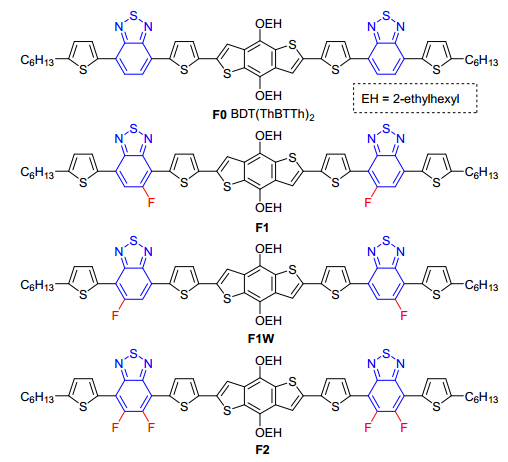
图 1.
化合物F0, F1, F1W和F2的分子结构
Figure 1.
Chemical structures of compounds F0, F1, F1W and F2

有机光电材料具有廉价、质轻、柔性和易于合成与加工等优点而被广泛应用于有机太阳能电池(OPV)[1]、有机场效应晶体管(OFET)[2]、有机发光二极管(OLED)[3]和生物传感器[4]中.近年来, 通过对材料的设计合成[5]、器件结构的优化[6]、活性层形貌的精细调控[7]以及工作机理的深入研究[8], 有机光电器件的性能得到了大幅度的提升.目前, OPV光电转换效率(PCE)已经超过了17.3%[9], 而OFET器件的性能也超过了1 cm2•V-1•s-1[10].然而, 为了解决进一步提升器件性能的巨大挑战, 开发新材料并进行材料结构与器件性能关系的研究是现阶段有机光电材料发展的关键所在.
在最近十几年里, 很多有效的分子设计策略被开发出来以调节光电材料的光电性质, 如给-受体(D-A)结构、侧链工程、官能团的调整等[11].其中, 氟化策略, 即将元素周期表中电负性最强的氟原子引入至有机光电材料的共轭基元, 是对光电材料分子设计和功能修饰的最有效策略[12].氟化赋予有机半导体分子很多独特的性质.首先, 氟化在未增加立体位阻的情况下降低了分子的最高电子占有轨道(HOMO)及最低未占有轨道(LUMO)能级[13]; 其次, 由于F—H、F—S等原子之间的非共价键作用, 有效的增强了分子内和分子间相互作用, 提高了光电材料的结晶度, 从而有利于电荷传输[14]; 第三, 氟化的分子有较高的偶极矩, 降低了电子与空穴之间的库伦力[15].在2009年之前, 已有不少含氟的有机光电材料被应用于OFET和OLED领域[16], 但在OPV中鲜有报道.直到2009年, Yu课题组[17]将氟原子引入受体基元并二噻吩(TT)中, 与给体基元苯并二噻吩(BDT)共聚, 制备了一系列PTB聚合物, 经器件优化, 氟化的聚合物PTB4光电转换性能要优于未被氟化的聚合物, 超过了6%.接着, 该课题组进一步尝试将氟原子引入给体基元BDT中, 制备了PTBF0, PTBF1, PTBF2和PTBF3.单氟化的PTBF1效率(7.2%)高于未被氟化的PTBF0 (PCE=5.1%), 但PTBF2和PTBF3的效率要远低于PTBF0, 仅仅为3.2%和2.7%[18].由此可见, 氟原子的引入能够提高器件性能, 但氟化位点及氟原子的数目对材料性能的研究有待深入研究.除PTB系列聚合物外, 氟原子也被成功引入其他给受体基元, 如苯并噻二唑(BT)[19]、二噻吩苯并噻二唑(DTBT)[20]、苯并三氮唑(BTA)[21]等, 氟化策略被广泛应用于D-A型共轭聚合物体系.
相比于聚合物材料, 氟化策略在小分子有机光电材料的应用却鲜有报道.小分子材料具有结构明确, 重复性好, 易于提纯等优点[22].因此合成含氟小分子并研究氟原子对材料性能的影响有至关重要的作用.在之前的工作中, 我们课题组合成了以BDT为中心BT为两边侧臂的A-D-A型小分子BDT(ThBTTh)2 (F0, 图 1), 其光电转换效率可达4.53%[23].在本工作中, 我们在其两个BT基元上, 引入不同数目的氟取代基, 获得了一系列可溶液加工的新型A-D-A光电小分子F1、F1W和F2(图 1), 系统性地研究了氟取代数目和位置对材料基本性质及其在OFET和OPV器件应用中的性能影响.
三种小分子的合成路线见Scheme 1.其中, 化合物1[24], 8[25], BDTSn2[26]和F0[23]是参照相关文献制备的.对于化合物F1和F1W, 由于含有单氟的二溴苯并噻二唑单元1在进行Suzuki偶联反应时, 远离氟取代基的溴原子具有更高的活性, 我们通过对单体反应顺序的控制, 分别以60%和64%的产率获得中间体2与5; 进一步再通过Suzuki偶联和N-溴代丁二酯酰亚胺(NBS)溴化, 两步得到中间体4与7.对于化合物F2, 首先通过碳-氢键活化偶联直接得到中间体9.然后再将化合物9通过Suzuki偶联及NBS溴化制得中间体11.最后, 采用Stille偶联将4, 7和11分别与BDTSn2反应, 制得F1, F1W和F2; 产率分别为63%, 79%和45%.所得化合物F0的溶解度较差, 但仍能以大于15 mg•mL-1的溶解度溶于热的含氯溶剂(氯仿、氯苯、二氯苯等溶剂)中, 满足器件制备加工的要求.而所制备的化合物F1与F1W的溶解度比F0略有降低, 但仍可溶于热的含氯溶剂. F2的溶解度最差, 须在80 ℃氯苯中才能达到10 mg•mL-1以上的溶解度.
利用热重分析(TGA)和差示扫描量热法(DSC)对三个化合物的热学性质进行表征(图 2).从图 2(a)的TGA曲线可以看出, 三个化合物的5%热失重温度(Td)分别为337.8, 339.4和343.4 ℃, 满足器件加工的温度要求.相比于未被氟化的化合物F0 (Td=335.6 ℃), 氟化之后, 分子的热稳定性略有提升.从图 2(b)的DSC曲线可以看出, 化合物F1和F1W在加热过程和降温过程中都出现了尖锐的吸热和放热峰, 熔点分别为230 ℃和250 ℃.相比于F0(熔点为197 ℃), 氟化之后, 分子的熔点显著提高, 表明氟原子的引入增强了分子间相互作用. F1W的熔点要高于其同分异构体F1, 表明氟化位点对分子间相互作用的影响存在差别.而化合物F2的熔点高于其分解温度, 无法用此方法测得.此外, 三个化合物在其熔点以下都存在宽峰, 表明三个化合物在固态是存在相转变, 可能是由于烷基侧链的相变引起的.
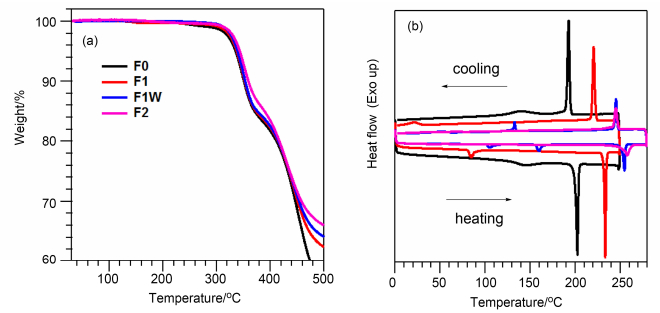
Heating and cooling rate: 10 ℃•min-1, under N2 atmosphere
图 3给出了四个化合物在氯苯溶液及薄膜状态下的紫外可见吸收光谱, 相关谱峰数据列于表 1中.溶液及薄膜状态下吸收光谱对比可以看出, 在化合物中引入氟原子使其吸收光谱略微变窄.对于D-A型结构的化合物来说, 在溶液状态下通常300~650 nm范围内展现出两个主要的吸收峰, 其中, 波长在300~400 nm区域的吸收峰是由于分子共轭骨架-*的跃迁吸收峰; 在这些分子内, 富电子的苯并二噻吩、噻吩与缺电子的BT单元连接在一起, 波长在400~650 nm的吸收峰可归结为分子内电荷转移(Intramolecular charge transfer, ICT)跃迁导致.在溶液状态下, 化合物F1和F1W的吸收光谱非常相似, 通过将一个氟原子引入到BT单元上, 相比与化合物F0均有2 nm的蓝移.而引入两个氟原子的F2相比于F0蓝移了12 nm. 图 3b是四个化合物在薄膜状态下的紫外可见吸收谱图. F1、F1W和F2三个化合物的峰型相近, 起始吸收波长分别是696、685和685 nm, 相比F0依次蓝移了4, 15和15 nm.由薄膜状态下的起始吸收波长位置(λonset), 利用公式可计算出化合物F1, F1W与F2的光学带隙(Eg, opt)分别为1.78, 1.81和1.81 eV.
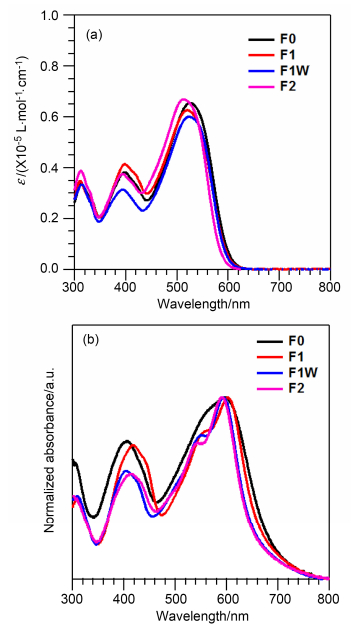
(a) In chlorobenzene solutions with a concentration of 1×10-5 mol/L, and (b) in film state
 下载:
导出CSV
下载:
导出CSV
| Compd. | λmax, solution/nm | λmax, film/nm | λonset, film/nm | Eox, onset/V | Experimental/eV | DFT/eV | |||||
| HOMO | LUMO | Eg, opt | HOMO | LUMO | Eg | ||||||
| F0 | 314, 398, 526 | 406, 596 | 700 | 0.52 | -5.17 | -3.40 | 1.77 | -5.17 | -3.08 | 2.09 | |
| F1 | 310, 398, 524 | 418, 600 | 696 | 0.57 | -5.22 | -3.44 | 1.78 | -5.15 | -3.11 | 2.05 | |
| F1W | 314, 394, 524 | 403, 596 | 685 | 0.63 | -5.28 | -3.47 | 1.81 | -5.30 | -3.18 | 2.12 | |
| F2 | 313, 394, 514 | 415, 592 | 685 | 0.64 | -5.29 | -3.48 | 1.81 | -5.28 | -3.18 | 2.10 | |
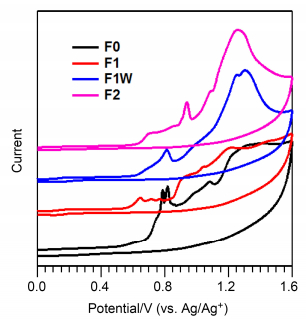
在有机光电材料领域, 循环伏安法被广泛应用于研究材料的电化学性质, 估算能级结构.测试采用三电极体系, 参比电极为Ag/Ag+电极, 工作电极为玻碳电极, 辅助电极为铂丝电极.测试方法是将化合物的氯苯溶液滴在玻碳电极上涂成薄膜, 然后将其浸入溶有0.1 mol/L Bu4NPF6的无水乙腈溶液, 以50 mV•s-1的扫描电压速度进行扫描. 图 4展示了化合物F1、F1W和F2的循环伏安曲线, 由此获得各化合物的起始氧化电位分别为0.57、0.63和0.64 V.在实验中, 我们利用二茂铁进行电极电位的标定, 在相同实验条件下, 测得它的氧化还原电势为0.15 V.根据二茂铁氧化还原电势的真空能级(-4.8 eV)[27], 计算可得Ag/Ag+参比电极的能级为-4.65 eV, 进一步通过公式: HOMO=-e•(Eox+4.65)算出化合物F1、F1W和F2最高占有轨道(HOMO)能级分别为-5.22、-5.28和-5.29 eV.结合光学带隙Eg, opt, 由公式LUMO=HOMO+Eg, opt算得F1、F1W和F2最低空轨道(LUMO)的能级分别为-3.44, -3.47和-3.48 eV.相关数据列于表 1中.
为了进一步了解电化学性质与分子能级结构的关系, 我们采用密度泛函理论(density functional theory, DFT)对四个分子的稳定结构及HOMO、LUMO能级分布进行计算(图 5).采用GAUSSIAN03软件包[28]在B3LYP/6-311+G**//B3LYP/6-31G*水平[29]下计算.为了简化计算过程, 分子中的长烷基链用甲基代替.计算表明, 四个分子都表现出较好的平面性, F原子的取代并没有影响化合物结构的平面型.化合物的HOMO能级电子云基本上分布于整个分子骨架, 而LUMO能级电子云则主要集中在吸电子的BT基团上.计算所得的HOMO和LUMO能级数据列于表 1中.数据表明F取代基本上是降低了化合物的HOMO和LUMO能级.但是, 其取代的位置对此有较大的影响.比如, 相比F0化合物, F1化合物并没有表现出降低的HOMO能级, 但其LUMO有所降低.而同样单F取代的F1W, HOMO和LUMO能级都降低较多.

采用常规的本体异质结有机太阳能电池结构来研究四个化合物的光伏性质, 器件结构为ITO/PEDOT: PSS/活性层/Ca/Al.活性层是以所研究化合物为给体材料, [6, 6]-phenyl-C61-butyric acid methyl ester (PC61BM)为受体材料, 分别对给受体比例、溶液浓度、转速、退火温度、添加剂等条件进行优化后获得.由于这些化合物室温溶解度差, 活性层的制备均在高温下迅速旋涂获得.为了获得最佳的器件效率, 化合物F1和F1W与PC61BM的混合比例为1:1, 而化合物F2与PC61BM的混合比例为1:2. 图 6a展示了四个优化后器件在模拟AM1.5太阳光100 mW•cm-2光强照射下的电流-电压(J-V)曲线, 相关数据列于表 2中.从中可以看出, 化合物F1、F1W与F2所制备的器件, 其开路电压(VOC)分别为0.92, 0.95和0.91 V, 均高于化合物F0器件的0.89 V.氟原子的引入降低了分子的HOMO能级, 提升了器件VOC, 测试结果与向分子内引入氟原子所预期的相一致.然而双氟取代的F2并未获得最高的VOC, 可能是由于活性层膜的形貌不佳, 从而影响了其与电极接触.在这些化合物器件中, F0器件展示出4.53%的最佳PCE, 其中VOC为0.89 V, 短路电流密度(JSC)为9.33 mA•cm-2, 填充因子(FF)为54.5%.通过向分子结构中引入氟原子, 器件性能在不同程度上稍有降低.有趣的是, 对于同分异构体F1和F1W, F1W器件拥有更高的光电转换效率3.6%, 这说明氟原子的取向也是一个非常重要的影响因素对于器件的性能.
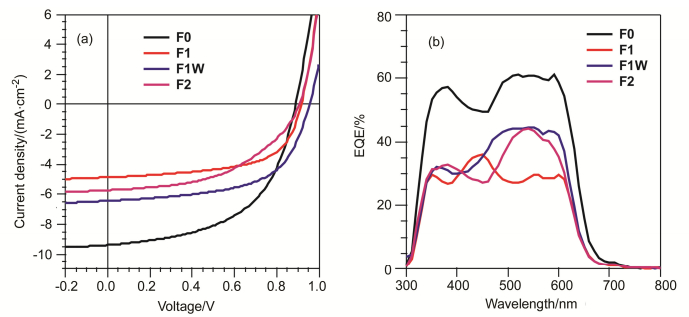 下载:
导出CSV
下载:
导出CSV
| Compound | Voc/V | Jsca/(mA•cm-2) | FF/% | PCEb/% |
| F0 | 0.89 | 9.33 (9.02) | 54.5 | 4.53 (4.45) |
| F1 | 0.92 | 4.85 (4.66) | 60.8 | 2.70 (2.63) |
| F1W | 0.95 | 6.42 (6.13) | 59.3 | 3.63 (3.31) |
| F2 | 0.91 | 5.71 (5.51) | 49.7 | 2.57 (2.36) |
| a括号内为由EQE谱图的计算值; b括号内为平均值. | ||||
图 6b为四个器件的外量子效率谱图(EQE).所有的曲线在350~700 nm范围内都有光响应, 与光吸收光谱相对应.由EQE谱图可以看出氟原子的引入对分子的光响应范围影响不大, 只有略微的变窄, 而光响应强度的降低是导致了器件JSC的下降的主要原因.由EQE计算得到的F1、F1W与F2器件的JSC分别为4.66、6.13与5.51 mA•cm-2, 与J-V曲线测定值相差都在5%以内.
有机太阳能电池的器件性能与活性层的空穴和电子迁移率有着密切联系.活性层中给体材料相传输空穴, 受体材料相传输电子.本工作中, 我们利用空间电荷限制电流法(Space charge-limited current, SCLC)测试三个化合物与PC61BM形成共混膜的空穴迁移率.由此, 我们制备了ITO/PEDOT: PSS/活性层/Au的单空穴传输器件, 所测J-V曲线见图 7所示.通过数据拟合处理, 得到F1、F1W和F2与PC61BM共混膜的空穴迁移率分别为2.0×10-5、4.2×10-5和1.0×10-5 cm2•V-1•s-1, 比F0/PC61BM共混膜的4.7×10-4 cm2•V-1•s-1低了一个数量级.由此可见, 氟原子的引入导致了活性层空穴迁移率的降低, 是氟化后分子的JSC低于F0器件的主要原因.

活性层的膜形貌对器件的性能有着至关重要的影响, 互穿网络结构和适当的相分离有助于提升器件的JSC和FF.在本工作中, 我们利用原子力显微镜(AFM)对膜形貌进行表征(图 8).从AFM图观察可知, 引入氟原子使活性层的膜形貌在一定程度上都变的比较粗糙.特别是引入两个氟原子的化合物F2, 可明显看出有颗粒析出.

Scanning range: 2 μm×2 μm, tapping mode
利用X射线衍射(XRD)对分子在纯物质膜状态及与PC61BM共混膜状态下的有序结构进行表征. 图 9给出了分子在纯物质膜状态及与PC61BM共混膜状态下的XRD谱图.由图可知, 氟化后分子的衍射峰位置相较与F0都有所变动.在3°~10°范围内, F1存在5.7°和6.9°两个强衍射峰, 分别对应1.59和1.28 nm的空间距离; 另外, 在10°~35°范围内也存在一些弱峰. F1W与F1衍射谱图相近, 两个衍射峰分别位于5.7°和6.8°. F2主要的衍射峰则位于5.6°, 6.8°和8.2°, 对应的空间距离分别为1.57, 1.30和1.08 nm.化合物F0在加入PC61BM后衍射峰位置基本没有改变, 说明共混膜的自组装形态受PC61BM影响较小.而F1, F1W和F2与PC61BM共混膜的衍射谱图相较于其纯物质膜衍射有一定的变化, 说明PC61BM的加入对其纯物质膜自组装形态有一定的影响, F1与F1W依然具有相似的衍射峰位置, 都位于6.1°和12.2°处, 具有相似的有序结构. F2除了在6.0°和12.0°处有与F1、F1W相近的衍射峰外, 还在3.85°, 5.8°, 7.2°和17.2°处有一些衍射峰.
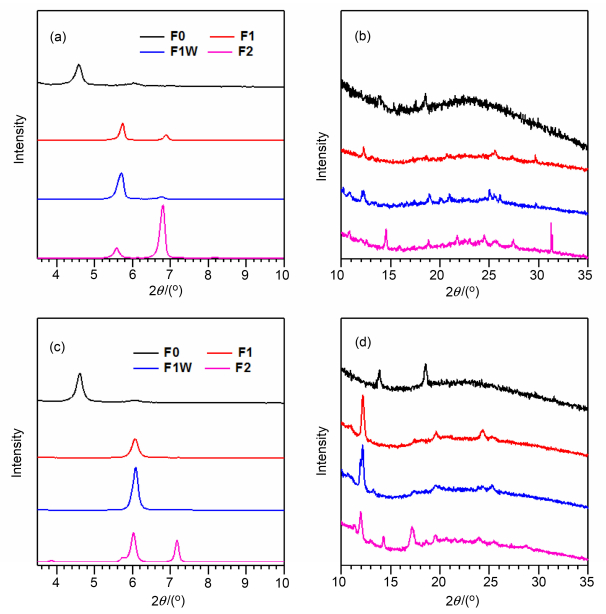
(a) Pure films, scan range is 3.5°~10°; (b) pure films, scan range is 10°~35°; (c) PC61BM-blended films, scan range is 3.5°~10°; (d) PC61BM-blended films, scan range is 10°~35°
三个化合物为P型材料, 制备成OFET器件测试其空穴迁移率.器件结构为Si/SiO2/OTS/active layer/Au, 其中OTS为十八烷基三氯硅烷.将化合物配制成5 mg• mL-1的氯仿溶液, 旋涂在OTS修饰的硅片上, 在真空条件下蒸镀上金电极, 制得OFET器件, 并在120 ℃退火15 min后测试其性能. 图 10给出了F1、F1W与F2的转移曲线与输出曲线, 表 3中列出了具体数据.由这些数据可知, 化合物F0与F1的FET器件空穴迁移率相接近, 约为0.1 cm2•V-1•s-1, 虽然氟原子的引入使器件的开关比略有降低, 但却有效地降低了器件的阈值电压. F1W的迁移率相比于其他化合物有所降低, 仅为0.023 cm2•V-1•s-1.而引入两个氟原子的F2则能获得一个比较好的空穴迁移率, 为0.27 cm2•V-1•s-1, 接近于F0的两倍.由此可见, 氟原子的引入及氟化位点的选择对OFET器件的性能有着不同程度的影响.
 下载:
导出CSV
下载:
导出CSV
| Compound | μOFET/(cm2•V-1•s-1) | VT/V | Ion/Ioff |
| F0 | 0.1 (0.15) | ―24.2 | 1.2×105 |
| F1 | 0.1 (0.14) | ―7.15 | 2.2×104 |
| F1W | 0.018 (0.023) | ―10.3 | 2.5×105 |
| F2 | 0.19 (0.27) | ―17.5 | 5.9×105 |
| a括号内为最大值. | |||
通过在受体单元BT不同位点上引入氟原子, 设计合成了四个具有相同骨架的以BDT为核BT为臂ADA型小分子个光电化合物.研究表明, 氟原子的引入位点及氟原子的数目能使化合物的HOMO能级下降, 溶解性降低, 热稳定性提高, 但化合物的光吸收范围变化不大.氟的引入对于OFET器件有较大的影响, 当氟位于BT外侧位点时, 化合物的空穴迁移率显著降低, 而双氟取代的化合物迁移率得到了明显提高, 达到0.27 cm2•V-1•s-1.对于有机太阳能电池, 氟的引入虽然提高了开路电压, 但由于与PC61BM的共混膜形貌变差, 结构有序性降低, 最终导致短路电流密度和电池效率都有所下降.
合成中所用的商品试剂均购于百灵威、TCI、阿拉丁、绍远、国药、安耐吉等化学试剂公司; 若无特殊说明, 均为直接使用.实验中所使用的溶剂, 如四氢呋喃、甲苯、氯苯等, 均由分析纯溶剂加入二苯甲酮和纳丝后, 在惰性气体氛围下回流制得, 现用现蒸.
实验所得1H NMR, 19F NMR和13C NMR谱在Varain Mercury 300或Bruker Avance 400型核磁共振仪上测定.质谱用HP5973、Saturn2000、Voyager-DE STR、IonSpec 4.7T MALDI傅里叶变换质谱仪或Shimadzu Biotech Axima MALDI基质辅助质谱仪测定.紫外可见光谱(UV-vis)在Hitachi U-3310紫外可见光谱测试仪上测定.循环伏安(CV)数据在上海晨华公司CHI660C电化学工作站上测定.采用三探针法, 玻碳电极作为工作电极, Ag/Ag+电极作为参比电极, 铂丝电极作为辅助电极.将测试化合物的溶液在玻碳电极上滴涂成膜, 浸在含有0.1 mol/L的Bu4NPF6的无水乙腈溶液中, 以50 mV•S-1的速度进行扫描.聚合物的分子量采用分析GPC测定, 采用配有2414 RI或2489紫外检测器的Waters 1515 HPLC系统, 流动相为THF.原子力电子显微镜(AFM)测试仪器为: Veeco Nanoscope Ⅲ测试仪, 轻敲模式.
250 mL的三口瓶中, 依次加入化合物1 (1.24 g, 4.0 mmol), 5-己基-2-噻吩频哪醇硼酸酯(1.18 g, 4.0 mmol), K2CO3水溶液(2 mol/L, 30 mL), Pd(PPh3)4 (230 mg, 0.2 mmol)和新蒸的THF (90 mL).置换气氩气保护后, 利用液氮冷冻-抽气-融化循环三次后, 充氩气保护.将反应体系加热回流反应过夜后降至室温, 加入冷水淬灭反应, 用二氯甲烷萃取三次, 水洗三次.合并有机相, 无水硫酸钠干燥, 过滤, 浓缩.利用柱层析色谱法分离提纯, 正己烷为流动相, 得粗产物后, 利用制备型GPC再次分离提纯, 得淡黄色固体959 mg, 产率为60%. 1H NMR (400 MHz, CDCl3) δ: 7.93 (d, J=3.6 Hz, 1H), 7.58 (d, J=10.4 Hz, 1H), 6.86 (d, J=3.6 Hz, 1H), 2.87 (t, J=7.6 Hz, 2H), 1.77~1.70 (m, 2H), 1.41~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―103.65; LRMS (EI) m/z: 398 (M+). HRMS (EI) m/z: [M+H]+ calcd for C16H16N2FS2Br: 397.9922, found 397.9919.
150 mL反应瓶中, 依次加入化合物2 (875.6 mg, 2.2 mmol), 2-噻吩硼酸(1.41 g, 11.0 mmol), K2CO3水溶液(2 mol/L, 16.5 mL), Pd(PPh3)4 (127 mg, 0.11mmol)和新蒸的THF (50 mL).置换气氩气保护后, 利用液氮冷冻-抽气-融化循环三次后, 充氩气保护.将反应体系加热回流反应过夜后降至室温, 加入冷水淬灭反应, 用氯仿萃取三次, 水洗三次.合并有机相, 无水硫酸钠干燥, 过滤, 浓缩.利用柱层析色谱法分离提纯, 正己烷为流动相, 得固体850 mg, 产率为96%. 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=3.6 Hz, 1H), 7.97 (d, J=4.0 Hz, 1H), 7.70 (d, J=8.8 Hz, 1H), 7.55 (d, J=5.2 Hz, 1H), 7.25~7.23 (m, 1H), 6.89 (d, J=3.6 Hz, 1H), 2.89 (t, J=7.6 Hz, 2H), 1.78~1.72 (m, 2H), 1.42~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3-d1) δ: ―108.51; LRMS (EI) m/z: 402 (M+). HRMS (EI) m/z: [M+H]+ calcd for C20H19N2FS3 402.0694, found 402.0692.
25 mL的三口反应瓶中, 依次加入化合物3 (402 mg, 1.0 mmol), NBS (180.5 mg, 1.02 mmol)和新蒸的THF (40 mL).将体系避光过夜搅拌, 加入水停止反应, 用氯仿萃取三次, 水洗三次.合并有机相, 无水硫酸钠干燥, 过滤, 浓缩.利用柱层析色谱法分离提纯, 正己烷为流动相, 得橙黄色固体400 mg, 产率为83.3%. 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=4.0 Hz, 1H), 7.95 (d, J=4.0 Hz, 1H), 7.64 (d, J=13.2 Hz, 1H), 7.16 (d, J=4.0 Hz, 1H), 6.87 (d, J=4.0 Hz, 1H), 2.88 (t, J=7.6 Hz, 2H), 1.78~1.72 (m, 2H), 1.42~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3-d1) δ: ―108.15; LRMS (EI) m/z: 482 (M+). HRMS (EI) m/z: [M+H]+ calcd for C20H18N2FS3Br 479.9800, found 479.9803.
合成方法同化合物2. 2-噻吩硼酸(382 mg, 2.98 mmol), 4, 7-二溴-5-氟苯并噻二唑(930 mg, 3 mmol), K2CO3水溶液(2 mol/L, 22.5 mL), Pd(PPh3)4 (173 mg, 0.15 mmol)和新蒸的THF (67.5 mL).最终得到600 mg浅黄色固体化合物, 产率64%. 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=3.6 Hz, 1H), 7.72 (d, J=10.0 Hz, 1H), 7.53 (d, J=4.8 Hz, 1H), 7.22 (t, J=4.4 Hz, 1H); 19F NMR (400 MHz, CDCl3) δ: ―103.5; LRMS (EI) m/z: 314 (M+). HRMS (EI) m/z: [M+H]+ calcd for C10H4N2FS2Br 313.8983, found 313.8979.
合成方法同化合物3.化合物5 (205 mg, 0.65 mmol), 5-己基-2-噻吩硼酸频哪醇硼酸酯(729 mg, 2.45 mmol), K2CO3水溶液(2 mol/L, 5.3 mL), Pd(PPh3)4 (80 mg, 0.07 mmol)和新蒸的THF (18 mL).最终得到固体230 mg, 产率为81.7%. 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=3.6 Hz, 1H), 8.08 (d, J=4.0 Hz, 1H), 7.77 (d, J=12.8 Hz, 1H), 7.49 (d, J=5.2 Hz, 1H), 7.23~7.21 (m, 1H), 6.92 (d, J=3.6 Hz, 1H), 2.91 (t, J=7.6 Hz, 2H), 1.80~1.74 (m, 2H), 1.43~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―109.4; LRMS (EI) m/z: 402 (M+). HRMS (EI) m/z: [M+H]+ calcd for C20H19N2FS3: 402.0694, found 402.0692.
合成方法同化合物4.化合物6 (120.6 mg, 0.3 mmol), NBS (54.2 mg, 0.306 mmol)和THF (10 mL).最终得到固体110 mg, 产率为76.4%. 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=4.0 Hz, 1H), 7.78 (d, J=4.0 Hz, 1H), 7.68 (d, J=13.2 Hz, 1H), 7.16 (d, J=4.0 Hz, 1H), 6.91 (d, J=4.0 Hz, 1H), 2.91 (t, J=7.6 Hz, 2H), 1.79~1.73 (m, 2H), 1.42~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―109.3; LRMS (MALDI) m/z: 482 (M+).
化合物9的合成参考文献[30]的方法合成. 1H NMR (400 MHz, CDCl3, 110 ℃) δ: 8.05 (d, J=3.6 Hz, 1H), 6.91 (d, J=3.6 Hz, 1H), 2.89 (d, J=7.6 Hz, 2H), 1.79~1.73 (m, 2H), 1.42~1.25 (br, 6H), 0.90 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―120. 4, ―127. 8.
合成方法同化合物3.化合物9 (832 g, 2.0 mmol), 2-噻吩硼酸(1.28 g, 10.0 mmol), K2CO3水溶液(2 mol/L, 15 mL), Pd(PPh3)4 (115.4 mg, 0.1 mmol)和新蒸的THF (45 mL).最终得到橙黄色固体750 mg, 产率为89.3%. 1H NMR (400 MHz, CDCl3) δ: 8.26 (d, J=3.6 Hz, 1H), 8.09 (d, J=4.0 Hz, 1H), 7.59 (d, J=4.8 Hz, 1H), 7.26~7.24 (m, 1H), 6.92 (d, J=3.6 Hz, 1H), 2.91 (d, J=7.2 Hz, 2H), 1.80~1.74 (m, 2H), 1.42~1.25 (br, 6H), 0.91 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―128.0, ―129.1; LRMS (EI) m/z: 420 (M+).
合成方法同化合物4.化合物10 (420.0 mg, 1.0 mmol), NBS (180.5 mg, 1.02 mmol)和新蒸的THF (25 mL).最终得到固体370 mg, 产率为74.3%. 1H NMR (400 MHz, CDCl3) δ: 8.07 (d, J=3.6 Hz, 1H), 7.96 (d, J=3.6 Hz, 1H), 7.17 (d, J=3.6 Hz, 1H), 6.90 (d, J=3.6 Hz, 1H), 2.89 (t, J=7.6 Hz, 2H), 1.77~1.71 (m, 2H), 1.42~1.25 (br, 6H), 0.89 (t, J=6.8 Hz, 3H); 19F NMR (400 MHz, CDCl3) δ: ―127.9, ―129.2; LRMS (EI) m/z: 500 (M+)
50 mL的三口瓶中, 依次加入单体4 (300 mg, 0.625 mmol), BDTSn2 (193.5 mg, 0.25 mmol), Pd(PPh3)4 (28.8 mg, 0.025 mmol)和新蒸的甲苯(25 mL).置换气氩气保护后, 利用液氮冷冻-抽气-融化循环三次后, 充氩气保护.将反应体系加热回流反应24 h后降至室温, 加入冷水淬灭反应, 用氯仿萃取三次, 水洗三次.合并有机相, 无水硫酸钠干燥, 过滤, 浓缩.利用柱层析色谱法分离提纯, 进一步利用凝胶柱提纯, 重结晶得到紫色固体210 mg, 产率为63.2%. m.p. 230 ℃; 1H NMR (400 MHz, C2D2Cl4, 110 ℃) δ: 8.21 (s, 2H), 7.98 (s, 2H), 7.67 (d, J=4.8 Hz, 2H), 7.62 (s, 2H), 7.42 (s, 2H), 6.88 (s, 2H), 4.26 (s, 4H), 2.89 (t, J=4.8 Hz, 4H), 1.92~1.86 (m, 2H), 1.80~1.20 (br, 30H), 1.09 (br, 6H), 1.00 (br, 6H), 0.91 (br, 6H). LRMS (MALDI) m/z: 1247 (M+). HRMS (MALDI) m/z: [M+H]+ calcd for C66H74N4O2F2S8 1246.3384, found 1246.3349.
合成方法同化合物F1.化合物7 (300 mg, 0.625 mmol), BDTSn2 (194 mg, 0.25 mmol), Pd(PPh3)4 (42 mg, 0.075 mmol)和新蒸的甲苯(12.5 mL).最终得到固体245 mg, 产率为78.6%. m.p. 250 ℃. 1H NMR (400 MHz, C2D2Cl4, 110 ℃) δ: 8.08 (d, J=2.4 Hz, 2H), 8.06 (d, J=2.4 Hz, 2H), 7.73 (d, J=8.8 Hz, 2H), 7.60 (s, 2H), 7.38 (s, 2H), 6.90 (s, 2H), 4.26 (d, J=3.6 Hz, 4H), 2.90 (t, J=4.8 Hz, 4H), 1.92~1.86 (m, 2H), 1.80~1.20 (br, 30H), 1.09 (t, J=8.8 Hz, 6H), 1.01 (t, J=8.8 Hz, 6H), 0.91 (t, J=8.8 Hz, 6H); LRMS (MALDI) m/z: 1247 (M+). HRMS (EI) m/z: [M+H]+ calcd for C66H72O2N4F2S8 1246.3384, found 1246. 3426.
合成方法同化合物F1.化合物11 (323 mg, 0.65 mmol), BDTSn2 (201.2 mg, 0.26 mmol), Pd(PPh3)4 (30 mg, 0.026 mmol)和新蒸的甲苯(26 mL).最终得到固体150 mg, 产率为45%. 1H NMR (400 MHz, C2D2Cl4, 110 ℃) δ: 8.23 (d, J=2.0 Hz, 2H), 8.11 (s, J=2.0 Hz, 2H), 7.61 (s, 2H), 7.42 (s, 2H), 6.91 (s, 2H), 4.27 (s, 4H), 2.91 (t, J=4.8 Hz, 4H), 1.92~1.88 (m, 2H), 1.80~1.20 (br, 30H), 1.09 (br, J=4.8 Hz, 6H), 1.00 (br, J=4.8 Hz, 6H), 0.91 (br, J=4.8 Hz, 6H). LRMS (MALDI) m/z: 1283 (M+).
有机太阳能电池器件结构为ITO/PEDOT:PSS (Heraeus Clevios P VP. Al 4083)/active layer/Ca (LiF)/Al.具体制备步骤为:在ITO玻璃上, 以6000 r/min的转速旋涂上PEDOT:PSS, 140 ℃加热15 min.迅速转移至手套箱内, 旋涂上配置好的溶液, 并按需求进行退火.最后, 利用真空蒸镀仓蒸镀上Ca (10 nm)或LiF (1 nm), 电极层Al (100 nm).活性层的面积为7 mm2, 膜厚通过Veeco Dektak 150 profilometer进行测试. J-V曲线采用Keithley 2420测试仪与AAA光源模拟器(Oriel 94043A, 450 W)进行测试; 外量子效率由Oriel单色仪(74125)测得.
OFET采用底栅极顶接触(bottom-gate top-contact, BGTC)的器件结构.掺杂的硅片作为底层栅极, 硅片表面覆盖SiO2 (300 nm, 电容为11 nF•cm-2)并用OTS修饰, 作为绝缘层.测试样品通过其溶液旋涂于基底, 并根据需要进行退火处理.金电极(50 nm)可通过掩膜板真空蒸镀沉积制得.器件效率是在常温空气环境中, 通过Keithley 4200半导体测试仪测试而得.沟道的长宽比(L/W)为8.95, 场效应迁移率可由公式IDS=(μWCi/2L)(VG―VT)2计算而得.其中, IDS是漏极电流, μ是载流子迁移率, W是沟道宽度, Ci绝缘体电容, L是沟道长度, VG栅极电压, VT是阈值电压.
辅助材料(Supporting Information) 化合物的1H NMR和19F NMR、低分辨和高分辨质谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Brabec, C. J.; Gowrisanker, S.; Halls, J. J. M.; Laird, D.; Jia, S.; Williams, S. P. Adv. Mater. 2010, 22, 3839.
(b) Facchetti, A. Chem. Mater. 2011, 23, 733.
(c) Ilmi, R.; Haque, A.; Khan, M. S. Org. Electr. 2018, 58, 53.
(d) Guo, H.; Li, W.; Chang, C.; Guo, X.; Zhang, M. Chin. J. Chem. 2018, 36, 502.
(e) Liang, X.; Wang, Z.; Wang, L.; Hanif, M.; Hu, D.; Su, S.; Xie, Z.; Gao, Y.; Yang, B.; Ma, Y. Chin. J. Chem. 2017, 35, 1559.
(a) Zhao, X.; Zhan, X. Chem. Soc. Rev. 2011, 40, 3728.
(b) Di, C.; Yu, G.; Liu, Y.; Zhu, D. J. Phys. Chem. B 2007, 111, 14083.
(c) Zaumseil, J.; Sirringhaus, H. Chem. Rev. 2007, 107, 1296.
(a) Reineke, S.; Lindner, F.; Schwartz, G.; Seidler, N.; Walzer, K.; Lüssem, B.; Leo, K. Nature 2009, 459, 234.
(b) Yang, X.; Zhou, G.; Wong, W. Y. Chem. Soc. Rev. 2015, 44, 8484.
(c) Jou, J. H.; Kumar, S.; Agrawal, A.; Li, T. H.; Sahoo, S. J. Mater. Chem. C 2015, 3, 2974.
陈红海, 陈玉哲, 李仲谨, 杨清正, 辛利, 有机化学, 2012, 32, 46. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340957.shtmlChem, H.; Chen, Y.; Li, Z.; Yang, Q.; Xin, L. Chin. J. Org. Chem. 2012, 32, 46(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract340957.shtml
(a) Zhang, G.; Zhang, K.; Yin, Q.; Jiang, X. F.; Wang, Z.; Xin, J.; Ma, W.; Yan, H.; Huang, F.; Cao, Y. J. Am. Chem. Soc. 2017, 139, 2387.
(b) Li, M.; Gao, K.; Wan, X.; Zhang, Q.; Kan, B.; Xia, R.; Liu, F.; Yang, X.; Feng, H.; Ni, W.; Wang, Y.; Peng, J.; Zhang, H.; Liang, Z.; Yip, H.-L.; Peng, X.; Cao, Y.; Chen, Y. Nat. Photonics 2017, 11, 85.
(c) Zhao, J.; Li, Y.; Yang, G.; Jiang, K.; Lin, H.; Ade, H.; Ma, W.; Yan, H. Nat. Energy 2016, 1, 15027.
(d) Deng, D.; Zhang, Y. J.; Zhang, J. Q.; Wang, Z. Y.; Zhu, L. Y.; Fang, J.; Xia, B. Z.; Wang, Z.; Lu, K.; Ma, W.; Wei, Z. X. Nat. Commun. 2016, 7, 13740.
(e) Li, Y. F. Acc. Chem. Res. 2012, 45, 723.
(f) Yang, F.; Li, C.; Lai, W.; Zhang, A.; Huang, H.; Li, W. Mater. Chem. Front. 2017, 1, 1389.
(g) Liu, W.; Zhou, Z.; Vergote, T.; Xu, S.; Zhu, X. Mater. Chem. Front. 2017, 1, 2349.
(a) Zhang, J.; Zhang, Y.; Fang, J.; Lu, K.; Wang, Z.; Ma, W.; Wei, Z. J. Am. Chem. Soc. 2015, 137, 8176.
(b) Li, G.; Chu, C. W.; Shrotriya, V.; Huang, J.; Yang, Y. Appl. Phys. Lett. 2006, 88, 253503.
(c) Kim, J. Y.; Lee, K.; Coates, N. E.; Moses, D.; Nguyen, T. Q.; Dante, M.; Heeger, A. J. Science 2007, 317, 222.
(d) He, Z.; Zhong, C.; Huang, X.; Wong, W.-Y.; Wu, H.; Chen, L.; Su, S.; Cao, Y. Adv. Mater. 2011, 23, 4636.
(a) Li, G.; Shrotriya, V.; Huang, J. S.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. Nat. Mater. 2005, 4, 864.
(b) Peet, J.; Kim, J. Y.; Coates, N. E.; Ma, W. L.; Moses, D.; Heeger, A. J.; Bazan, G. C. Nat. Mater. 2007, 6, 497.
(c) Zimmerman, J. D.; Xiao, X.; Renshaw, C. K.; Wang, S.; Diev, V. V.; Thompson, M. E.; Forrest, S. R. Nano Lett. 2012, 12, 4366.
(d) Wang, J. L.; Xiao, F.; Yan, J.; Wu, Z.; Liu, K. K.; Chang, Z. F.; Zhang, R. B.; Chen, H.; Wu, H. B.; Cao, Y. Adv. Funct. Mater. 2016, 26, 1803.
(e) Liu, X.; Ye, L.; Zhao, W.; Zhang, S.; Li, S.; Su, G. M.; Wang, C.; Ade, H.; Hou, J. Mater. Chem. Front. 2017, 1, 2057.
(a) Neugebauer, H.; Brabec, C.; Hummelen, J. C.; Sariciftci, N. S. Sol. Energy Mater. Sol. Cells 2000, 61, 35.
(b) Lu, L.; Kelly, M. A.; You, W.; Yu, L. Nat. Photonics 2015, 9, 491.
(c) Min, J.; Luponosov, Y. N.; Gasparini, N.; Richter, M.; Bakirov, A. V.; Shcherbina, M. A.; Chvalun, S. N.; Grodd, L.; Grigorian, S.; Ameri, T.; Ponomarenko, S. A.; Brabec, C. J. Adv. Energy Mater. 2015, 5, 1500386.
Meng, L.; Zhang, Y.; Wan, X.; Li, C.; Zhang, X.; Wang, Y.; Ke, X.; Xiao, Z.; Ding, L.; Xia, R.; Yip, H.-L.; Cao, Y.; Chen, Y. Science 2018, 361, 1094. doi: 10.1126/science.aat2612
(a) Lei, Y.; Deng, P.; Zhang, Q.; Xiong, Z.; Li, Q.; Mai, J.; Lu, X.; Zhu, X.; Ong, B. S. Adv. Funct. Mater. 2018, 28, 1706372.
(b) Kang, I.; Yun, H. J.; Chung, D. S.; Kwon, S. K.; Kim, Y. H. J. Am. Chem. Soc. 2013, 135, 14896.
(a) Xiao, S.; Zhang, Q.; You, W. Adv. Mater. 2017, 29, 1601391.
(b) Zhang, Z.; Li, Y. Sci. China: Chem. 2015, 58, 192.
(c) Chen, H.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G. W.; Yang, Y.; Yu, L.; Wu, Y.; Li, G. Nat. Photonics 2009, 3, 649.
(a) Tang, M. L.; Bao, Z. A. Chem. Mater. 2011, 23, 446.
(b) Oh, J.; Kranthiraja, K.; Lee, C.; Gunasekar, K.; Kim, S.; Ma, B.; Kim, B. J.; Jin, S.-H. Adv. Mater. 2016, 28, 10016.
(c) Stuart, A. C.; Tumbleston, J. R.; Zhou, H.; Li, W.; Liu, S.; Ade, H.; You, W. J. Am. Chem. Soc. 2013, 135, 1806.
(a) Zhang, M.; Guo, X.; Zhang, S.; Hou, J. Adv. Mater. 2014, 26, 1118.
(b) Kawashima, K.; Fukuhara, T.; Suda, Y.; Suzuki, Y.; Koganezawa, T.; Yoshida, H.; Ohkita, H.; Osaka, I.; Takimiya, K. J. Am. Chem. Soc. 2016, 138, 10265.
Reichenbacher, K.; Suss, H. I.; Hulliger, J. Chem. Soc. Rev. 2005, 34, 22. doi: 10.1039/B406892K
Lu, L.; Yu, L. Adv. Mater. 2014, 26, 4413. doi: 10.1002/adma.v26.26
(a) Yoon, M.-H.; DiBenedetto, S. A.; Facchetti, A.; Marks, T. J. J. Am. Chem. Soc. 2005, 127, 1348.
(b) Heidenhain, S. B.; Sakamoto, Y.; Suzuki, T.; Miura, A.; Fujikawa, H.; Mori, T.; Tokito, S.; Taga, Y. J. Am. Chem. Soc. 2000, 122, 10240.
Liang, Y.; Feng, D.; Wu, Y.; Tsai, S.-T.; Li, G.; Ray, C.; Yu, L. J. Am. Chem. Soc. 2009, 131, 7792. doi: 10.1021/ja901545q
Son, H. J.; Wang, W.; Xu, T.; Liang, Y.; Wu, Y.; Li, G.; Yu, L. J. Am. Chem. Soc. 2011, 133, 1885. doi: 10.1021/ja108601g
(a) Zhang, Y.; Chien, S.-C.; Chen, K.-S.; Yip, H.-L.; Sun, Y.; Davies, J. A.; Chen, F.-C.; Jen, A. K.-Y. Chem. Commun. 2011, 47, 11026.
(b) Schroeder, B. C.; Huang, Z.; Ashraf, R. S.; Smith, J.; D¢ Angelo, P.; Watkins, S. E.; Anthopoulos, T. D.; Durrant, J. R.; McCulloch, I. Adv. Funct. Mater. 2012, 22, 1663.
(a) Zhou, H.; Yang, L.; Stuart, A. C.; Price, S. C.; Liu, S.; You, W. Angew. Chem., Int. Ed. 2011, 50, 2995.
(b) Peng, Q.; Liu, X.; Su, D.; Fu, G.; Xu, J.; Dai, L. Adv. Mater. 2011, 23, 4554.
Hu, J.; Wang, X.; Chen, F.; Xiao, B.; Tang, A.; Zhou, E. Polymers 2017, 9, 516. doi: 10.3390/polym9100516
(a) Roncali, J. Acc. Chem. Res. 2009, 42, 1719.
(b) Li, Y.; Guo, Q.; Li, Z.; Pei, J.; Tian, W. Energy Environ. Sci. 2010, 3, 1427.
(c) Walker, B.; Kim, C.; Nguyen, T.-Q. Chem. Mater. 2011, 23, 470.
(d) Mishra, A.; Bauerle, P. Angew. Chem., Int. Ed. 2012, 51, 2020.
Liang, L.; Wang, J.-T.; Xiang, X.; Ling, J.; Zhao, F.-G.; Li, W.-S. J. Mater. Chem. A 2014, 2, 15396. doi: 10.1039/C4TA03125C
van der Poll, T. S.; Love, J. A.; Nguyen, T.-Q.; Bazan, G. C. Adv. Mater. 2012, 24, 3646. doi: 10.1002/adma.v24.27
He, C.-Y.; Wu, C.-Z.; Zhu, Y.-L.; Zhang, X. Chem. Sci. 2014, 5, 1317. doi: 10.1039/C3SC53119H
Mei, C.-Y.; Liang, L.; Zhao, F.-G.; Wang, J.-T.; Yu, L.-F.; Li, Y.-X.; Li, W.-S. Macromolecules 2013, 46, 7920. doi: 10.1021/ma401298g
(a) Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864.
(b) Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133.
(c) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. 1988, B37, 785.
(d) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(a) Hohenberg. P.; Kohn, W.; Phys. Rev. 1964, 136, B864(b) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A. J.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Gaussian, Inc., Wallingford CT, 2004.
Wang, J.-L.; Chang, Z.-F.; Song, X.-X.; Liu, K.-K.; Jing, L.-M. J. Mater. Chem. 2015, 3, 9849.

图 1 化合物F0, F1, F1W和F2的分子结构
Figure 1 Chemical structures of compounds F0, F1, F1W and F2

图 2 化合物F0, F1, F1W和F2的(a) TGA曲线和(b)第二循环DSC曲线
Figure 2 (a) TGA profiles and (b) the second circle of DSC traces of compounds F0, F1, F1W and F2
Heating and cooling rate: 10 ℃•min-1, under N2 atmosphere

图 3 化合物F0, F1, F1W和F2的紫外可见吸收光谱
Figure 3 UV-Vis absorption spectra of compounds F0, F1, F1W and F2
(a) In chlorobenzene solutions with a concentration of 1×10-5 mol/L, and (b) in film state

图 4 化合物F0, F1, F1W和F2的循环伏安曲线.
Figure 4 Cyclic voltammograms of compounds F0, F1, F1W and F2

图 5 DFT计算的化合物F0, F1, F1W和F2稳定构象及HOMO与LUMO能级电子云分布图.
Figure 5 DFT computed molecular geometries and electron wave functions of HOMO and LUMO orbits for compounds F0, F1, F1W and F2

图 6 在AM 1.5G模拟太阳光100 mW•cm-2照明下化合物F0, F1, F1W和F2的(a) J-V曲线和(b) EQE谱图
Figure 6 (a) J-V curves and (b) EQE spectra of OSC devices based on compounds F0, F1, F1W and F2

图 7 化合物F1, F1W和F2与PC61BM共混膜的ln J与ln V曲线及SCLC法拟合曲线(膜厚度分别为75, 80和50 nm)
Figure 7 ln J vs. ln V and fitted SCLC curves of PC61BM- lended films of compounds F1, F1W and F2 with thickness of 75, 80 and 50 nm, respectively

图 8 化合物F0, F1, F1W和F2与PC61BM共混膜的AFM谱图
Figure 8 AFM pictures of PC61BM-blended films of compounds F0, F1, F1W and F2
Scanning range: 2 μm×2 μm, tapping mode

图 9 化合物F0, F1, F1W和F2的XRD谱图
Figure 9 XRD profiles of compounds F0, F1, F1W and F2
(a) Pure films, scan range is 3.5°~10°; (b) pure films, scan range is 10°~35°; (c) PC61BM-blended films, scan range is 3.5°~10°; (d) PC61BM-blended films, scan range is 10°~35°

图 10 基于F1、F1W和F2的OFET器件的转移曲线(左)和输出曲线(右)
Figure 10 Transfer (left) and output curves (right) of OFET devices based on compounds F1, F1W and F2
表 1 化合物F0, F1, F1W和F2的光谱吸收性质、起始氧化电位和材料能级
Table 1. Light-absorption properties, onset oxidation potential, and molecular energy levels of compounds F0, F1, F1W and F2
| Compd. | λmax, solution/nm | λmax, film/nm | λonset, film/nm | Eox, onset/V | Experimental/eV | DFT/eV | |||||
| HOMO | LUMO | Eg, opt | HOMO | LUMO | Eg | ||||||
| F0 | 314, 398, 526 | 406, 596 | 700 | 0.52 | -5.17 | -3.40 | 1.77 | -5.17 | -3.08 | 2.09 | |
| F1 | 310, 398, 524 | 418, 600 | 696 | 0.57 | -5.22 | -3.44 | 1.78 | -5.15 | -3.11 | 2.05 | |
| F1W | 314, 394, 524 | 403, 596 | 685 | 0.63 | -5.28 | -3.47 | 1.81 | -5.30 | -3.18 | 2.12 | |
| F2 | 313, 394, 514 | 415, 592 | 685 | 0.64 | -5.29 | -3.48 | 1.81 | -5.28 | -3.18 | 2.10 | |
 下载: 导出CSV
下载: 导出CSV
表 2 化合物F0, F1, F1W和F2太阳能电池器件参数
Table 2. Device parameters of OSCs based on compounds F0, F1, F1W and F2
| Compound | Voc/V | Jsca/(mA•cm-2) | FF/% | PCEb/% |
| F0 | 0.89 | 9.33 (9.02) | 54.5 | 4.53 (4.45) |
| F1 | 0.92 | 4.85 (4.66) | 60.8 | 2.70 (2.63) |
| F1W | 0.95 | 6.42 (6.13) | 59.3 | 3.63 (3.31) |
| F2 | 0.91 | 5.71 (5.51) | 49.7 | 2.57 (2.36) |
| a括号内为由EQE谱图的计算值; b括号内为平均值. | ||||
下载: 导出CSV
表 3 化合物F0, F1, F1W和F2的空穴迁移率参数
Table 3. Hole mobility parameters of compounds F0, F1, F1W and F2
| Compound | μOFET/(cm2•V-1•s-1) | VT/V | Ion/Ioff |
| F0 | 0.1 (0.15) | ―24.2 | 1.2×105 |
| F1 | 0.1 (0.14) | ―7.15 | 2.2×104 |
| F1W | 0.018 (0.023) | ―10.3 | 2.5×105 |
| F2 | 0.19 (0.27) | ―17.5 | 5.9×105 |
| a括号内为最大值. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

