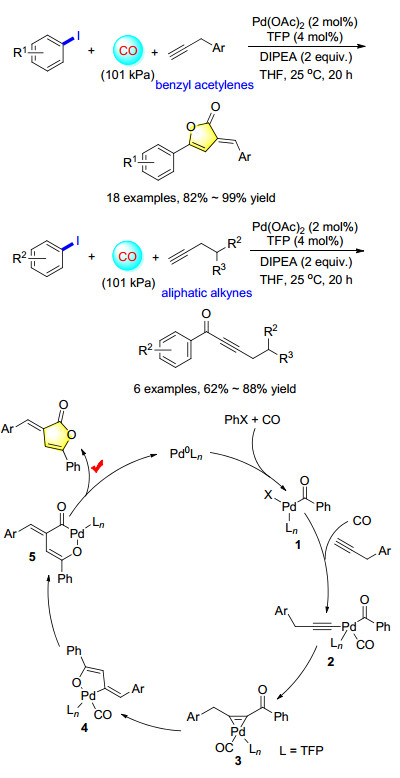
Scheme 1.
Palladium-catalyzed carbonylative coupling of aryl iodides and acetylenes

Heterocyclic compounds are an integral part of many biologically active molecules and many currently marketed drugs hold heterocycles as their core structure. Hence, extensive efforts have been focused on the development of efficient approaches for the synthesis of heterocycles. Considering the synthetic value of carbonylation reactions and the importance of heterocycles preparation, the merging of these two topics offers interesting possibilities for organic synthesis. Although a number of reviews on catalytic carbonylation reactions as well as on the synthesis of heterocycles already exist, no general summary on transition metal catalyzed carbonylation synthesis of heterocycles via activation of C—H or C—X bond by using CO gas and CO surrogates have been published so far.[1] Here we report a summary of our group's recent progress in this area, and this account mainly divided into four parts according to the different bonds activated (C—H or C—X) and the different CO sources applied (CO gas or CO surrogates).
Furanones are important compounds with various biological activities, such as anti-inflammatory, cardiotonic, analgesic, anticonvulsant and antiviral activity. In 1993, Eaton group[2a] reported a catalytic iron-catalyzed [4+1] cycloaddition with allenyl ketones, aldehydes, and carbon monoxide to give alkylidenebutenolides. Our group developed a general and efficient palladium-catalyzed carbonylation coupling reaction of aryl iodides with benzyl acetylenes under CO atmosphere. Various furanones were prepared in excellent yields from the corresponding benzyl acetylenes. Interestingly, when aliphatic alkynes were used as substrates, the corresponding alkynones were formed in good yields, which might come from the fact that the benzylic hydrogen atoms are more activated than the aliphatic hydrogen atoms.
Meanwhile, density functional theory (DFT) calculations were also carried out to understand the reaction pathway. The most probable reaction mechanism is proposed (Scheme 1). The reaction begins with the oxidative addition of PhX to the Pd0 center, followed by CO coordination and insertion to form acyl complex 1. Then complex 2 are formed through a ligand exchange of X with the alkynyl moiety, which undergoes reductive C-C coupling to afford complex 3. The isomerization of complex 3 forms complex 4. Further CO insertion forms complex 5 and subsequent reductive CO coupling forms the product and regenerate the catalyst.[2b]
For the synthesis of some other furanone derivatives, a new palladium-catalyzed double carbonylation procedure by coupling of aryl halides, CO and terminal alkenes was developed later (Scheme 2). Different aryl bromides and iodides transformed into the corresponding furanones in modest and good yields. Using this new atom-efficient carbonylation reaction, a total of 18 different 4-aryl- furanones were synthesized in up to 87% yield.[3]
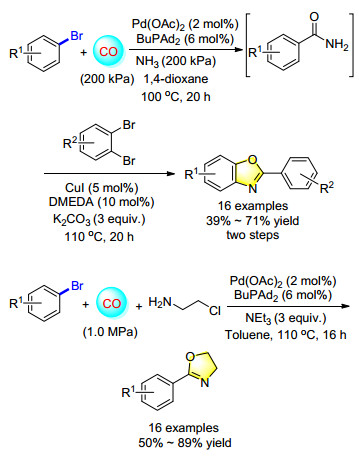
Meanwhile, a one-pot synthesis of benzoxazoles from aryl bromides and 1, 2-dibromobenzenes (Scheme 3) was developed. The first step is a palladium-catalyzed aminocarbonylation of bromobenzene under 200 kPa of NH3 and CO in 1, 4-dioxane, which gave the desired product benzamide successfully. Then treated this crude mixture with CuI/DMEDA/K2CO3 under 110 ℃, various 2-aryl- substituted benzoxazoles were produced in moderate to good yield.[4] In addition, a general palladium-catalyzed carbonylation synthesis of different 2-aryloxzolines was also represented. Bromobenzene and 2-chloroethylamine were selected as the substrates, various five-membered- ring oxazolines were synthesized successfully under 1.0 MPa CO in the presence of Pd(OAc)2/BuPAd2/NEt3 with the yield up to 89%.[5]
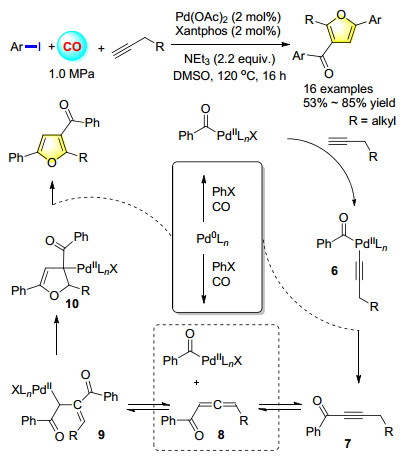
Due to our ongoing interest in the carbonylation reactions and the crucial of furans, a palladium-catalyzed double carbonylation coupling of aryl halides and aliphatic alkynes to give furan heterocycles was developed (Scheme 4). The most sensible mechanism based on detailed DFT calculation results is proposed. The first step is the Sonogashira-type formation of alkynone intermediate 7, and then acyl insertion to the isomerized allene 8 from the formed alkynone 7. The crucial step is a concerted ring- closure and Pd shift, which is also the rate-determining step. The last step is β-H elimination with the formation of furan product and the regeneration of the catalyst by the base.[6]
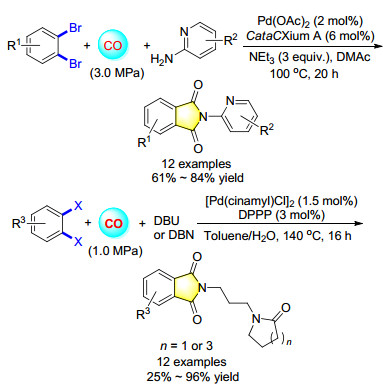
Imide derivatives, especially phthalimides, are an important class of compounds with various biological activities. A convenient double carbonylation of o-dibromoben- zenes with various 2-amino pyridines by using Pd(OAc)2 and cataCXium A as the catalyst system was estabilished (Scheme 5). More importantly, this procedure is also suitable for the synthesis of biological compounds containing phthalimide groups.[7] Interestingly, after changing the base from NEt3 to 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 1, 5-diazabicyclo[4.3.0]non-5-ene (DBN), they were found act as both base and nucleophile in the reaction. Hydrolysis leading to the ring opening reaction of DBU or DBN, different caprolactam and butyrolactam were synthesized in one-pot with the yield up to 96%.[8]
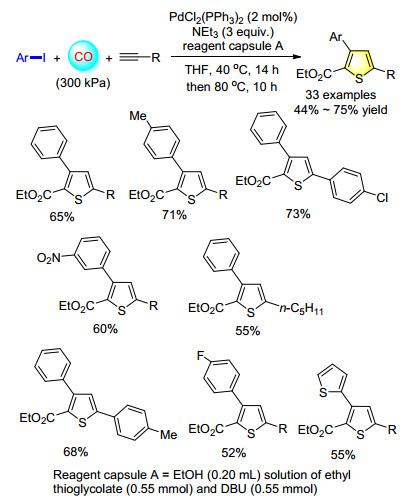
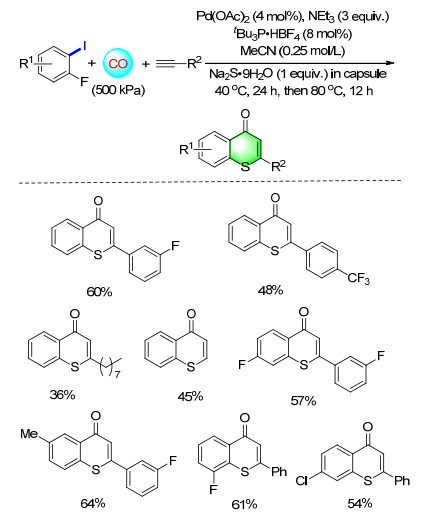
The application of encapsulation has been extended from pharmacy to other realms. In organic synthesis, reagent capsules can help to resolve the compatibility problem of reagents in multicomponent processes. A facile method for encapsulating smelly liquid sulfur chemicals and its application for the synthesis of thiophenes and benzothiophenes was estabilished (Scheme 6). In this process, the capsule plays a crucial role in solving the selectivity of multicomponent reactions as well as avoiding the deactivation of catalysts.[9]
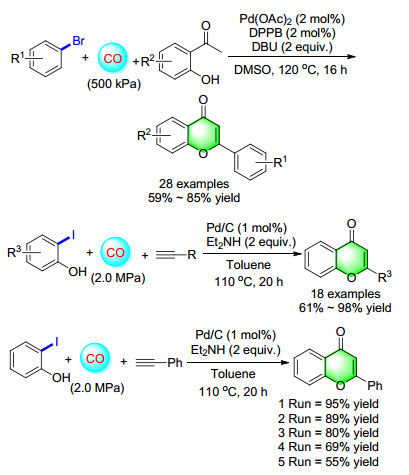
Flavones are an important class of products with a range of biological activities. In view of the structure of flavones, carbonylation reactions should be an efficient method for their construction. Actually, the application of carbonylation reaction in flavone synthesis has been shown by different groups, [10] 2-iodophenol derivatives and terminal acetylenes were used as start materials in these reactions. In 2012, the first carbonylative reaction of bromobenzene and hydroxyacetophenone was reported, the yield of the desired products can be up to 85% when DPPB and DBU were used as the ligand and base (Scheme 7). Different bromobenzenes including hetero-bromobenzenes were transformed into the corresponding flavones in moderate to good yields.[11] Considering for the advantages of heterogeneous catalysts, we also developed a highly efficient and selective Pd/C catalyzed cyclo-carbonylation reaction of 2-iodophenol and ethynylbenzene. Et2NH was the best base and gave the desired product up to 98% yield. Both aromatic terminal acetylenes and aliphatic terminal acetylenes worked well in this condition, gave the corresponding flavones in 61%~98% yields. Furthermore, Pd/C as the heterogeneous catalysts, the reusability experiment was tested and the yield was 55% after five times running.[12]
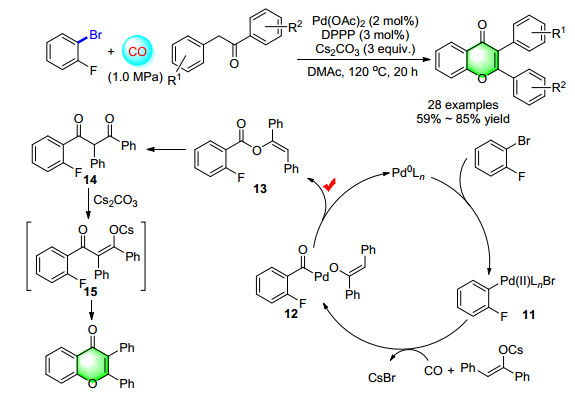
Regarding the synthesis of 2, 3-disubstituted chromones, we also discovered an unexpected palladium-catalyzed carbonylation reaction from 2-bromofluorobenzenes and ketones (Scheme 8). According to the results of control experiments, the possible reaction mechanism was proposed. The first step is a palladium catalyzed carbonylative arylation of deoxybenzoin generated an enol ester 13 instead of a 1, 3-diketone. Then this carbonylation event was followed by the Claisen-Hasse rearrangement and followed an intramolecular SNAr reaction to give the product chromones. The reaction displays good tolerance to various substituent groups at different positions on the start materials in moderate to good yield.[13]
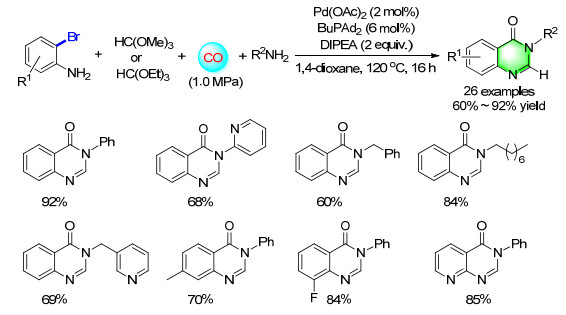
Quinazolinones are an important class of heterocycle, and many synthetic procedures have been developed for their preparation. Among them, 2-aminobenzamides with benzyl alcohols, acyl chlorides or their derivatives are the typical methodologies. From a synthetic point of view and the significant of carbonylation transformations, our group established a series of elegant palladium-catalyzed carbonylation coupling system for the synthesis of different quinazolinones. In 2014, a four-component method for the synthesis of quinazolinones was developed (Scheme 9). By using N, N-diisopropylethylamine (DIPEA), Pd(OAc)2/ BuPAd2 as the catalyst system, 2-bromoaniline, aniline, triethyl orthoformate and CO proceeded smoothly in 1.4-dioxane gave 3-phenyl-4(3H)-quinazolinone up to 92% yield. Aromatic amines and hetero-aromatic amines with different substitutes participated smoothly in our condition. Notably, the benzylamines and aliphatic amines were also successfully transformed into the corresponding quinazolinones.[14]
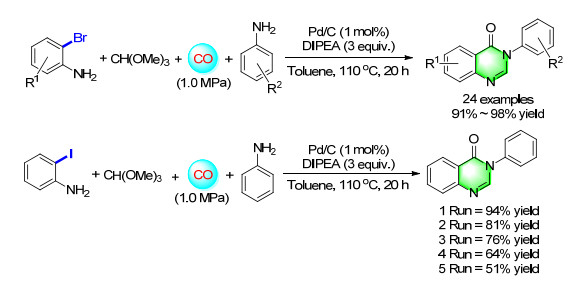
Given the advantage of heterogeneous catalysts, the application of Pd/C as a heterogeneous catalyst for the four-component carbonylation reaction was studied. To our delight, when 2-iodoaniline was selected as model substrate, in the presence of DIPEA, Pd/C and toluene the desired product was provided in 96% yield (Scheme 10). The result of substrates scoping showed a good tolerance of various aromatic amine derivatives and aliphatic amines. Moreover, Pd/C was found to be effectively recycled for four consecutive cycles in our recycling experiments.[15]
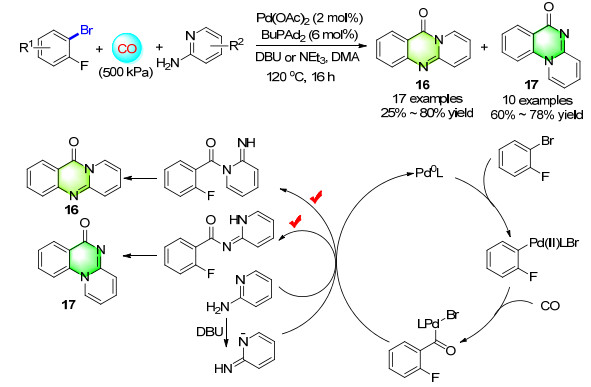
Following the same idea, an efficient novel palladium catalyzed carbonylation/intramolecular nucleophilic aromatic substitution reaction to give pyridoquinazolones has been developed (Scheme 11). The reaction showed good regioselectivity for the formation of both linear and angular fused isomers, which could simply be controlled by the choice of base (NEt3 or DBU). Whereas in the presence of DBU the linear products 16 were obtained, NEt3 gave the angular derivatives 17. The strength of the base may play a pivotal role in this process (the acidities of the conjugate acids are pKa=12 and 10.8 for DBU and NEt3 (triethylamine), respectively). Indeed, NMR spectroscopic experiments confirmed that DBU deprotonates 2-aminopyridine to a major extent to afford the 2-imino-2H-pyridin-1-ide which was not observed in the presence of NEt3.[16]
Combining the same substrate 1-bromo-2-fluoro- benzenes with amidines under the catalytic palladium system, the corresponding quinazolinones can also be formed in moderate to excellent yields (Scheme 12). Different types of double nucleophiles, such as N-N, N-C, and O-C, can be effectively applied as coupling partners. The corresponding six-membered heterocycles were isolated in moderate to good yields.[17]
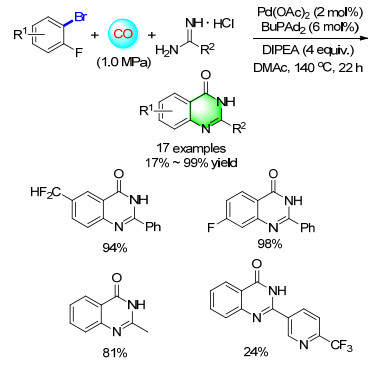
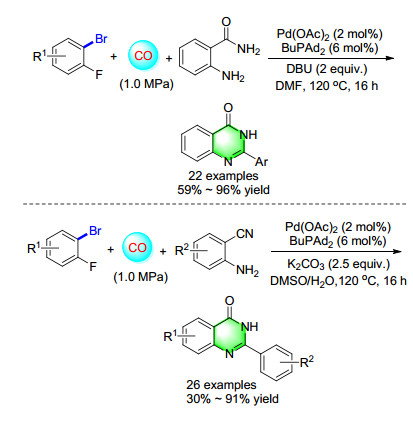
Continuing to expand the protocols for the synthesis of quinazolinones, an interesting reaction was carried out with 2-aminobenzamides, bromobenzenes, DBU, and Pd(OAc)2/BuPAd2 in dimethylformamide (DMF) under 1.0 MPa of CO at 120 ℃. Various substitutes at different positions of bromobenzenes were transformed into the corresponding quinazolinones (Scheme 13).[18] Since the variation of 2-aminobenzamide is very limited, a methodology with widely available substitutes is more attractive and necessary. In 2014, a cascade synthesis of quinazolinones from 2-aminobenzonitriles and aryl bromides via palladium-catalyzed carbonylation reaction was developed in our group. The reactions go through aminocarbonylation of aryl bromides-hydration of nitriles-cyclization sequence.[19]
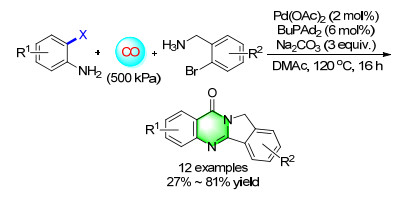
In contrast, starting from commercially available 2-bromoanilines and 2-bromobenzyl amines, with the assistance of a palladium catalyst, isoindolo[1, 2-b]- quinazolin-10(12H)-ones can be isolated in good yields (Scheme 14). Notably, this procedure proceeded in a highly selective manner and atom economy; two molecules of CO were incorporated into the substrates selectively.[20]
The reagent capsule was essential to solve problems related to the poisoning of the transition-metal catalyst as well as the compatibility of the various reagents in the one-pot process. An efficient palladium-catalyzed carbonylation method for preparing thiochromenones in a four-component reaction that makes use of a reagent capsule has been developed based on these merits (Scheme 15). With substituted phenylacetylenes and aliphatic alkynes, moderate to good yields of the desired products were achieved. This is the first example of applying a reagent capsule for preventing catalyst poisoning and undesired side reactions in a multicomponent reaction.[21]
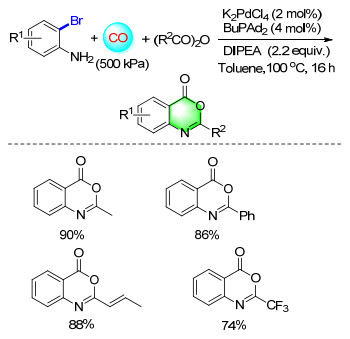
Benzoxazinones represent a class of annulated nitrogen heterocycles that are of interest in organic synthesis due to their various biological activities. By starting from readily available 2-bromoanilines, inexpensive acid anhydrides, CO and K2PdCl4 as the catalyst precursor, various 2-alkylbenzoxazinones were synthesized in good yields (Scheme 16).[22]
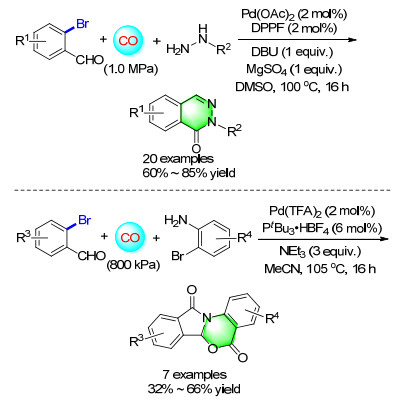
A general and straightforward methodology for the carbonylative synthesis of pharmacologically interesting phthalazinones has been established by using Pd(OAc)2/ DPPF as the catalysts. MgSO4 was used as the additive to facilitate the condensation step in the domino sequence. Starting from readily accessible 2-bromobenz-aldehydes or 2-bromoacetophenone and hydrazines, twenty different phthalazinones were synthesized in moderate to good isolated yields (Scheme 17).[23] When simply changing the hydrazines with 2-bromoanilines, a concise and highly versatile method for the synthesis of functionalized isoindolinones was estabilished. Various 2-bromoanilines undergo palladium-catalyzed carbonylation with 2-formyl- benzoic acid under a convenient and mild procedure to give good to excellent yields of the corresponding isoindolinones.[24]
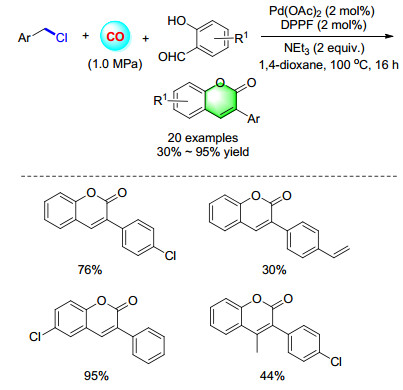
In 2013, our group reported a palladium-catalyzed carbonylative synthesis of coumarins from salicylic aldehydes and benzyl chlorides (Scheme 18). Various coumarins were produced in good to excellent yields. Concerning the reaction pathway, we proposed a palladium catalyzed oxyl- carbonylative reaction and followed an intramolecular condensation.[25]
Through palladium-catalyzed carbonylative coupling of 2-aminobenzylamine with aryl bromides, we reported an efficient method for the synthesis of quinazolines (Scheme 19). The reactions followed an aminocarbonylation- condensation-oxidation sequence in a one-pot one-step manner. The preliminary investigation showed DMSO serves as both solvent and oxidant in this procedure.[26] By simply changing the electrophile bromobenzene to o-di- bromo-benzene, an interesting and double carbonylation reaction for the synthesis of isoindoloquinazolinones has been developed. Several kinds of isoindoloquinazolinones were produced and isolated in moderate to good yields. This procedure enriched the synthesis method of isoindoloquinazolinones and helped in exploring the biological applications of batracylin derivatives.[27]
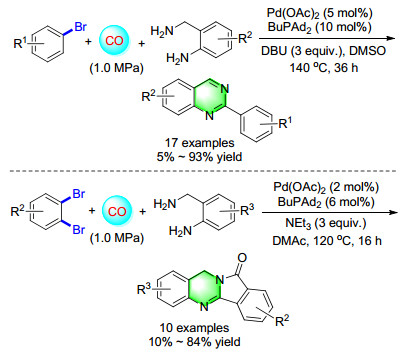
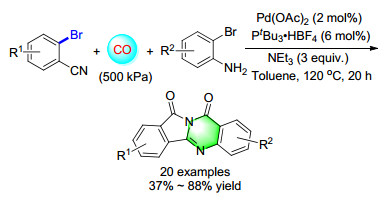
In 2014, a palladium-catalyzed double-carbonylation process for the synthesis of quinazolinediones has been developed (Scheme 20). Starting from commercially available 2-bromobenzonitriles and 2-bromoanilines, a series of isoindolo[1, 2-b]quinazoline-10, 12-diones were synthesized in a straightforward manner with good isolated yields. At least five different C—C and/or C—N bonds are selectively formed in this 3-component reaction, which likely proceeds through sequential carbonylation-cycliza- tion-isomerisation-carbonylation steps.[28]
Seven-membered heterocycles are received continuing attention as their skeletons are widely present in numerous pharmaceuticals and natural products. Among these seven-membered heterocycles, benzoxazepinones represent a class of versatile compounds owing to their promising pharmaceutical and biological activities. Conventional routes to benzoxazepinones and their derivatives usually involve several steps, and isolation of intermediates. In 2015, our group[29] reported a highly-efficient one-pot palladium-catalyzed amino-carbonylation/SNAr approach to dibenzoxazepinones and pyridobenzoxazepinones (Scheme 21). 2-Aminophenol was employed as model substrates to react with 2-bromofluorobenzene or 3-bromo-2-chloro- pyridine.
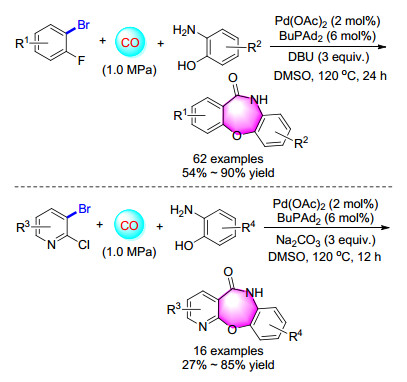
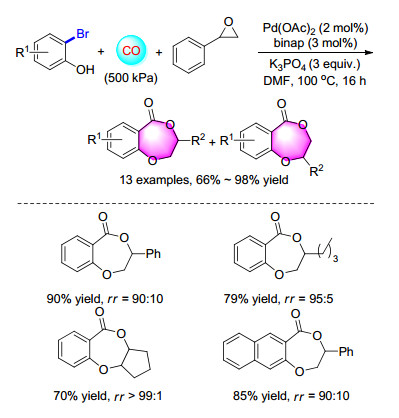
A cascade procedure for the synthesis of 2, 3-dihydro- benzodioxepinone from 2-bromophenols and epoxides was estabilished as well. Starting from commercially available substrates, moderate to good yields of versatile desired products were obtained in a good regioselectivity (major product > 90%) under mild conditions (Scheme 22). The reactions went through nucleophilic ring-opening of epoxides and subsequent palladium-catalyzed intramolecular alkoxylcarbonylation.[30]
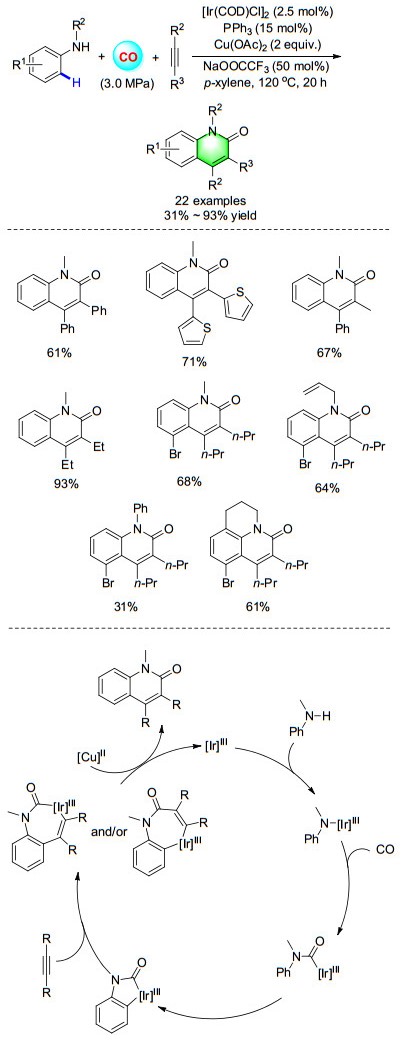
Among all the heterocycles, quinolin-2(1H)-ones are an important class of natural products with a wide range of biological activities, such as anticancer, antibacterial, antiviral, antibiotic activity and so on. Additionally, quinolin- 2(1H)-ones have also been used as valuable synthetic intermediates in organic synthesis. In 2016, a novel and efficient iridium-catalyzed carbonylative annulation of simple anilines with internal alkynes for the straightforward synthesis of halogen-containing quinolin-2(1H)-ones was developed (Scheme 23). The reaction proceeds without pre-activation and directing groups through direct N—H and C—H bond activation with a broad substrate scope, high efficiency, and excellent selectivity. Remarkably, halogen functional groups can be well tolerated here, and this is the first example of iridium-catalyzed carbonylative C—H activation of anilines.[31] The first step is the oxidation of IrⅠ to IrⅢ which then get ready for the catalytic cycle. Then, a nitrogen bonded iridium complex will be formed after ligand exchange with aniline derivative which will produce iridium formamide intermediate after CO insertion. A five-membered iridium cycle through ortho C—H bond activation will be produced from the iridium formamide complex. After the insertion of internal alkyne, seven-membered iridium cycle will be formed which then give the final quinolin-2(1H)-ones after reductive elimination and the together formed IrⅠ will be reoxidized into IrⅢ by copper salt.
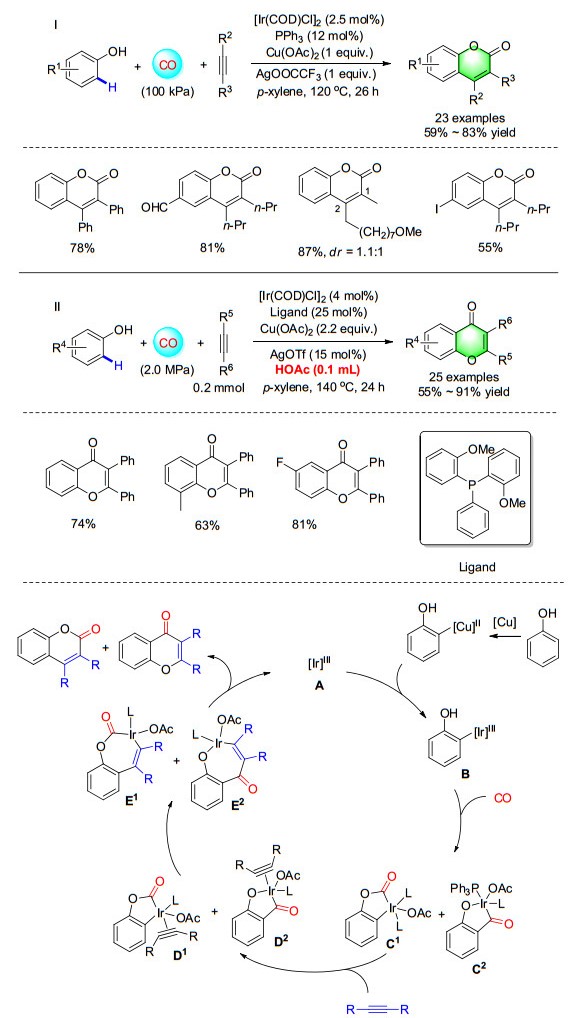
Inspired by this positive result, we also expand the iridium-catalyzed carbonylation reactions. In 2016, the first carbonylative annulation reaction for the direct synthesis of coumarins from phenols and alkynes was reported (Scheme 24). With iridium as the catalyst and copper as the promotor, various coumarins were prepared in good to excellent yields and with good selectivity at atmospheric pressure. Interestingly, when AcOH and bis(2-methoxy- phenyl)(phenyl)phosphane were used, the isomer product flavones was got. Various flavones were also isolated in moderate to good yields with excellent regioselectivity and functional group tolerance by using the iridium catalyst system. This is the first example of direct carbonylation annulation of non-preactivated phenols and alkynes to produce flavones, with the choice of ligand proving to be critical for the success of this transformation.[32] More recently, with palladium as the catalyst, the carbonylative cyclization of terminal alkynes with phenols was realized as well.[32c]
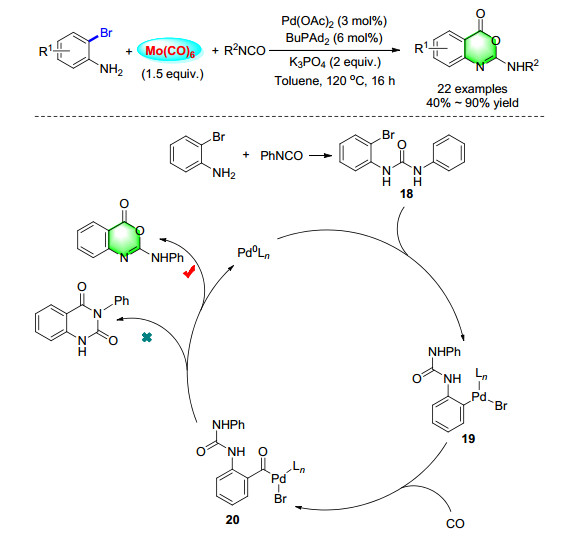
Since the highly toxic and flammable properties of carbon monoxide, performing carbonylation reactions without the use of CO are highly desired and will contribute to the further improvement of sustainable chemistry. Among the candidates, Mo(CO)6 as an air-stable and non-toxic solid is an attractive CO source. Therefore, a general and convenient methodology for 2-amino benzoxazinone synthesis using 2-bromoanilines and isocyanates as substrates and Mo(CO)6 as a solid CO surrogate was developed (Scheme 25). This reaction can tolerate both electron-donating and electron-withdrawing groups and give the corresponding benzoxazinones up to 90% yield. Remarkably, 3-phenyl- quinazoline-2, 4(1H, 3H)dione as one of the most possible products is not detected in this transformation. We propose that Mo(CO)6 may act as a Lewis acid and coordinate with 20 to assist in the formation of 2-amino- benzoxazinones.[33]
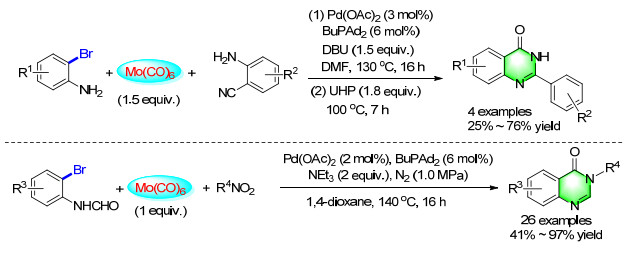
With Mo(CO)6 as a solid CO surrogate, a novel and convenient way to synthesize N-phenylquinazolinones through aminocarbonylation of 2-aminobenzonitriles with bromoarenes was developed (Scheme 26). Aryl bromides and 2-aminobenzonitrile derivatives were selected as the substrates, various quinazolinones can be formed by the assistance of urea hydroperoxide (UHP) in 25%~76% yields of two steps.[34] Later on, a procedure for palladium-catalyzed carbonylation synthesis of quinazolinones from 2-bromoformanilides and nitro compounds in 41%~97% yields was also reported. Selective reduction of the nitro group in the presence of other sensitive competing functionalities was a challenging problem, and the compatibility between the conditions of nitro reduction and aminocarbonylation was hard to be achieved. In this transformation, both aromatic nitros and aliphatic nitros were found to be suitable substrates. Moreover, Mo(CO)6 was not only a CO source but also a nitro compound reducing reagent and a cyclization promoter.[35]
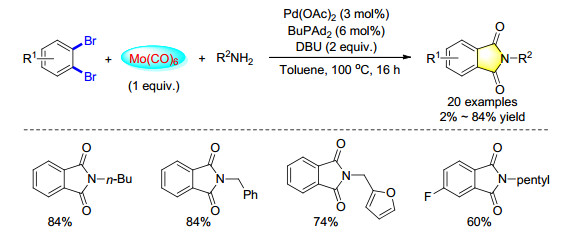
N-Substituted phthalimides represented an important class of biologically active molecules. Regarding their importance, a palladium-catalyzed carbonylation synthesis of phthalimides from 1, 2-dibromoarenes with Mo(CO)6 was demonstrated (Scheme 27). The reaction tolerated various functional groups on the aromatic rings and gave the corresponding phthalimides up to 84% yield.[36]
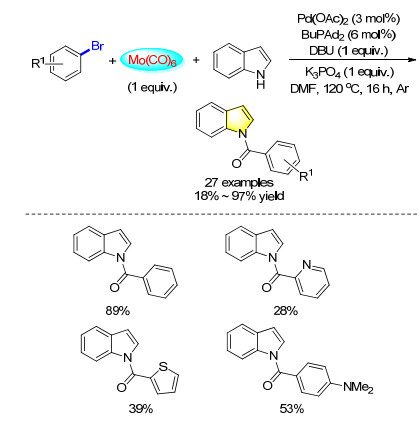
Indoles were important skeletons in material science and pharmaceuticals. Recently, a variety of methodologies have been published. Our group[37] has elaborated a versatile method to form different kinds of N-benzoylindoles via palladium-catalyzed aminocarbonylation of aryl bromides with Mo(CO)6 (Scheme 28). In this reaction, DBU was used as the base due to its ability to coordinate at the molybdenum to promote the in situ release of CO. K3PO4 can increase the yield of the reaction as a base.
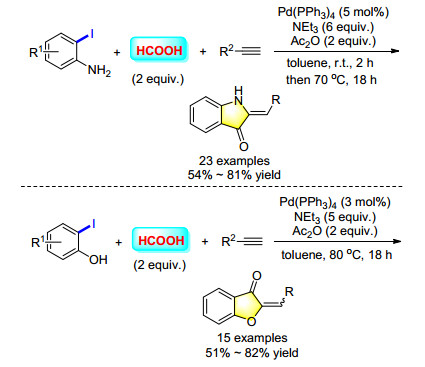
Formic acid represents an interesting and potential bio-renewable compound that can act as a carbon monoxide precursor for the production of value-added chemicals. With formic acid as the CO source and Ac2O as the activator, a novel method for the synthesis of a variety of 2-benzylideneindolin-3-ones was reported (Scheme 29). A plausible reaction mechanism was proposed, a palladium catalyzed carbonylation reaction of 2-iodoanilines and alkynes gave the alkynyl ketone intermediate, which formed the final product 2-benzylideneindolin-3-ones through a ring closing reaction with the assistance of PPh3. Furthermore, using the same condition, different 2-iodophenols and terminal alkynes transformed into the corresponding aurones in moderate to good yield.[38]
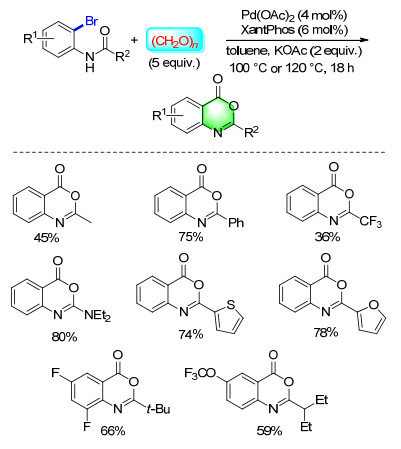
Compared to many other CO surrogates, paraformaldehyde is more desirable since it is a solid, cheap, stable, easily-to-handle and exhibits low toxicity. In 2014, our group established a simple and general method for the synthesis of benzoxazinone derivatives from N-(o-bromo- aryl)amides and paraformaldehyde (Scheme 30). The starting materials can be easily prepared from 2-bromo- anilines and acid chlorides or anhydrides. This method provided a practical pathway for the carbonylation synthesis of various benzoxazinone heterocycles in moderate to good yields.[39]
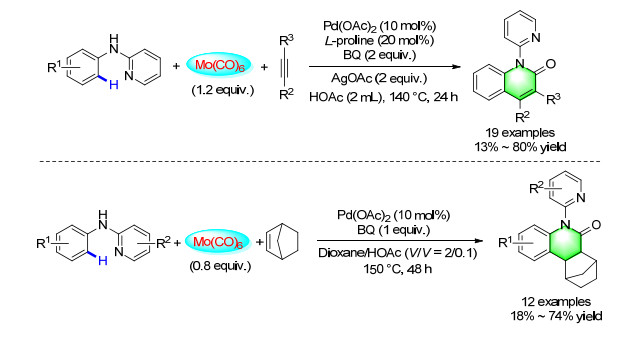
The transition-metal-catalyzed direct functionalization of inert C—H bonds for the synthesis of complex molecules matched well with the concept of sustainable development as it avoids tedious and sluggish pre-activations. In 2014, a novel procedure for the synthesis of highly substituted 2-quinolinones by carbonylation cyclization of N-aryl-pyridine-2-amines and internal alkynes through C—H activation was reported. Mo(CO)6 was applied as a solid CO source, the reaction proceeded in an atom economic manner, and various 2-quinolinone derivatives were prepared in moderate to good yields.[40] When we chose norbornene as the coupling partner, an interesting transformation on palladium-catalyzed carbonylative C—H activation of arenes with norbornene has been developed. Various 5-(pyridin-2-yl)-hexahydro-7, 10-methanophenan- thridin-6(5H)-ones were produced in moderate yields. The product can also be oxidized to the corresponding 5- (pyridin-2-yl)-tetrahydro-7, 10-methanophenanthridin- 6(5H)-one by using 2, 3-dichloro-5, 6-dicyano-p-benzo- quinone (DDQ) as the oxidant (Scheme 31).[41]
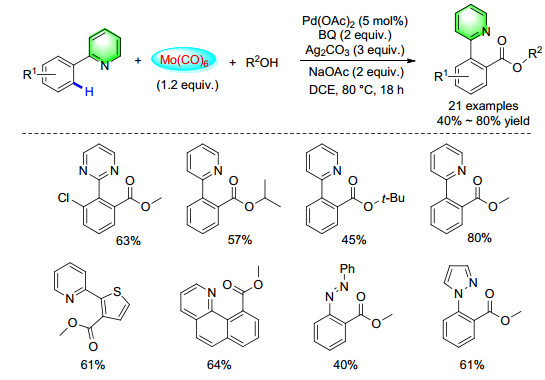
Apart from using N-aryl-pyridine-2-amines as the directing group, we also tested other directing groups for the functionalization or synthesis of the heterocycles. For example, in 2016, a general palladium-catalyzed carbonylation transformation of the C—H bond on aromatic rings to produce esters by using 2-arylpyridine derivatives as the substrates was reported (Scheme 32). Good yields of the corresponding products have been obtained with wide functional group tolerance and excellent regioselectivity. A variety of aliphatic alcohols are suitable reactants here.[42]
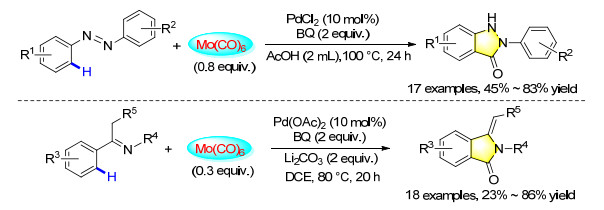
Furthermore, two interesting palladium-catalyzed carbonylation protocols for the intramolecular cyclization of azoarenes and ketimines were also established. With Mo(CO)6 as the solid CO source and through C(sp2)—H bond activation, a series of azoarenes and ketimines were transformed into the corresponding 2-aryl-indazolones and 3-methyleneisoindolin-1-ones in moderate to good yields (Scheme 33).[43]
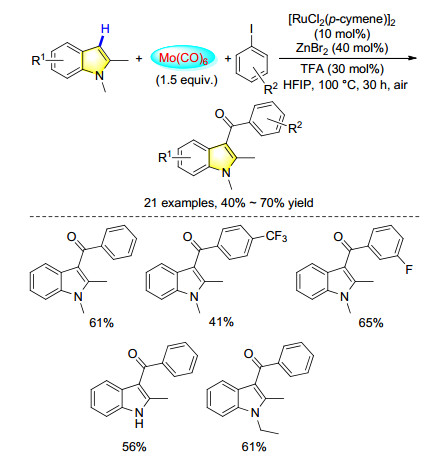
Among the various noble transition-metal catalysts, ruthenium catalysts are attractive due to their relative low cost and high reaction selectivity. Several challenging transformations have been realized with ruthenium complex as the catalyst, however, the reports about ruthenium catalyzed carbonylation reactions are still very limited. In 2017, our group developed a convenient procedure for the synthesis of 3-acylindoles from simple indoles and aryl iodides. Through ruthenium-catalyzed carbonylative C—H functionalization of indoles, with Mo(CO)6 as the solid CO source, the desired indol-3-yl aryl ketones were isolated in moderate to good yields. Not only N-alkyl indoles but also N-H indoles can be applied in this reaction (Scheme 34).[44]
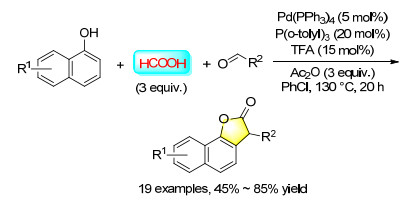
Benzofuranones and isobenzofuranones are very important structural moieties which present in numerous pharmaceutical compounds and biological molecules. Thus the development of new synthetic methods for such moieties is always under demand. In 2016, a convenient palladium-catalyzed carbonylation synthesis of benzofuran-2(3H)-ones by using commercially available phenols and aldehydes as the substrates with formic acid as the CO precursor was reported, and benzofuran-2(3H)-ones have been prepared in moderate to good yields. Both aromatic and aliphatic aldehydes are applicable in this reaction (Scheme 35).[45]
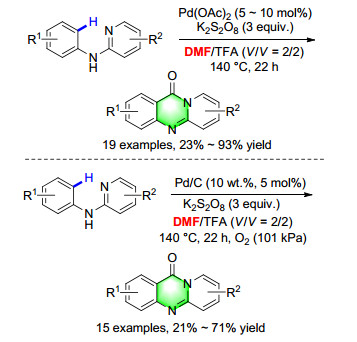
Among all the possible CO candidates, DMF is an attractive one because it is cheap and easily accessible. The first general palladium-catalyzed carbonylation cyclization of N-arylpyridin-2-amines by C—H activation using DMF as the CO surrogate was developed. Various quinazolinones were formed in moderate to good yields with good functional group tolerance. A 13CO-labelled DMF study and other control experiments indicated that the carbonyl group of DMF is the CO source in this methodology. The kinetic isotope effect (KIE) value suggested that the C—H activation step might not be involved in the rate-determining step under our conditions.[46] Meanwhile, the heterogeneous catalyst Pd/C was also applicable in this reaction (Scheme 36).[47]
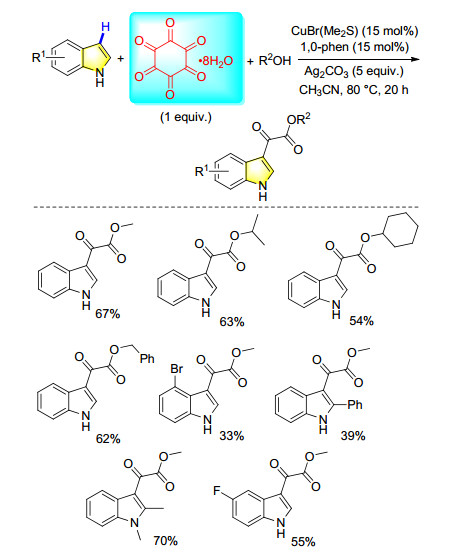
Hexaketocyclohexane octahydrate (C6O6•8H2O) is a kind of nontoxic stable solid. It was originally produced by oligomerization of carbon monoxide, and can be considered to be six-fold of carbon monoxide with almost 100% atom efficiency. Because the ring of hexaketocyclohexane is highly strained, it can potentially decompose to CO in the reaction solution. Very recently, our group developed a novel procedure on copper-catalyzed double carbonylation of indoles with alcohols via C—H bond functionalization (Scheme 37).[48] Using alcohols as reaction partners, moderate to good yields of the desired double carbonylation products have been obtained. Wide functional group tolerance and substrate scope can be observed.
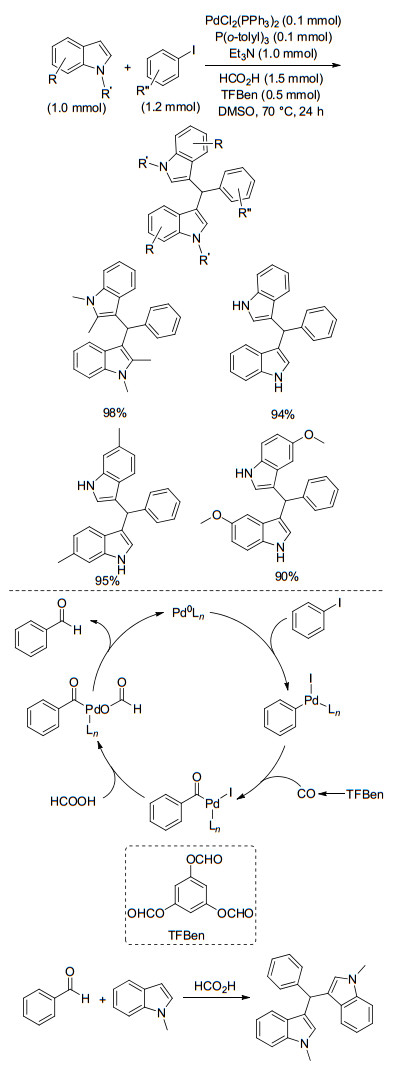
Benzene-1, 3, 5-triyl triformate (TFBen) is a stable and non-reacting CO surrogate was developed in our group.[49] Recently, we achieved a CO gas-free palladium-catalyzed carbonylative procedure for the synthesis of bis(indolyl)- methanes.[50] With TFBen as the solid CO source, aryl iodides and indoles were transformed into the corresponding bis(indolyl)methane derivatives in moderate to excellent yields (Scheme 38). Based on our mechanistic studies, the reaction system can be divided into two parts: the first part is the reductive carbonylation of aryl iodides to produce benzaldehydes in situ; the second part is the formic acid promoted nucleophilic addition of indoles to benzaldehydes to give the final products.
Transition metal catalyzed carbonylation reactions already became the most important methods for organic synthesis in academic as well as industry. Despite various kinds of novel carbonylation reactions have been developed over the past decades, several challenge problems still exist, which need to be solved. Generally, catalyst efficiency in such reactions is still low, relatively harsh reaction conditions and additional additives are needed to activate the alternative CO surrogates, which mean additional cost and waste. Besides, the regioselectivity and sustainability in most cases were inefficient, and most of those cases need relatively high-pressure CO. All of these disadvantages limited the widely application of transition metal catalyzed carbonylation reactions.
What are the goals for the future years in carbonylation reactions? Regarding the CO sources, the inexpensive, easily available, and less toxic CO surrogates should be discovered and applied more in organic chemistry. Apart from metal carbonyl compounds, formic acid and aldehyde, the in situ reduction of CO2 to CO is the most promising method since the greenhouse effect. In addition, biomass, which is also a readily available and renewable resource, offers potential opportunities for the future research. In the case of catalysts, up until now, palladium catalysts dominate the most cases in the carbonylation reactions. The other cheap catalysts like iron, cobalt, manganese, and copper have not yet been systemically explored.
For selected recent reviews, see: (a) Wu, X.-F.; Neumann, H.; Beller, M. Chem. Soc. Rev. 2011, 40, 4986.
(b) Gabriele, B.; Mancuso, R.; Salerno, G. Eur. J. Org. Chem. 2012, 6825.
(c) Peng, J.-B.; Qi, X.; Wu, X.-F. Synlett 2017, 28, 175.
(d) Wu, X.-F. RSC Adv. 2016, 6, 83831.
(e) Peng, J.-B.; Qi, X.; Wu, X.-F. ChemSusChem 2016, 9, 2279.
(f) Peng, J.-B.; Wu, X.-F. Angew. Chem., Int. Ed. 2018, 57, 1152.
(g) Peng, J.-B.; Wu, F.-P.; Wu, X.-F. Chem. Rev. 2019, 119, 2090.
(a) Sigman, M. S.; Kerr, C. E.; Eaton, B. E. J. Am. Chem. Soc. 1993, 115, 7545.
(b) Wu, X. F.; Jiao, H. J.; Neumann, H.; Beller, M. Chem. Eur. J. 2012, 18, 16177.
Schranck, J.; Wu, X. F.; Tlili, A.; Neumann, H.; Beller, M. Chem. Eur. J. 2013, 19, 12959. doi: 10.1002/chem.v19.39
Wu, X. F.; Neumann, H.; Neumann, S.; Beller, M. Tetrahedron Lett. 2013, 54, 3040. doi: 10.1016/j.tetlet.2013.03.053
Wu, X. F.; Neumann, H.; Neumann, S.; Beller, M. Chem.-Eur. J. 2012, 18, 13619. doi: 10.1002/chem.201202652
Wu, X. F.; Zhang, M.; Jiao, H. J.; Neumann, H.; Beller, M. Asian J. Org. Chem. 2013, 2, 135. doi: 10.1002/ajoc.v2.2
Chen, J. B.; Natte, K.; Spannenberg, A.; Neumann, H.; Beller, M.; Wu, X. F. Org. Biomol. Chem. 2014, 12, 5578. doi: 10.1039/C4OB00796D
Chen, J. B.; Natte, K.; Wu, X. F. Tetrahedron Lett. 2015, 56, 342. doi: 10.1016/j.tetlet.2014.11.091
Shen, C. R.; Spannenberg, A.; Auer, M.; Wu, X. F. Adv. Synth. Catal. 2017, 359, 941. doi: 10.1002/adsc.201601343
(a) Yang, Q.; Alper, H. J. Org. Chem. 2010, 75, 948. (b) Awuah, E.; Capretta, A. Org. Lett. 2009, 11, 3210. (d) Miao, H.; Yang, Z. Org. Lett. 2000, 2, 1765.
Wu, X. F.; Neumann, H.; Beller, M. Chem.-Eur. J. 2012, 18, 12595. doi: 10.1002/chem.201202141
Zhu, F. X.; Li, Y. H.; Wang, Z. C.; Wu, X. F. Catal. Sci. Technol. 2016, 6, 2905 doi: 10.1039/C6CY00613B
Shen, C. R.; Li, W. F.; Yin, H. F.; Spannenberg, A.; Skrydstrup, T.; Wu, X. F. Adv. Synth. Catal. 2016, 358, 466. doi: 10.1002/adsc.201500858
He, L.; Li, H. Q.; Neumann, H.; Beller, M.; Wu, X. F. Angew. Chem., Int. Ed. 2014, 53, 1420. doi: 10.1002/anie.v53.5
Natte, K.; Neumann, H.; Wu, X. F. Catal. Sci. Technol. 2015, 5, 4474. doi: 10.1039/C5CY00907C
Chen, J. B.; Natte, K.; Spannenberg, A.; Neumann, H.; Langer, P.; Beller, M.; Wu, X. F. Angew. Chem., Int. Ed. 2014, 53, 7579. doi: 10.1002/anie.201402779
Chen, J. B.; Natte, K.; Neumann, H.; Wu, X. F. Chem.-Eur. J. 2014, 20, 16107. doi: 10.1002/chem.v20.49
Wu, X. F.; He, L.; Neumann, H.; Beller, M. Chem.-Eur. J. 2013, 19, 12635. doi: 10.1002/chem.201302182
Li, H. Q.; He, L.; Neumann, H.; Beller, M.; Wu, X. F. Green Chem. 2014, 16, 1336. doi: 10.1039/C3GC42089B
Shen, C.; Man, N. Y. T.; Stewart, S.; Wu, X. F. Org. Biomol. Chem. 2015, 13, 4422. doi: 10.1039/C5OB00368G
Shen, C. R.; Spannenberg, A.; Wu, X. F. Angew. Chem., Int. Ed. 2016, 55, 5067. doi: 10.1002/anie.v55.16
Wu, X. F.; Neumann, H.; Beller, M. Chem.-Eur. J. 2012, 18, 12599. doi: 10.1002/chem.201202142
Wu, X. F.; Neumann, H.; Neumann, S.; Beller, M. Chem.-Eur. J. 2012, 18, 8596. doi: 10.1002/chem.v18.28
Natte, K.; Chen, J. B.; Li, H. Q.; Neumann, H.; Beller, M.; Wu, X. F. Chem.-Eur. J. 2014, 20, 14184. doi: 10.1002/chem.v20.44
Wu, X. F.; Wu, L. P.; Jackstell, R.; Neumann, H.; Beller, M. Chem.-Eur. J. 2013, 19, 12245. doi: 10.1002/chem.201301774
Chen, J. B.; Natte, K.; Neumann, H.; Wu, X. F. RSC Adv. 2014, 4, 56502. doi: 10.1039/C4RA11303A
Chen, J. B.; Neumann, H.; Beller, M.; Wu, X. F. Org. Biomol. Chem. 2014, 12, 5835. doi: 10.1039/C4OB01103A
Li, H. Q.; Li, W. F.; Spannenberg, A.; Baumann, W.; Neumann, H.; Beller, M.; Wu, X. F. Chem.-Eur. J. 2014, 20, 8541. doi: 10.1002/chem.201403417
(a) Shen, C. R.; Wu, X. F. Catal. Sci. Technol. 2015, 5, 4433.
(b) Shen, C. R.; Neumann, H.; Wu, X. F. Green Chem. 2015, 17, 2994.
Li, H. Q.; Spannenberg, A.; Neumann, H.; Beller, M.; Wu, X. F. Chem. Commun. 2014, 50, 2114. doi: 10.1039/c3cc48490d
Zhu, F. X.; Li, Y. H.; Wang, Z. C.; Wu, X. F. Adv. Synth. Catal. 2016, 358, 3350. doi: 10.1002/adsc.v358.21
(a) Zhu, F. X.; Li, Y. H.; Wang, Z. C.; Wu, X. F. Angew. Chem., Int. Ed. 2016, 55, 14151. (b) Zhu, F. X.; Wang, Z. C.; Li, Y. H.; Wu, X. F. Chem.-Eur. J. 2017, 23, 3276. (c) Zhu, F. X.; Wu, X. F. Org. Lett. 2018, 20, 3422.
Wu, X. F.; Sharif, M.; Shoaib, K.; Neumann, H.; Pews-Davtyan, A.; Langer, P.; Beller, M. Chem.-Eur. J. 2013, 19, 6230. doi: 10.1002/chem.201300537
Wu, X. F.; Oschatz, S.; Sharif, M.; Beller, M.; Langer, P. Tetrahedron 2014, 70, 23. doi: 10.1016/j.tet.2013.11.055
(a) He, L.; Sharif, M.; Neumann, H.; Beller, M.; Wu, X. F. Green Chem. 2014, 16, 3763.
(b) Peng, J.-B.; Geng, H.-Q.; Wang, W.; Qi, X.; Ying, J.; Wu, X.-F. J. Catal. 2018, 365, 10.
(a) Wu, X. F.; Oschatz, S.; Sharif, M.; Flader, A.; Krey, L.; Beller, M.; Langer, P. Adv. Synth. Catal. 2013, 355, 3581.
(b) Xu, J.-X.; Wu, X.-F. Adv. Synth. Catal. 2018, 360, 3376.
(c) Zhu, F.; Li, Y.; Wang, Z.; Wu, X.-F. ChemCatChem 2016, 8, 3710.
Wu, X. F.; Oschatz, S.; Sharif, M.; Langer, P. Synthesis 2015, 47, 2641. doi: 10.1055/s-00000084
(a) Li, R.; Qi, X. X.; Wu, X. F. Org. Biomol. Chem. 2017, 15, 6905.
(b) Qi, X. X.; Li, R.; Wu, X. F. RSC Adv. 2016, 6, 62810.
(c) Qi, X.; Li, R.; Li, H.-P.; Peng, J.-B.; Ying, J.; Wu, X.-F. ChemCatChem 2018, 10, 3415.
Li, W. F.; Wu, X. F. J. Org. Chem. 2014, 79, 10410. doi: 10.1021/jo5020118
Chen, J. B.; Natte, K.; Spannenberg, A.; Neumann, H.; Beller, M.; Wu, X. F. Chem.-Eur. J. 2014, 20, 14189. doi: 10.1002/chem.v20.44
Chen, J. B.; Natte, K.; Wu, X. F. J. Organomet. Chem. 2016, 803, 9. doi: 10.1016/j.jorganchem.2015.12.010
Wang, Z. C.; Li, Y. H.; Zhu, F. X.; Wu, X. F. Adv. Synth. Catal. 2016, 358, 2855. doi: 10.1002/adsc.201600395
(a) Wang, Z. C.; Yin, Z. P.; Zhu, F. X.; Li, Y. H.; Wu, X. F. ChemCatChem 2017, 9, 3637.
(b) Wang, Z. C.; Zhu, F. X.; Li, Y. H.; Wu, X. F. ChemCatChem 2017, 9, 94.
(a) Wang, Z. C.; Yin, Z. P.; Wu, X. F. Org. Lett. 2017, 19, 4680.
(b) Yin, Z.; Wang, Z.; Wu, X.-F. Org. Biomol. Chem. 2018, 16, 3707.
Qi, X. X.; Li, H. P.; Wu, X. F. Chem. Asian J. 2016, 11, 2453. doi: 10.1002/asia.201600873
Chen, J. B.; Feng, J. B.; Natte, K.; Wu, X. F. Chem.-Eur. J. 2015, 21, 16370. doi: 10.1002/chem.v21.46
Chen, J. B.; Natte, K.; Wu, X. F. Tetrahedron Lett. 2015, 56, 6413. doi: 10.1016/j.tetlet.2015.09.142
Wang, Z.; Yin, Z.; Wu, X.-F. Chem. Commun. 2018, 54, 4798. doi: 10.1039/C8CC01784K
(a) Ying, J.; Zhou, C.; Wu, X.-F. Org. Biomol. Chem. 2018, 16, 1065.
(b) Jiang, L.-B.; Qi, X.; Wu, X.-F. Tetrahedron Lett. 2016, 57, 3368.
(c) Li, C.-L.; Zhang, W.-Q.; Qi, X.; Peng, J.-B.; Wu, X.-F. J. Organomet. Chem. 2017, 838, 9.
(d) Wang, H.; Ying, J.; Lai, M.; Qi, X.; Peng, J.-B.; Wu, X.-F. Adv. Synth. Catal. 2018, 360, 1693.
Qi, X.; Ai, H.-J.; Zhang, N.; Peng, J.-B.; Ying, J.; Wu, X.-F. J. Catal. 2018, 362, 74. doi: 10.1016/j.jcat.2018.03.028

Scheme 19 Carbonylation reactions of aminobenzylamine with bromobenzene or o-dibromobenzene

Scheme 21 Carbonylation reactions of 2-aminophenols with 2-bromofluorobenzenes or 3-bromo-2-chloropyridines

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


































