烯丙基自由基E-22b可以发生双键的异构得到Z-22b, Z-22b进一步地进行质子化变成Z-22a, 最终经历反向的单电子转移被还原成最终顺式产物Z-22.另一种则是双自由基的机理:激发态的光敏剂通过能量转移(energy transfer, ET)过程将能量传递给底物E-22, 生成双自由基中间体23, 该中间体可以围绕C—C键进行90 ℃的旋转得到具有临界立体构型的中间体, 由于第一步的速率常数更大, 所以进一步通过系间窜越(Intersystem Crossing, ISC)得到以顺式构型为主的产物Z-22.
除了对胺类取代的烯烃进行异构化研究外, Weaver等[7]还对其他类型取代的烯烃, 包括烃类(E-24a, E- 25a)、醇类(E-26a)和酯类(E-27a)等进行了尝试, 均可得到有用的选择性与收率.这也证明了从热力学稳定的E构型烯烃出发, 通过光催化直接获得热力学不稳定的Z构型烯烃的可行性, 底物的范围也可以进一步地拓宽.
|
|
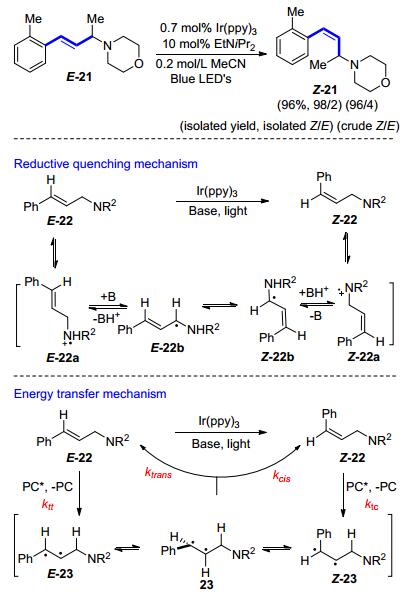
Reiser等[24]使用fac-Ir(ppy)3作为光催化剂, 同样实现了化合物E-28的E→Z异构化反应(Scheme 12).他们推断此反应仍然是通过双自由基中间体的转换而来.有趣的是, 他们发现此异构化反应在较高的温度(90 ℃)中进行并不会导致最终的Z/E比例发生改变.
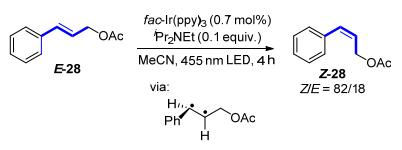
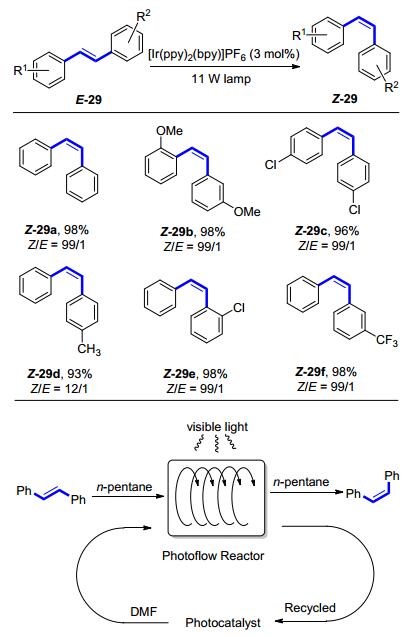
2015年, Rueping等[25]报道了一种反应条件温和的, 可见光诱导的二苯乙烯类化合物E-29 E→Z异构化反应(Scheme 13).该反应使用3 mol%的铱配合物作为光催化剂, 通过11 W荧光灯照射, 在氧气的氛围下实现完全的构型反转, Z/E选择性高达99/1, 而反应的收率也接近定量.此反应最大的优势在于其操作的简便性与异构化效率的高效性.此外, Rueping等[25]还极具创造性地将此异构化反应应用于有机-离子液体两相溶液的非均相体系.光催化剂由于所带电荷而溶于离子液体中, 反应的底物则溶于有机相中, 反应完成后, 有机相可以直接进行分离, 而离子液体相则可以继续循环使用, 实验结果表明, 在循环使用8次后, 此反应体系仍然可以保证89%的收率.最后他们又将此异构化过程设计成一种连续流动化学的反应.该方法的建立可以使得流速控制在10~20 mL/h的底物在大功率的蓝灯照射下, 实现定量的异构化反应, 并且可以放大到克级规模.
2018年, Gilmour等[26]在前期对极化烯烃异构化工作的理论基础上, 发展了可见光诱导的烯基硼酸酯反热力学的E→Z异构化反应(Scheme 14).该反应是通过能量转移过程实现的.共轭的π系统被激发态的铱配合物活化, 得到的瞬时双自由基的中间体围绕着C—C键进行旋转, 产生顺式和反式两种异构体.其中, 顺式异构体中烯丙位的张力使得π系统产生去共轭效应, 而不能再次被活化.促使整个平衡趋向于顺式产物的生成.该反应条件温和, 操作简便, 芳环上吸电子基团与供电子基团都可以容忍, 尤其是对β-位具有取代基的烯基硼酸酯也可以很好的调控.
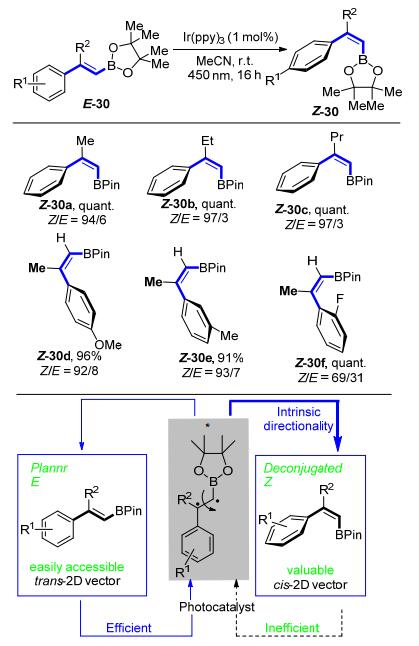
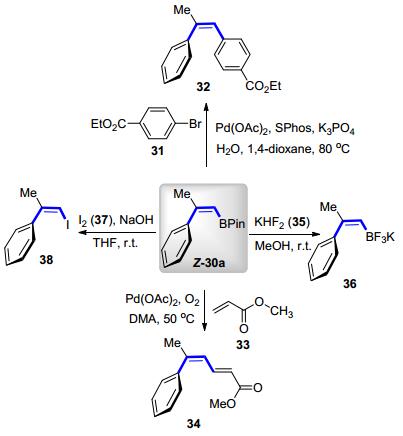
为了进一步凸显此异构化在合成中的应用价值, Gilmour等[26]进行了相应的硼酸酯的衍生化.使用Z-30a作为起始原料, 顺利地与4-溴苯甲酸乙酯31偶联得到Suzuki-Miyaura反应的产物32; 在氧化偶联的条件下, 与丙烯酸甲酯33生成烯基化产物34.碘的取代反应以及氟硼酸钾物种36的制备都可以顺利地进行(Scheme 15).
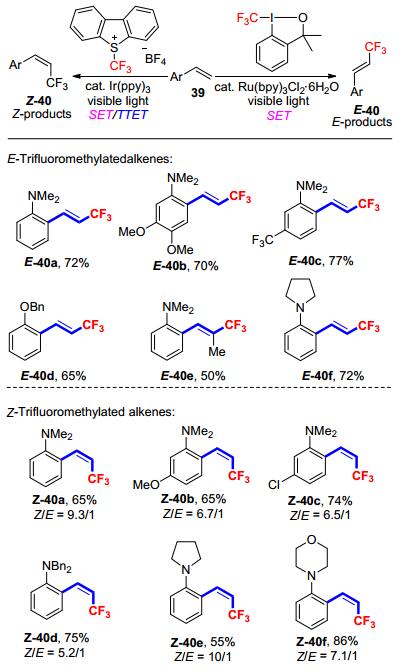
2014年, 卿凤翎等[27]在此前使用过渡金属介导和催化的三氟甲基化反应的研究基础上, 开发了一种新型的可见光诱导的苯乙烯的直接三氟甲基化方法, 更重要的是, 此方法可以通过选择特定的光催化剂来实现三氟甲基化后烯烃的E/Z构型的控制(Scheme 16).热力学稳定的E-异构体可以通过钌配合物催化的SET循环来直接构建, 而热力学不稳定的Z-异构体则是依赖于铱配合物催化的SET/TTET“一锅法”的过程来制备.这是第一例可调节的立体选择性合成三氟甲基化烯烃的方法.研究表明, 在苯乙烯芳环上引入给电子基团, 使得氧化淬灭循环中碳自由基更容易被氧化成碳正离子中间体.此外, 给电子基团处于苯乙烯的邻位, 也可以有效地防止碳正离子中间体受到分子间的亲核进攻.这两点可以有效地避免副产物的生成, 以提高反应收率.
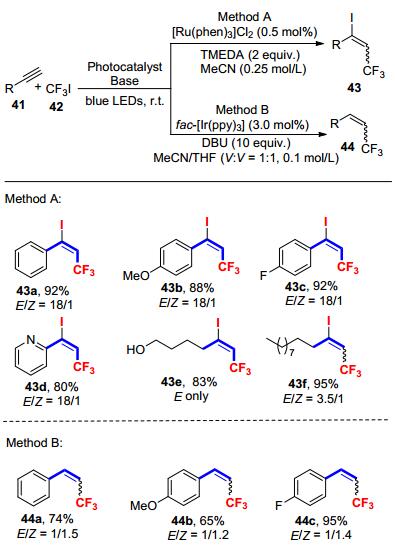
同年, Cho等[28]报道了光催化的炔烃的双官能团化反应(Scheme 17).研究发现, 反应条件中催化剂、碱和溶剂的差别, 决定了产物Z/E的选择性.当使用[Ru(phen)3]- Cl2作为光催化剂时, 炔烃41与三氟碘甲烷42主要得到的是炔烃双官能团化的E-产物43, 选择性可以高达18:1, 收率也较高.当使用fac-Ir(ppy)3作光催化剂, 1, 8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)作碱, 在乙腈/四氢呋喃的混合溶剂中反应时, 则发生炔烃的氢三氟甲基化反应, 产物44以Z-式为主, 但是选择性有待提高.
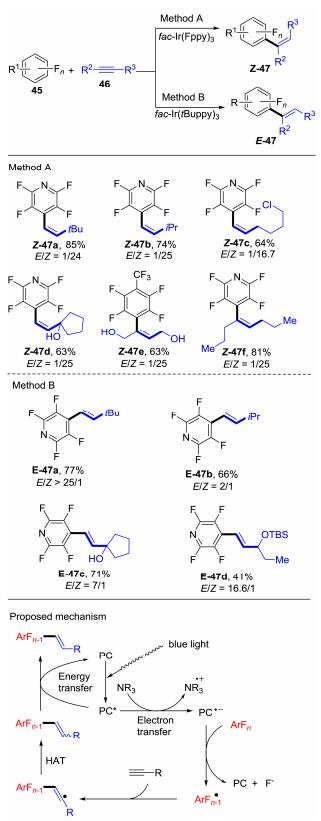
Weaver等[29]从2014年开始就对光诱导的C—F键活化进行了一系列的研究.在前期成功实现多氟芳烃烷基化和芳基化[30]的基础上, 他们使用相同的策略, 在可见光的激发下, 使用多氟芳烃自由基对取代炔烃46加成得到骨架多样的烯基化多氟芳烃化合物, 并且通过对光催化剂的筛选来实现对烯基化产物立体选择性的控制[31].该反应也是电子转移与能量转移在同一个反应中共同发生的鲜有报道例子之一(Scheme 18).光催化剂在可见光的照射下, 吸收一个光子后变成激发态, 经过还原淬灭的过程产生中间体, 接着发生单电子转移, 多氟芳烃45得到一个电子后脱去一个氟负离子以产生多氟芳烃自由基中间体, 该中间体会进一步对炔烃进行自由基加成得到乙烯基自由基, 乙烯基自由基再经过氢原子转移(HAT)过程攫取氢原子形成苯乙烯类似物, 其顺反的选择性取决于炔烃上R基团的大小.最后, 苯乙烯类似物与激发态的光催化发生能量交换, 有趣的是, 研究表明能量转移的速率与光催化剂体积的大小有着密切的关系, 大体积的光催化剂能量转移的速率较慢, 所以异构化的平衡最终趋向于反式构型, 反之亦然.
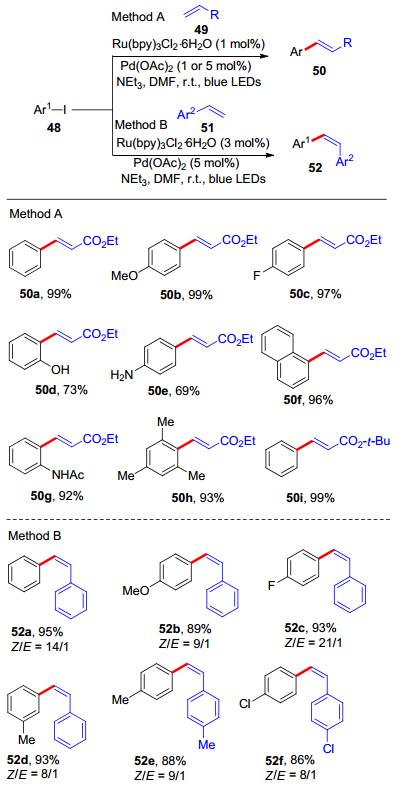
2016年, 黄学良等[32]报道了可见光诱导的钯/钌协同催化芳基碘化物48和烯烃的Heck偶联反应(Scheme 19).该反应无需添加配体, 条件温和, 室温下即可进行, 同时也避免了使用紫外光等高能量光源.通过大量的条件筛选以及控制实验的验证, 他们发现使用Ru(bpy)3Cl2•6H2O与Pd(OAc)2协同催化效果最为理想, 可以以最高99%的收率得到相应的E-肉桂酸酯50.研究表明, 碘化物芳环上的电性不会对反应性造成影响(50a~50c).芳环上存在未保护的羟基或氨基的底物同样可以顺利地反应(50d~50e), 虽然收率有所降低, 但是反应整体底物范围广泛, 对官能团的容忍度较好, 取代基的位阻也不会影响产率与立体选择性.值得注意的是, 当使用苯乙烯类化合物51与芳基碘化物48偶联时, 产物会出现E→Z异构化现象, 通过增加Ru(bpy)3Cl2•6H2O与Pd(OAc)2, 可以成功地控制产物的立体构型, 最终得到高选择性的Z-二芳基乙烯52.
王磊等[33]报道了一种可见光控制的三组分参与的合成菲啶类化合物的自由基串联反应(Scheme 20).该反应是通过芳基亚磺酸53与联芳基异氰55形成电子给体受体(EDA)复合物后被光化学激发所驱动.研究发现, 室温下使用3 W的蓝灯照射可以得到热力学稳定的E-式菲啶类化合物, 而使用3 W的紫外灯进行照射, 产物的构型则以Z-式为主.该串联反应显示出了很高的区域和立体选择性, 反应条件温和, 实验操作也很简便, 具有广泛的底物适用性, 并以较高的收率得到预期产物.
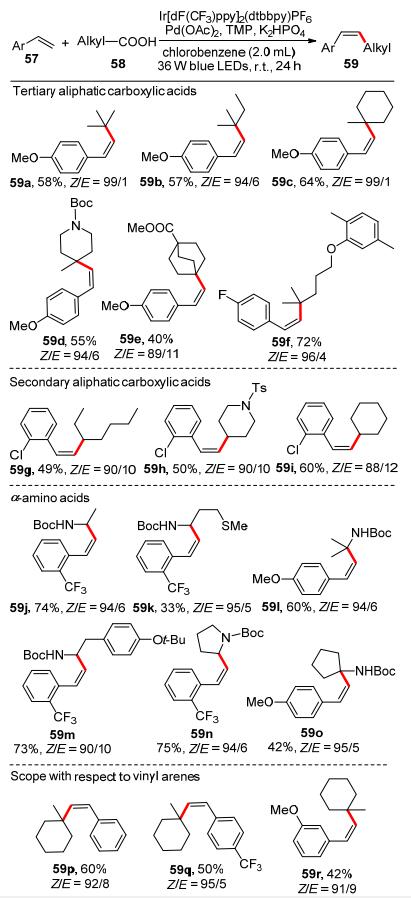
2018年, 尚睿等[34]报道了一种光催化Heck脱羧偶联合成顺式烯烃化合物的方法(Scheme 21).他们采用将过渡金属催化剂与光氧化还原催化剂合并的策略, 利用生物质衍生物烷基羧酸58作为原料, 在Heck交叉偶联反应中得到了良好Z-选择性的取代苯乙烯衍生物59.该反应展示了很好的烷基羧酸的底物范围, 二级、三级烷基羧酸以及α-氨基酸都可以很好的兼容(59a~59o).此外, 富电子和缺电子的取代苯乙烯也都有较好的反应结果(59p~59r).这一新型的可见光促进的光催化烷基羧酸脱羧偶联反应具有条件温和、绿色环保等优点, 为脱羧偶联的发展提供了新的机遇.
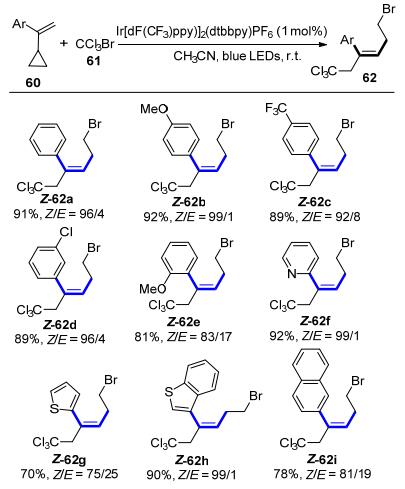
程旭等[35]报道了一种取代苯乙烯的1, 5-溴三氯甲基化反应, 并对产物的构型进行了立体选择性控制(Scheme 22).通过量子产率实验和荧光淬灭实验, 他们提出了一个可见光引发的串联反应的历程. α-环丙烷苯乙烯首先通过自由基链式反应机理进行1, 5-溴三氯甲基化反应, 生成的E构型三取代苯乙烯类化合物进一步在光照条件下发生构型翻转, 最终获得Z构型产物, 产物的Z/E比例最高可达99/1.
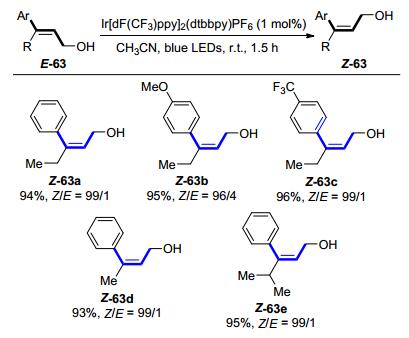
在此基础上, 程旭等[35]进一步探讨了三取代的反式芳基烯丙醇E-63的E→Z异构化反应(Scheme 23).研究发现, 芳基取代基的电性以及α位的空间位阻对异构化效率的影响不大, 都可以获得高收率和高选择性的Z异构体.
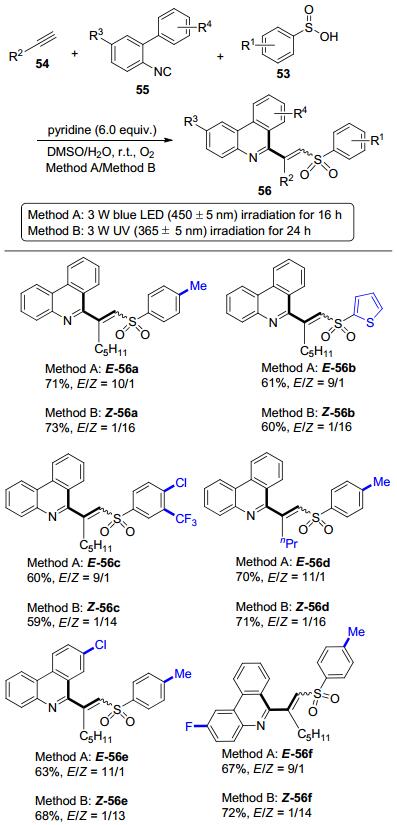
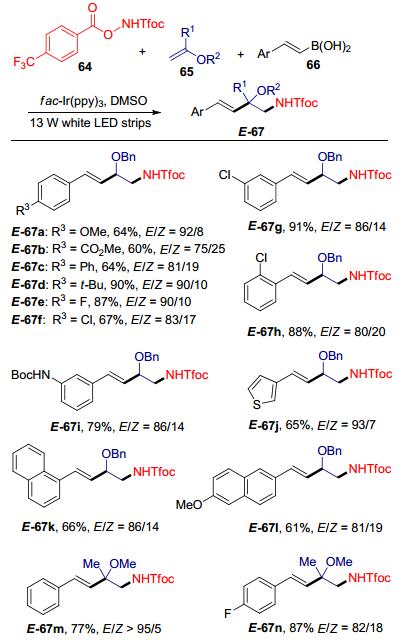
近期, 俞寿云等[36]报道了可见光催化的三组分参与的自由基接力反应来制备α-氨甲基肉桂基醚67, 并对其立体选择性进行了控制(Scheme 24).发现当使用DMSO作为溶剂, 白色LED灯作为光源时, 可以快速地得到E构型为主的α-氨甲基肉桂基醚(E-67), 产物的立体选择性良好.该反应底物范围广泛, 芳基乙烯基硼酸以及杂环乙烯基硼酸都可以很好地兼容.此外, 富电子和缺电子取代的芳基乙烯基硼酸也都有较好的反应结果, 并且对各种官能团如酯、卤素、胺等皆有很好的容忍度.值得注意的是, 包含季碳中心的产物同样具有良好的收率与选择性.
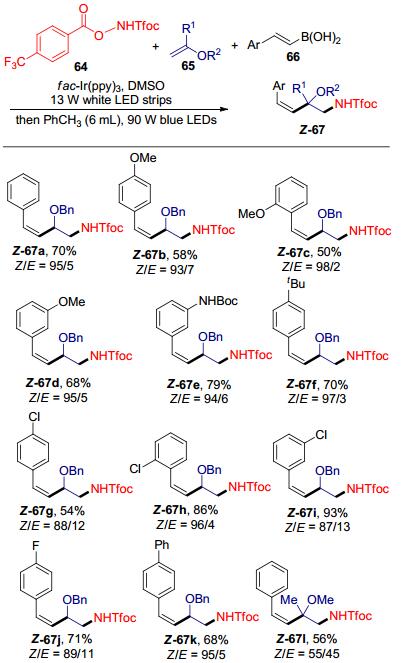
在此研究基础上, (E)-α-氨甲基肉桂基醚(E-67)的E→Z异构化反应也被研究(Scheme 25).研究发现, 光源以及溶剂的选择对异构化反应至关重要.经过条件筛选, 我们发现将第一步自由基接力反应得到的E构型为主的产物直接加入甲苯溶剂中, 在蓝色LED灯照射下, 即可实现E→Z异构化.结果显示芳基乙烯基硼酸取代基的电性以及空间位阻对异构化效率的影响都很小.遗憾的是, 包含季碳中心的产物顺式选择性并不理想.
对烯烃光化学异构化的策略以及促进光反应所需的光催化剂类型进行分类并总结, 同时对相应的反应机理进行了讨论.光催化烯烃的异构化反应, 最早使用紫外光为外加光源, 但是由于紫外光能量较高, 极易导致副反应的发生, 降低了反应收率, 因此大大地限制了其在有机合成中的应用.近年来, 随着有机光化学的不断发展, 可见光的引入以及三线态光敏剂的不断开发, 使得可见光催化的策略成为一种新的有机合成的方法, 解决了众多有机合成中的难点问题.在烯烃的异构化, 尤其是热力学不稳定的Z-式烯烃的合成上取得了一系列的突破.反应操作简便, 条件温和, 大多数情况下室温即可反应, 原子经济性高.
可见光促进的烯烃异构化还存在一些问题, 需要克服和解决.首先, 绝大部分的烯烃底物都是苯乙烯的衍生物, 非共轭的烯烃底物很难异构化; 其次, E→Z异构化研究较多, 反向异构化研究较少; 再次, 很多反应中, 异构化的选择性不够理想, 还需要进一步提高.随着这些问题的解决, 可以预见, 可见光促进的烯烃异构化反应这一策略将成为立体选择性地构建烯烃的重要方法之一.
(a) Pearson, C. M.; Snaddon, T. N. ACS Cent. Sci. 2017, 3, 922.
(b) Cai, W.; Fan, H.; Ding, D.; Zhang, Y.; Wang, W. Chem. Commun. 2017, 53, 12918.
(c) Singh, K.; Staig, S. J.; Weaver, J. D. J. Am. Chem. Soc. 2014, 136, 5275.
(d) Li, X.; Song, S.; Jiao, N. Acta Chim. Sinica 2017, 75, 1202 (in Chinese).
(李昕伟, 宋颂, 焦宁, 化学学报, 2017, 75, 1202.)
(e) Zhao, M.; Ji, Y. Chin. J. Org. Chem. 2018, 38, 401 (in Chinese).
(赵明, 纪原, 有机化学, 2018, 38, 401.)
(a) Wittig, G.; Geissler, G. Justus Liebigs Ann. Chem. 1953, 580, 44.
(b) Bergelson, L. D.; Shemyakin, M. M. Tetrahedron 1963, 19, 149.
(c) Dong, D. J.; Li, H.-H.; Tian, S.-K. J. Am. Chem. Soc. 2010, 132, 5018.
(a) Julia, M.; Paris, J.-M. Tetrahedron Lett. 1973, 14, 4833.
(b) Yao, C.-Z.; Li, Q.-Q.; Wang, M.-M.; Ning, X.-S.; Kang, Y.-B. Chem. Commun. 2015, 51, 7729.
(a) Peterson, D. J. J. Org. Chem. 1968, 33, 780.
(b) Staden, L. F. V.; Gravestock, D.; Ager, D. J. Chem. Soc. Rev. 2002, 31, 195.
(a) Koh, M. J.; Khan, R. K. M.; Torker, S.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2014, 53, 1968.
(b) Quigley, B. L.; Grubbs, R. H. Chem. Sci. 2013, 5, 501.
(c) Gottumukkala, A. L.; Madduri, A. V. R.; Minnaard, A. J. ChemCatChem 2012, 4, 462.
(d) Meek, S. J.; O'Brien, R. V.; Llaveria, J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 471, 461.
(e) Endo, K.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8525.
(a) Lindlar, H. Helv. Chim. Acta 1952, 35, 446.
(b) Li, X.; Song, W.; Tang, W. J. Am. Chem. Soc. 2013, 135, 16797.
(c) Song, W.; Li, X.; Yang, K.; Zhao, X.-L.; Glazier, D. A.; Xi, B.-M.; Tang, W. J. Org. Chem. 2016, 81, 2930.
Singh, K.; Staig, S. J.; Weaver, J. D. J. Am. Chem. Soc. 2014, 136, 5275. doi: 10.1021/ja5019749
Cai, W.; Fan, H.; Ding, D.; Zhang, Y.; Wang, W. Chem. Commun. 2017, 53, 12918. doi: 10.1039/C7CC07984B
(a) Wald, G. J. Gen. Physiol. 1935, 19, 351.
(b) O'Leary, B.; Duke, B.; Eilers, J. E. Nature 1973, 246, 166.
(c) Lion, F.; Rotmans, J. P.; Daemen, F. J. M.; Bonting, S. L. Biochim. Biophys. Acta 1975, 384, 283.
(d) Gai, F.; Hasson, K. C.; Cooper McDonald, J.; Anfinrud, P. A. Science 1998, 279, 1886.
(e) Rando, R. R. Chem. Rev. 2001, 101, 1881.
(f) Strauss, O. Physiol. Rev. 2005, 85, 845.
(g) Redmond, T. M.; Poliakov, E.; Yu, S.; Tsai, J.-Y.; Lu, Z.; Gentleman, S. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13658.
Metternich, J. B.; Gilmour, R. Synlett 2016, 27, 2541. doi: 10.1055/s-0036-1588621
(a) Metternich, J. B.; Gilmour, R. J. Am. Chem. Soc. 2015, 137, 11254.
(b) Metternich, J. B.; Artiukhin, D. G.; Holland, M. C.; Bremen-Kühne, M. V.; Neugebauer, J.; Gilmour, R. J. Org. Chem. 2017, 82, 9955.
Walker, A. G.; Radda, G. K. Nature 1967, 215, 1483.
Metternich, J. B.; Gilmour, R. J. Am. Chem. Soc. 2015, 137, 11254. doi: 10.1021/jacs.5b07136
Metternich, J. B.; Gilmour, R. J. Am. Chem. Soc. 2016, 138, 1040. doi: 10.1021/jacs.5b12081
Metternich, J. B.; Artiukhin, D. G.; Holland, M. C.; Bremen-Kühne, M. V.; Neugebauer, J.; Gilmour, R. J. Org. Chem. 2017, 82, 9955. doi: 10.1021/acs.joc.7b01281
Metternich, J. B.; Sagebiel, S.; Lückener, A.; Lamping, S.; Ravoo, B. J.; Gilmour, R. Chem.-Eur. J. 2018, 24, 4228. doi: 10.1002/chem.v24.17
Hammond, G. S.; Saltiel, J.; Lamola, A. A.; Turro, N. J.; Bradshaw, J. S.; Cowan, D. O.; Counsell, R. C.; Vogt, V.; Dalton, C. J. Am. Chem. Soc. 1964, 86, 3197. doi: 10.1021/ja01070a002
(a) Hammond, G. S.; Saltiel, J. J. Am. Chem. Soc. 1962, 84, 4983.
(b) Hammond, G. S.; Saltiel, J. J. Am. Chem. Soc. 1963, 85, 2515.
(c) Hammond, G. S.; Saltiel, J.; Lamola, A. A.; Turro, N. J.; Bradshaw, J. S.; Cowan, D. O.; Counsell, R. C.; Vogt, V.; Dalton, C. J. Am. Chem. Soc. 1964, 86, 3197.
(a) Arai, T.; Sakuragi, H.; Tokumaru, K. Chem. Lett. 1980, 9, 261.
(b) Arai, T.; Sakuragi, H.; Tokumaru, K. Bull. Chem. Soc. Jpn. 1982, 55, 2204.
Zhao, Y.-P.; Yang, L.-Y.; Liu, R. S. H. Green Chem. 2009, 11, 837. doi: 10.1039/b819207c
Cai, W.; Fan, H.; Ding, D.; Zhang, Y.; Wang, W. Chem. Commun. 2017, 53, 12918. doi: 10.1039/C7CC07984B
Faßbender, S. I.; Metternich, J. B.; Gilmour, R. Org. Lett. 2018, 20, 724. doi: 10.1021/acs.orglett.7b03859
Osawa, M.; Hoshino, M.; Wakatsuki, Y. Angew. Chem., Int. Ed. 2001, 40, 18.
Rackl, D.; Kreitmeier, P.; Reiser, O. Green Chem. 2016, 18, 214. doi: 10.1039/C5GC01792K
Fabry, D. C.; Ronge, M. A.; Rueping, M. Chem. Eur. J. 2015, 21, 5350. doi: 10.1002/chem.201406653
Molloy, J. J.; Metternich, J. B.; Daniliuc, C. G.; Watson, A. J. B.; Gilmour, R. Angew. Chem., Int. Ed. 2018, 57, 3168. doi: 10.1002/anie.201800286
Lin, Q.-Y.; Xu, X.-H.; Qing, F.-L. J. Org. Chem. 2014, 79, 10434. doi: 10.1021/jo502040t
Iqbal, N.; Jung, J.; Park, S.; Cho, E. J. Angew. Chem., Int. Ed. 2014, 53, 539. doi: 10.1002/anie.v53.2
Senaweera, S. M.; Singh, A.; Weaver, J. D. J. Am. Chem. Soc. 2014, 136, 3002. doi: 10.1021/ja500031m
(a) Singh, A.; Kubik, J. J.; Weaver, J. D. Chem. Sci. 2015, 6, 7206.
(b) Senaweera, S.; Weaver, J. D. J. Am. Chem. Soc. 2016, 138, 2520.
Singh, A.; Fennell, C. J.; Weaver, J. D. Chem. Sci. 2016, 7, 6796. doi: 10.1039/C6SC02422J
Zhang, H.; Huang, X. Adv. Synth. Catal. 2016, 358, 3736. doi: 10.1002/adsc.201600704
Li, Y.; Miao, T.; Li, P.; Wang, L. Org. Lett. 2018, 20, 1735. doi: 10.1021/acs.orglett.8b00171
Zheng, C.; Cheng, W.-M.; Li, H.-L.; Na, R.-S.; Shang, R. Org. Lett. 2018, 20, 2559. doi: 10.1021/acs.orglett.8b00712
李进, 陈靖之, 黄文浩, 程旭, 有机化学, 2018, 38, 1507. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346458.shtmlLi, J.; Chen, J.; Huang, W.; Cheng, X. Chin. J. Org. Chem. 2018, 38, 1507. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346458.shtml
An, X.-D.; Zhang, H.; Xu, Q.; Yu, L.; Yu, S. Chin. J. Chem. 2018, 36, 1147. doi: 10.1002/cjoc.v36.12

图式 2 光催化烯烃异构化的机理
Scheme 2 Conceptual framework for photocatalytic isomerization of alkenes

图式 3 核黄素介导的全反式视黄醇异构化为11-顺式视黄醇
Scheme 3 (-)-Riboflavin-mediated isomerization of all-trans- retinol to 11-cis-retinol

图式 4 核黄素催化的活化烯烃的异构化
Scheme 4 (-)-Riboflavin-catalyzed isomerization of activated olefins

图式 5 光介导的核黄素催化合成香豆素
Scheme 5 Photocatalysis route to coumarins catalyzed by (-)-riboflavin

图式 7 稠环化合物或酮类化合物实现的E→Z异构化反应
Scheme 7 E→Z isomerization reaction used condensed nucleus compounds or ketones

图式 8 光催化的二苯乙烯类衍生物E→Z异构化
Scheme 8 Photocatalytic E→Z isomerization of stilbene derivatives

图式 10 可见光诱导的4-氰基二苯乙烯的E→Z异构化
Scheme 10 Photocatalytic E-to-Z isomerization of 4-cyano- stilbene

图式 11 两种可能的可见光催化的烯丙基胺的E→Z异构化的机理
Scheme 11 Two possible mechanism of the photocatalytic E-to-Z isomerization of allylic amine

图式 13 催化的二苯乙烯类化合物E→Z异构化
Scheme 13 Photocatalytic E-to-Z isomerization of diverse stilbene derivatives

图式 14 光催化的反式-α-取代的烯基硼酸酯的E→Z异构化
Scheme 14 Photocatalytic E→Z isomerization of trans-α- substituted styrenyl BPins

图式 17 可见光催化的炔烃的三氟甲基化反应
Scheme 17 Trifluoromethylation reactions of alkynes through visible light photoredox catalysis

图式 19 可见光诱导的无配体的Heck反应
Scheme 19 Visible light-mediated Pd-Ru-catalyzed ligand-free Heck reactions

图式 20 光驱动的官能团化的菲啶类化合物的合成
Scheme 20 Photo-driven synthesis of functionalized phenanthridines

图式 21 光氧化还原/钯协同催化的Z-选择性脱羧Heck反应
Scheme 21 Z-Selective decarboxylative Heck reactions through photoredox/palladium catalysis

图式 22 1, 5-溴三氯甲基化产物的E→Z异构化
Scheme 22 E→Z isomerization of 1, 5-bromotrichloromethyla- tion reaction products

图式 24 自由基接力反应制备(E)-α-氨甲基肉桂基醚
Scheme 24 Radical relay reaction for synthesizing (E)-α-aminomethyl cinnamyl ethers

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:







