引用本文:
廖旭, 蒋岩, 赖石林, 刘源岗, 王士斌, 熊兴泉. 化学-酶多步串联反应及其在高附加值手性化合物高效、绿色合成中的应用[J]. 有机化学,
2019, 39(3): 668-678.
doi:
10.6023/cjoc201807038

Citation: Liao Xu, Jiang Yan, Lai Shilin, Liu Yuangang, Wang Shibin, Xiong Xingquan. Chemoenzymatic Relay Reaction and Its Applications in Highly Efficient and Green Synthesis of High-Value Chiral Compounds[J]. Chinese Journal of Organic Chemistry, 2019, 39(3): 668-678. doi: 10.6023/cjoc201807038
Citation: Liao Xu, Jiang Yan, Lai Shilin, Liu Yuangang, Wang Shibin, Xiong Xingquan. Chemoenzymatic Relay Reaction and Its Applications in Highly Efficient and Green Synthesis of High-Value Chiral Compounds[J]. Chinese Journal of Organic Chemistry, 2019, 39(3): 668-678. doi: 10.6023/cjoc201807038
化学-酶多步串联反应及其在高附加值手性化合物高效、绿色合成中的应用
摘要:
与化学催化或酶催化合成相比,化学-酶相结合的多步串联反应是一种更简单、高效、经济的方法,兼具酶催化剂的高效、高选择性等优点,且合成原料价廉易得,合成工艺简捷高效,生产环境绿色友好,所得产品光学纯度高,使其在高附加值手性化合物的开发和合成方面得到了广泛的应用.近年来,化学家们通过改变催化剂和设计更为合理的反应方式,致力于将化学-酶催化反应条件变得更为容易,从而将其应用于更多反应领域.综述了近年来国内外化学-酶多步串联反应,如酶与金属催化、酶与有机催化、酶与新型反应技术等相结合多步串联合成手性醇类、环氧类、杂环类以及其他手性化合物的研究进展,并对该领域的发展趋势进行了展望.
English
Chemoenzymatic Relay Reaction and Its Applications in Highly Efficient and Green Synthesis of High-Value Chiral Compounds
Abstract:
Compared with traditional chemical catalysis or enzymatic synthesis, chemoenzymatic relay reaction is a simpler, more efficient and economical method. It not only has the advantages of high efficiency and selectivity of enzyme catalysis, but also has the advantages of low price of synthetic raw materials, simple and high efficiency of synthetic process, green and friendly production environment, and excellent optical purity of the obtained product. Thus, chemoenzymatic relay synthesis methods have been widely used in the synthesis of high-value chiral compounds. In recent years, chemists have been committed to making chemoenzymatic catalytic conditions easier by changing catalysts and designing more reasonable ways, which could be used in more fields. In this review, the recent progress in the synthesis of chiral alcohols, epoxides, heterocyclics and other chiral compounds by using chemoenzymatic relay synthesis, such as enzyme and metal catalysis, enzyme and organic catalysis, enzyme and new reaction techniques, is reviewed, and the development trends of this field are also prospected.
-
近年来, 生物酶催化在精细化学品和药物合成中的应用有了长足的发展, 与传统化学催化相比, 生物酶催化具有反应条件温和、区域选择性及立体选择性好以及所得产品光学纯度高等优点, 已经成为获得手性药物分子的有效途径之一[1, 2].一般而言, 前手性化合物可利用生物酶不对称催化转化为手性化合物.化学合成与生物催化相结合的化学-酶多步串联反应, 即反应过程中不经过任何分离提纯, 对一般合成步骤则采用化学合成法, 在涉及手性中心的生成或转化步骤中采用生物酶催化法, 不仅能克服化学合成反应步骤繁琐及生产成本高等不足, 又可有效避免生物转化量较少、反应单一等缺点, 实现优势互补, 从而获得光学纯度高的手性化合物[3, 4].
20世纪80年代, van Bekkum课题组[5]报道了化学催化和生物催化结合的首个例子, 通过非均相金属催化氢化反应和酶催化异构化反应相结合, 以价廉易得的天然化合物D-葡萄糖为原料制备出相应的D-甘露糖醇.随后, Williams课题组[6, 7]成功突破经典外消旋体动力学拆分最高收率为50%的限制, 提出了酶与金属催化剂相结合的概念并加以广泛应用.该研究一个典型的例子是, 将脂肪酶催化的仲醇酰化反应与Pd或Rh催化的外消旋化反应结合, 通过可逆转移氢化实现动态动力学拆分(DKR).后来, Turner团队[8]将单胺氧化酶催化的亚胺形成与化学还原相结合, 通过去除异氰化过程从而使合成产率几乎接近100%的理论产率.
利用化学催化-酶法相结合多步串联反应制备手性化合物主要的瓶颈, 是化学反应和酶催化体系相容性问题难以解决.酶催化的首选溶剂是水, 而大多数化学反应无法在水中有很好的反应效果.然而, 随着酶工程设计的发展以及与各种化学催化剂的改进, 许多限制已一一被突破.近年, 化学-酶催化相结合多步串联成功的例子显著增加[9~11].此外, 还可通过使用两相或封装技术分隔催化剂, 依次加入催化剂和反应底物, 或使用相容性催化剂均可有效克服上述困难.另外, 在合成高附加值手性化合物方面, 酶除了与金属催化结合, 也可与有机分子催化相结合, 或与离子液体、微波催化或连续流等其他新技术的结合进行多步串联反应, 也取得明显进展并已经被广泛应用.
1. 酶催化与金属催化剂相结合
从实验室合成和工业生产的角度来看, 金属催化剂和生物催化剂多步串联组合对于开发高光学纯度的手性化合物是高效且很有应用价值的, 特别是过渡金属, 其在金属与酶催化的结合中一直占主导地位, 过渡金属与酶相结合的催化反应是研究最深入的化学-酶串联反应之一[12, 13].利用过渡金属催化剂的广泛反应性和酶的高选择性的优势来进行一系列的反应, 具有反应适应性强、选择性高和合成效率好等显著优势.
1.1 酶与金属钯基催化剂相结合
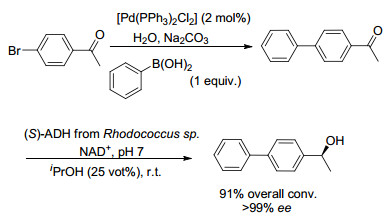
钯催化的交叉偶联反应在金属催化领域具有明显的优势, 酶促还原反应是生物催化制备手性化合物的主要手段.因此, 将酶与金属钯催化剂相结合进行化学-酶多步串联反应有着极其重要的意义. 2008年, Groger课题组[14]报道了在含水反应介质中, 钯催化的交叉偶联反应与生物催化结合的第一个实例, 通过Suzuki交叉偶联和随后的不对称酶促还原合成了手性二芳基醇(Scheme 1), 具有高达91%的转化率和出色的立体选择性(>99% ee).研究表明, 与酶催化相容的Suzuki交叉偶联反应的先决条件包括几点: (1)不可使用磷酸添加剂; (2)硼酸不可过量使用; (3)必须使用水作为反应介质.
图式 1
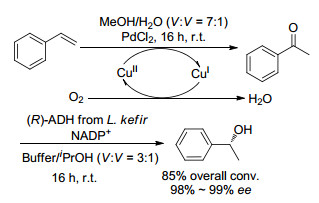
然而, 对于化学-酶催化相结合多步反应, 由于催化体系的不相容, 导致生物酶催化剂易失去活性. 2014年, Groger课题组[15]通过将钯和铜共同催化的Wacker氧化和酶促还原进行区隔化实现了烯烃不对称水合, 反应在聚二甲基硅氧烷(PDMS)套管内部进行Wacker氧化, Wacker氧化所得到的苯乙酮产物则通过PDMS膜扩散到外部发生生物转化.由于PDMS膜的隔离作用, 使钯和铜与酶彼此不接触, 所以它们在各自的隔离区域里面均展示出高的催化活性, 制备出一系列高转化率和ee可达99%的1-芳基乙醇(Schemes 2, 3).此外, Wacker氧化的催化剂循环15次后, 转化率无显著下降.
图式 2

图式 3

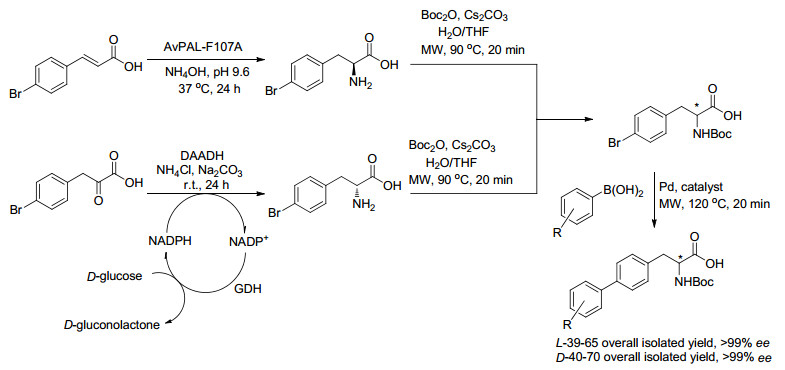
2015年, Turner课题组[16]报道了运用化学-酶多步串合成了一系列N-保护的非天然L-和D-二芳基丙氨酸衍生物.苯丙氨酸解氨酶(PAL)和D-氨基酸脱氢酶(DAADH)分别催化4-溴肉桂酸和4-溴苯基丙酮酸, 得到4-溴苯丙氨酸的两种对映体, 随后在温和的含水条件和芳基硼酸衍生物进行钯催化的Suzuki-Miyaura偶联反应, 以良好的收率和高的光学纯度得到L-和D-二芳基丙氨酸衍生物(Scheme 4).
图式 4

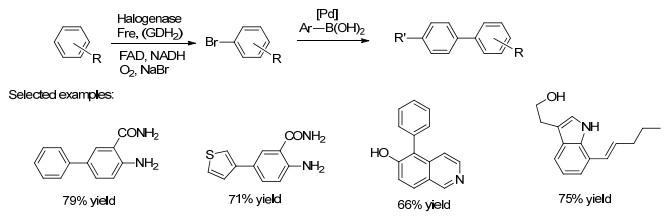
近年来, C—H位置的选择性活化已经改变了有机合成发展. 2016年Latham课题组[17]通过卤化酶与钯催化的交叉偶联相结合多步串联实现了吲哚的5-, 6-和7-位C—H区域控制芳基化, 在温和的含水条件下, 得到了一系列功能多样的芳基化产物(Scheme 5), 从而解决了两种相似的未活化C—H位置间的选择性活化的难题.此外, 还应用膜区室化克服了酶和过渡金属相容性的问题, 使得高效的生物催化剂可循环使用多次, 且优化的过程不需要任何中间处理或纯化步骤.
图式 5

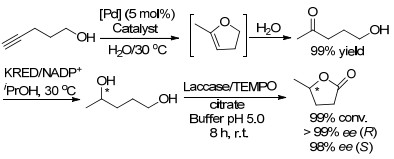
2017年, González-Sabín课题组[18]报道了钯催化的环异构化, 反应使用亚氨基正膦烷-Pd配合物作为催化剂, 将戊炔-1-醇转化为5-羟基-2-戊酮.研究表明, 金属Pd催化下的水合过程与酮还原酶(KRED)催化的生物还原步骤串联组合反应24 h后, 能够以良好的收率(高达90%)得到1, 4-戊二醇两种可能的对映异构体.最后, 通过将上述合成的手性1, 4-戊二醇与漆酶/2, 2, 6, 6-四甲基哌啶氮氧化合物(TEMPO)催化下进行氧化反应, 反应高产率地得到两种不同立体构型的γ-戊内酯(Scheme 6).
图式 6

1.2 酶催化与钌基催化剂相结合
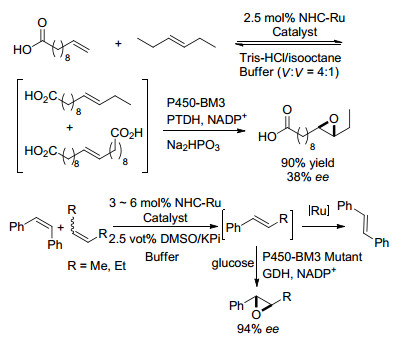
2015年, Hartwig课题组[19]报道了通过酶P450-BM3和烯烃复分解催化剂相结合将长链烯烃的混合物转化为单一环氧化物(Scheme 7).此后又报道了采用烯烃易位-酶促环氧化反应相结合的方式将烯烃类混合物选择性地转化为单一环氧化物, 且ee值大大提高, 证明了烯烃复分解和酶促环氧化可用于串联反应, 并且可得到具有高对映选择性的芳基环氧化物[20].
图式 7

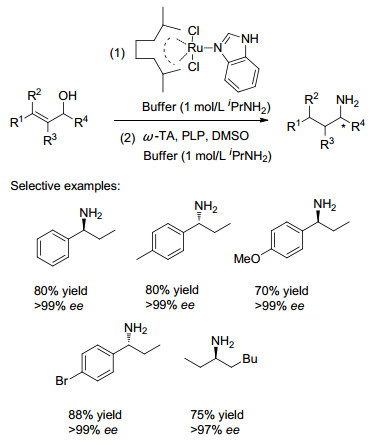
2015年, Garcia-Alvarez课题组[21]报道了一种高效、高立体选择性以及操作简单的化学-酶相结合一锅多步串联反应, 选用绿色无污染的水作为溶剂.在水性介质中, 通过将钌基催化剂催化的烯丙醇异构化和与ω-转氨酶(ω-TA)催化的转胺化反应串联起来, 以烯丙醇为原料可以高效绿色地合成出不同结构的手性胺, 且串联反应具有高的总收率和优异的ee值(Scheme 8).反应使用酮还原酶(KREDs)作为生物催化剂, 即使在非常低的辅因子浓度(NADP+或NADPH)条件下, 仍能正常反应, 但该过程必须以连续的方式才能完成, 因为ω-TA和辅因子等都会使金属催化剂失活.该方法对于在水相介质中金属和生物催化反应的一锅法过程具有很好的参考价值.
图式 8

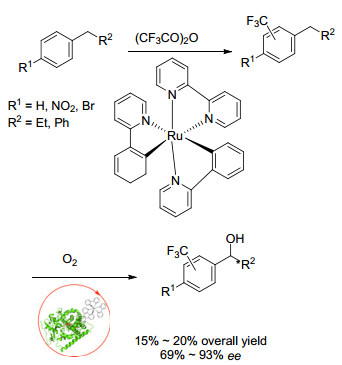
最近, Cheruzel课题组[22]通过杂化的P450-BM3酶与Ru(Ⅱ)-二亚胺共同作用, 这种光驱动的化学-酶方法能够通过利用Ru(Ⅱ)-二亚胺光敏剂和杂化P450-BM3酶的光化学性质来选择性地进行取代芳烃的三氟甲基化/羟基化, 在光氧化还原条件下, 由金属配合物促进的CF3自由基可引入到芳烃中(Scheme 9).在杂化P450- BM3酶中, 共价连接的Ru(Ⅱ)-二亚胺光敏剂提供必要的电子, 在可见光激活时在三氟甲基化底物上进行P450氧化官能化.由于P450-BM3突变体具有好的选择性, 所以能区分三氟甲基化异构体, 并产生区域选择性产物和立体选择性产物.
图式 9

1.3 酶催化与铱基催化剂相结合
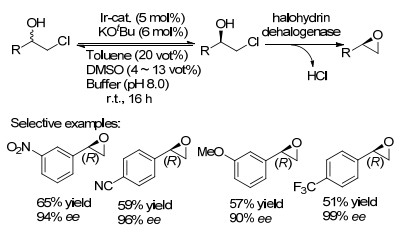
邻位卤代醇作为手性环氧化物或α-和γ-氨基醇的前体化合物, 在合成化学中是非常有价值的起始原料. 2008年, Janssen课题组[23]首次报道了以外消旋的α-卤代醇为原料通过化学-酶促动态动力学拆分(DKR)直接制备出相应的光学纯度的环氧化物.具体过程是, 反应用二甲亚砜(DMSO)作为助溶剂(5.0 vot%), 在甲苯和缓冲液组成的双相系统中进行反应, 在铱基催化剂的催化下反应16 h, 然后使用卤代醇脱卤素酶HheC(一种催化卤代醇和环氧化物互换的酶)催化, 得到相应的对映异构环氧化物(Scheme 10).此外, 当使用多种芳香族底物时, 该反应均具有良好的收率和出色的对映选择性.
图式 10
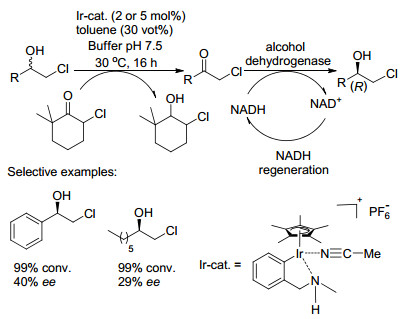
2010年Kroutil课题组[24]首次报道了铱基催化剂催化氧化与酶催化还原相结合的例子.研究表明, 当反应使用卤代醇为原料, 依次对卤代醇进行铱基催化剂催化氧化反应及对氧化所得中间体进行醇脱氢酶催化的对映选择性生物还原.金属铱基催化剂被用来通过氢转移进行原料卤代醇的氧化制备出关键中间体α-氯酮, 此催化剂在含水缓冲液存在下稳定存在, 然后使用来自Rhodococcus ruber的醇脱氢酶ADH-A立体选择性地将所获得的α-氯酮还原为相应的光学纯度的卤代醇(Scheme 11), 之所以选择ADH-A, 因为它对有机溶剂具有优异的耐受性.
图式 11

1.4 酶催化与金基催化剂相结合
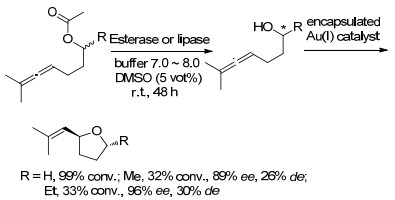
2013年, Bergman课题组[25]报道了通过包裹在Ga4L6四面体超分子簇中的脂肪酶或醇脱氢酶和一价金配合物共同进行催化反应, 一价金催化剂被封装在四面体超分子簇中以防止生物酶催化剂的失活.结果表明, 将该超分子包封用于丙二酰乙酰化底物的酶促水解和中间醇的进一步环化, 以在水性缓冲液中产生取代的四氢呋喃, 随后在外消旋底物上成功进行串联酶促动力学拆分/环化(Scheme 12).同样, 该方法也在Ru催化的烯丙醇的异构化/酶促还原中实现.利用超分子方法解决串联催化问题具有许多优点[26, 27].首先, 超分子组装体稳定反应性金属中心形成主客体复合物的能力可能会延长有机金属配合物的寿命.其次, 表现出高水溶性的超分子主体可将具有相对疏水的有机配体的金属络合物“拉”到水溶液中, 从而使传统上在有机溶剂中进行的反应可在水中实现[28].另外, 该团簇本身可以防止反应性金属催化剂扩散到溶液中, 在那里它可直接与蛋白质上的残基相互作用.
图式 12

最近Mihovilovic课题组[29]通过将金催化的水合反应与酶促还原相结合, 报道了一种简单且原子经济高适用于合成具有优异光学纯度的仲醇的化学-酶方法.该方法以苯乙炔及其衍生物为原料, 先以AuCl3为催化剂在水相条件下制备出苯乙酮及其衍生物, 再采用(S)-或(R)-选择性的醇脱氢酶(S-ADH或R-ADH)不对称催化合成出(S)-或(R)-芳基烷基醇类化合物.结果表明, 采用该方法合成的(S)-和(R)-芳基烷基醇类化合物光学纯度高且转化率高达91%以上(Scheme 13).由于在金催化步骤中使用iPrOH作Au(Ⅲ)催化水合的溶剂, 反应中金属催化步骤的反应混合物可用于随后的酶促还原反应.
图式 13

1.5 酶催化与其他金属催化剂相结合
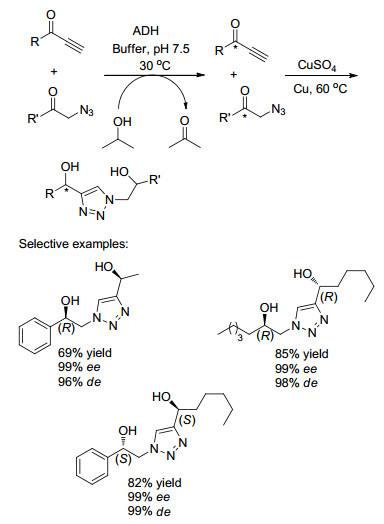
除了以上金属催化剂与酶催化相结合进行多步串联反应外, 其他过渡金属, 如Cu、Rh等也用来催化不对称转移氢化(ATH), 并与酶相结合合成光学活性化合物. 2013年, Lavandera课题组[30]报道了在温和的反应条件下, 在水性介质中通过组合单一醇脱氢酶(ADH)与Cu催化的“点击”反应的结合, 高收率、高对映选择性地合成出不同结构的手性1, 2, 3-三唑衍生的二醇(Scheme 14).产物的最终构型由生物催化剂控制, 且(R, S), (R, R)和(S, S)-异构体都能以高收率和光学纯度获得.
图式 14

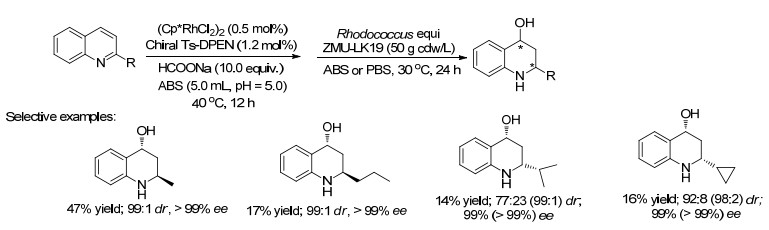
2017年, 陈永正课题组[31]报道了一种新颖的不对称“金属-生物催化体系”, 反应将Rh催化的ATH过程与全细胞介导的不对称羟基化相结合, 可得到一系列有两个立体中心的手性2-取代的-四氢喹啉-4-醇(47%的产率, 99:1的dr, >99% ee, Scheme 15).
图式 15

2. 酶催化与有机分子催化相结合
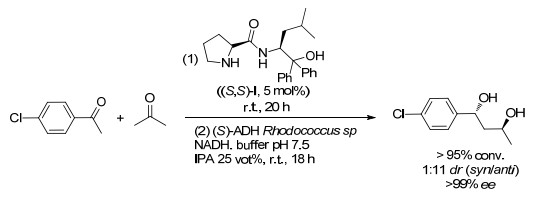
近年来, 不对称有机分子催化的快速发展使得它们与生物催化反应相结合得以实现.实际上, 不对称有机催化和生物催化反应也是互补的, 因此在组合时使得一系列独特的一锅多步串联成为可能.例如, 在2009年, Berkessel课题组[32]报道了在含水介质中进行了手性1, 3-二醇的一锅多步串联反应, 通过有机催化醛醇缩合反应和酶催化酮还原反应相结合制备出了四种同分异构的1, 3-二醇, 具有优异的结果(>95%转化率>99% ee和1:11 dr, Scheme 16).值得一提的是, 由有机催化反应路线产生的反应混合物与直接随后的酶促还原相容, 无需处理中间产物.
图式 16

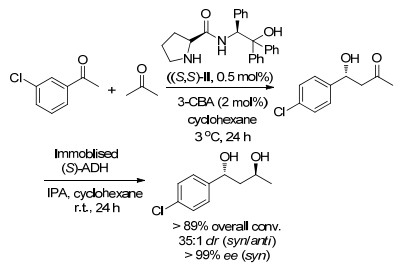
此后, 该课题组[33]又将底物范围扩展至间位取代的芳香醛, 他们报道了采用间氯苯甲醛、丙酮为原料, 通过将β-氨基醇衍生物催化的醛醇缩合反应与酶催化的生物催化反应相结合, 高效、绿色地制备出具有手性结构的1, 3-二醇(dr高达>25:1, ee>99%) (Scheme 17). 2014年, 该课题组[34]再次报道了使用另一种未固定化的手性有机小分子催化剂(S, S)-Ⅱ用于手性二醇的合成.研究表明, 当以间氯苯甲醛、丙酮为起始原料, 用(S, S)-Ⅱ作手性催化剂, 反应24 h后以95%的总转化率和95% ee制备出(R)-β-羟基酮.在该过程中, 为克服(S, S)-Ⅱ在非水相条件下低的活性, 需使用3-氯苯甲酸(3-CBA, 2 mol%)作为助催化剂, 并发现在3 ℃效果最佳, 在有机溶剂如环己烷中能保持良好的不对称选择性.随后再采用醇脱氢酶(ADH)通过对醛醇加成产物进行催化还原反应, 该反应过程中不需要任何中间体分离, 在酶催化还原步骤之前可通过倾析和蒸发有机层来循环回收固定的催化剂.结果表明, 该方法最终以高的转化率(89%)和优异的选择性(dr>35:1, >99% ee)制备出光学纯度的二醇化合物(Scheme 18).
图式 17

图式 18
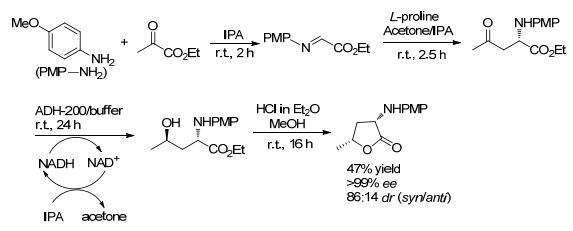
α-氨基丁内酯是存在于天然化合物和药物中的有价值的结构单元. 2014年, Simon课题组[35]报道了在异丙醇(IPA)中, 以茴香胺与乙醛酸乙酯为原料合成醛亚胺, 再将所得醛亚胺先后在L-脯氨酸的催化下进行Mannich反应、羰基还原酶催化的还原反应, 制备出非对映异构体氨基醇.所得氨基醇在自发或在酯交换条件下(HCl-MeOH)发生环化反应, 合成出高光学纯度的顺-(3S, 5R)-内酯, 产率47%, ee值高达99% (Scheme 19).
图式 19

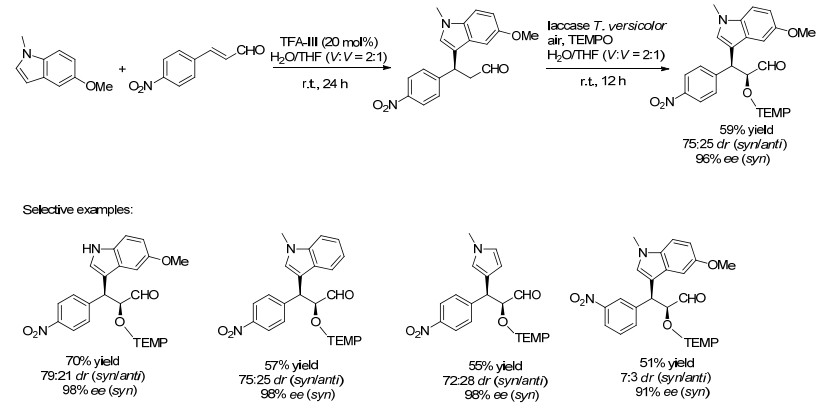
另外, 树脂固定化的肽也可用在含水条件下进行的有机催化反应, 这些催化剂与来自Trametes versicolor的漆酶结合已被用于功能化吲哚的多步串联合成[36, 37]. 2012年, Kudo课题组[38]报道了应用树脂固载的肽Ⅲ (TFA-Ⅲ)为催化剂, 以α, β-不饱和醛与吲哚衍生物为起始原料进行不对称Friedel-Crafts烷基化, 再将所得中间体在漆酶和2, 2, 6, 6-四甲基哌啶氮氧化物(TEMPO)的共同催化下进行选择性氧化.反应在含水介质中进行, 当使用4-硝基肉桂醛时, 最终产物的顺式/反式比率为75:25, ee值为96% (Scheme 20).结果表明, 当该反应扩展到其他取代的吲哚, 也可获得理想结果.
图式 20

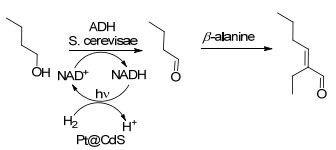
最近, Cha等[39]报道了将光催化、有机催化和生物催化相结合, 以正丁醇为原料, 多步串联合成2-乙基己烯醛[37].反应中使用NAD+作为辅因子催化正丁醇氧化成正丁醛, 然后中间体正丁醛在β-丙氨酸的催化下进行羟醛缩合反应, 最终合成出2-乙基己烯醛.过程中采用光催化剂铂-硫化镉量子点(Pt@CdS)可以使辅因子再生.结果表明, 在丙酮-丁醇-乙醇(ABE)溶液中, 将三种催化剂和NAD+加入到该溶液中, 当使用量为15.0 mmol丙酮, 30.0 mmol丁醇和5.00 mmol乙醇时, 3 h光照后, 即可得到含有2-乙基己烯醛的混合物(Scheme 21).
图式 21

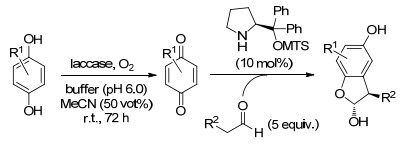
2015年, Pietruszka课题组[40]报道了先采用漆酶将1, 4-对苯二酚衍生物氧化为相应的对苯二醌衍生物, 再将所得中间体在(S)-2-[二苯基(三甲基甲硅烷基氧基)甲基]吡咯烷的催化下, 与醛类化合物进行立体选择性的α-芳基化反应.结果表明, 该反应在10 mol%的大位阻四氢吡咯衍生物(S)-2-[二苯基(三甲基甲硅烷基氧基)甲基]吡咯烷的催化下, 在1:1 (V:V)混合物MeCN/含水缓冲液中进行, 得到α-芳基化的醛, 再环化成相应的半缩醛.由于环化的可逆性, 产物的ee值在延长反应时间后略微降低.通过提供过量的醛(5.0 equiv.), 并将该反应应用到各种羟基醌和醛的组合上, 均能以高的产量和优异的ee值制备出相应的手性产物(Scheme 22).
图式 22

含有手性结构的吲哚衍生物代表了一类生物活性化合物.采用Friedel-Crafts型不对称烷基化(FCAA)反应, 以非手性吲哚衍生物为起始原料, 可合成出多功能化的手性吲哚衍生物.迄今为止, 已报道了金属催化剂或有机催化剂的多种FCAA反应[41].
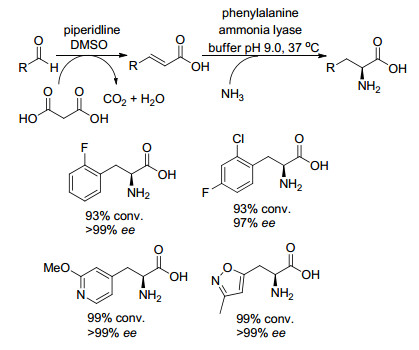
2016年, Turner课题组[42, 43]报道了从价廉易得的取代的苯甲醛开始, 通过化学-酶多步串联, 以高的收率和光学纯度合成出L-苯基-和L-吡啶基-丙氨酸衍生物.该方法利用了Knoevenagele-Doebner缩合反应(采用微波照射在短的反应时间内实现完全转化)和生物催化苯丙氨酸氨裂解酶介导的加氢胺化(用于立体选择性引入氨基), 得到L-苯基-和L-吡啶基-丙氨酸衍生物.除了苯甲醛和不同的吡啶醛之外, 还制备了其他具有杂环体系的产物, 如异噁唑、噻吩和喹啉, 并得到了高的转化率和ee值(Scheme 23).
图式 23

3. 其他
2017年, 李辉课题组[44]报道了通过使用化学-酶催化剂组合, 在微波辅助下成功地进行了芳香族仲醇的一锅动态动力学拆分(DKR).在微波辐射下, 固定在介孔二氧化硅上的Pd纳米颗粒Pd@SBA-15和Novozym 435组合作为各种外消旋仲醇的DKR的强有力的催化剂体系, 得到具有良好收率和优异的对映选择性的光学纯度的酯类化合物.当1-苯基乙醇的DKR经受微波加热时, Pd表面上的诱导局部“超热”点加速了外消旋作用, 因此大大缩短了DKR时间.
2011年, Arends课题组[45]报道了在离子液体中, 采用H2O2和脂肪酶相结合, 系统研究了脂肪族烯烃环氧化反应以及Baeyer-Villiger反应, 并发现离子液体对上述氧化反应有很好的促进作用.在室温下在Novozym 435(载体吸附的CalB)存在下研究了环己烯、环辛烯和苯乙烯的环氧化(室温, 24 h), 选择过辛酸作为氧化剂.对于环己烯和环辛烯, 在将离子液体从非配位的咪唑基ILs改变为配位和氢键供给[HOPMIm]+[NO3]-时观察到明显的差异.使用环己烯在后一种溶剂的情况下, 环氧化物的产率达到71%, 比[BMIm]+[BF4]-高9%.对于环辛烯, 差异甚至更大, 高出了23%.一系列对比实验表明, 每一种反应基质都有其对应的最好的ILs, 可以设计不同的ILs以获得最佳结果, 因此, 新一代提供氢键的离子液体的应用是寻求生物催化转化中的替代环境友好解决方案的一个进步.
除了离液体, 2017年王永华课题组[46]报道了将低共熔溶剂(DES)氯化胆碱/山梨糖醇用于环氧化物的合成.首先, 使用1-十八碳烯的化学酶促环氧化效率作为模型反应, 评估了不同EDS对反应的影响, 发现氯化胆碱/山梨糖醇作为溶剂有很好的效果, 原料转化率超过了70%.此后对其他底物进行了扩展, 24 h以内, 反应转化率在72%~90%之间.与ILs相比, 该系统所用DES对环境更加友好且系统的实际适用性和效率也表现突出.
4. 总结与展望
在含水介质中, 将生物催化和化学催化相结合, 通过串联反应的方式制备出高附加值手性化合物, 从学术和工业角度来看都是一个非常有吸引力的研究领域.它虽仍是一个新兴的研究领域, 但近年来发展了一系列基于化学-酶多步串联反应, 已经成功地证明了生物转化与化学催化剂这两个“世界”的结合可相互兼容.此外, 化学-酶与离子液体、微波、和光/电的联用使得该领域的发展更加多元化.一些新的概念和想法, 如使用金属有机框架, 模仿生物膜或蛋白质容器等, 为化学-酶串联反应的应用奠定了基础.
除了扩大适用于水相化学催化和生物催化相结合的多步串联反应的范围外, 还要解决的主要挑战是化学试剂和化学催化剂的使用条件与酶催化反应条件的不兼容性.幸运的是, 近年来蛋白质工程的巨大进步大大促进了酶工程设计, 可让作为生物催化剂的酶“接受”更苛刻的反应条件.此外, 除寻找更加稳定和更相容的化学和生物催化剂之外, 反应工程问题(如固定化的化学催化剂和酶催化剂在不同隔离区域中的使用)的设计与优化将在未来发展中发挥重要作用.总之, 对化学-酶相结合的多步串联反应的研究不仅可以帮助人们深入了解生物酶催化与化学催化反应的机制与历程, 同时也可为设计更绿色、高效的化学-酶相结合的多步串联反应催化体系提供丰富的参考信息和理论依据.
-
-
[1]
Katja, G.; Kirsten, S.; Stephan, L.; Andreas, L. Appl. Microbiol. Biotechnol. 2007, 76, 24.
-
[2]
Ni, Y.; Li, C. C.; Ma, H. M.; Zhang, J.; Xu, J. H. Appl. Mierobiol. Biotechnol. 2010, 89, 1111.
-
[3]
Patel, R. N. Coord. Chem. Rev. 2008, 252, 659. doi: 10.1016/j.ccr.2007.10.031
-
[4]
Wang, H.; Zong, M. H.; Wu, H. J. Biotechnology 2007, 129, 689.
-
[5]
Makkee, M.; Kieboom, A. P. G.; van Bekkum, H. J. Chem. Soc., Chem. Commun. 1980, 930. https://www.researchgate.net/publication/255767941_Combined_action_of_enzyme_and_metal_catalyst_applied_to_the_preparation_of_D-mannitol
-
[6]
Allen, J. V.; Williams, J. M. J. Tetrahedron Lett. 1996, 37, 1859. doi: 10.1016/0040-4039(96)00136-0
-
[7]
Dinh, P. M.; Howarth, J. A.; Hudnott, A. R.; Williams, J. M. J.; Harris, W. Tetrahedron Lett. 1996, 37, 7623. doi: 10.1016/0040-4039(96)01677-2
-
[8]
Carr, R.; Alexeeva, M.; Dawson, M. J.; Fernandez, V. G.; Humphrey, C. E.; Turner, N. J. ChemBioChem 2005, 6, 637. doi: 10.1002/cbic.v6:4
-
[9]
Bisogno, F. R.; Lopez-Vidal, M. G.; de Gonzalob, G. Adv. Synth. Catal. 2017, 359, 2026. doi: 10.1002/adsc.v359.12
-
[10]
Schrittwieser, J. H.; Velikogne, S.; Hall, M.; Kroutil, W. Chem. Rev. 2018, 118, 270. doi: 10.1021/acs.chemrev.7b00033
-
[11]
Rudroff, F.; Mihovilovic, A. D.; Gröger, H.; Radka, S.; Hans, I.; Uwe, T. B. Nat. Catal. 2018, 1, 12. doi: 10.1038/s41929-017-0010-4
-
[12]
Allen, J. V.; Williams, J. M. J. Tetrahedron Lett. 1996, 37, 1859. doi: 10.1016/0040-4039(96)00136-0
-
[13]
Pamies, O.; Backvall, J. E. Chem. Rev. 2003, 103, 3247. doi: 10.1021/cr020029g
-
[14]
Burda, E.; Hummel, W.; Groger, H. Angew. Chem., Int. Ed. 2008, 47, 9551. doi: 10.1002/anie.v47:49
-
[15]
Sato, H.; Hummel, W.; Groger, H. Angew. Chem., Int. Ed. 2015, 54, 4488. doi: 10.1002/anie.201409590
-
[16]
Ahmed, S. T.; Parmeggiani, F.; Weise, N. J.; Flitsch, S. L.; Turner, N. J. ACS Catal. 2015, 5, 5410. doi: 10.1021/acscatal.5b01132
-
[17]
Latham, J.; Henry, J, M.; Sharif, H, H.; Menon, B. R. K.; Shepherd, S. A.; Greaney, M. F.; Micklefield, J. Nat. Commun. 2016, 7, 11873. doi: 10.1038/ncomms11873
-
[18]
Rodriguez-Alvarez, M. J.; Rios-Lombardia, N.; Schumacher, S.; Perez-Iglesias, D.; Moris, F.; Cadierno, V.; Garcia-Alvarez, J.; Gonzalez-Sabín, J. ACS Catal. 2017, 7, 7753. doi: 10.1021/acscatal.7b02183
-
[19]
Denard, C. A.; Bartlett, M. J.; Wang, Y. J.; Lu, L.; Hartwig, J. F.; Zhao, H.-M. ACS Catal. 2015, 5, 3817. doi: 10.1021/acscatal.5b00533
-
[20]
Denard, C. A.; Huang, H.; Bartlett, M. J.; Lu, L.; Tan, Y.; Zhao, H.-M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2014, 53, 465. doi: 10.1002/anie.v53.2
-
[21]
Rios-Lombardia, N.; Vidal, C.; Cocina, M.; Moris, F.; Garcia-Alvarez, J.; Gonzalez-Sabin, J. Chem. Commun. 2015, 51, 10937. doi: 10.1039/C5CC03298A
-
[22]
Sosa, V.; Melkie, M.; Sulca, C.; Li, J.; Tang, L.; Li, J.; Faris, J.; Foley, B.; Banh, T.; Kato, M.; Cheruzel, L. E. ACS Catal. 2018, 8, 2225. doi: 10.1021/acscatal.7b04160
-
[23]
Haak, R. M.; Berthiol, F.; Jerphagnon, T.; Gayet, A. J. A.; Tarabiono, C.; Postema, C. P.; Ritleng, V.; Pfeffer, M.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L.; de Vries J. G. J. Am. Chem. Soc. 2008, 130, 135008. https://www.ncbi.nlm.nih.gov/pubmed/18800793
-
[24]
Mutti, F. G.; Orthaber, A.; Schrittwieser, J. H.; de Vries, J. G.; Pietschnig, R.; Kroutil, W. Chem. Commun. 2010, 46, 8046. doi: 10.1039/c0cc02813d
-
[25]
Wang, Z. J.; Clary, K. N.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Nat. Chem. 2013, 5, 100. doi: 10.1038/nchem.1531
-
[26]
Fiedler, D.; Bergman, R. G.; Raymond, K. N. Angew. Chem., Int. Ed. 2006, 45, 745. doi: 10.1002/(ISSN)1521-3773
-
[27]
Merlau, M. L.; Mejia, M. D. P.; Nguyen, S. T.; Hupp, J. T. Angew. Chem., Int. Ed. 2001, 40, 4239. doi: 10.1002/1521-3773(20011119)40:22<>1.0.CO;2-D
-
[28]
Oshovsky, G. V.; Reinhoudt, D. N.; Verboom, W. Angew. Chem., Int. Ed. 2007, 46, 2366. doi: 10.1002/(ISSN)1521-3773
-
[29]
Schaaf, P.; Gojic, V.; Bayer, T.; Rudroff, F.; Schnurch, M.; Mihovilovic, M. ChemCatChem 2018, 10, 920. doi: 10.1002/cctc.201701752
-
[30]
Cuetos, A.; Bisogno, F. R.; Lavandera, I.; Gotor, V. Chem. Commun. 2013, 49, 2625. doi: 10.1039/c3cc38674k
-
[31]
Wang, J.-X.; Li, K.; Zhou, X.-J.; Han, W.-Y.; Wan, N.-W.; Cui, B.-D.; Wang, H.-H.; Yuan, W.-C.; Chen, Y.-Z. Tetrahedron Lett. 2017, 58, 2252. doi: 10.1016/j.tetlet.2017.04.074
-
[32]
Baer, K.; Krauber, M.; Burda, E.; Hummel, W.; Berkessel, A.; Grçger, H. Angew. Chem., Int. Ed. 2009, 48, 9355. doi: 10.1002/anie.v48:49
-
[33]
Rulli, G.; Dunangdee, N.; Baer, K.; Hummel, W.; Berkessel, A.; Grçger, H. Angew. Chem., Int. Ed. 2011, 50, 7944. doi: 10.1002/anie.v50.34
-
[34]
Heidlindemann, M.; Rulli, G.; Berkessel, A.; Hummel, W.; Grçger, H. ACS Catal. 2014, 4, 1099. doi: 10.1021/cs4010387
-
[35]
Simon, R. C.; Busto, E.; Schrittwiesser, J. H.; Sattler, J. H.; Pietruszka, S. J.; Faber, K.; Kroutil, W. Chem. Commun. 2014, 50, 15669. doi: 10.1039/C4CC06230B
-
[36]
Akagawa, K.; Kudo, K. Adv. Synth. Catal. 2011, 353, 843. doi: 10.1002/adsc.v353.6
-
[37]
Akagawa, K.; Fujiwara, T.; Sakamoto, S.; Kudo, K. Chem. Commun. 2010, 46, 8040. doi: 10.1039/c0cc02301a
-
[38]
Akagawa, K.; Umezawa, R.; Kudo, K. Beilstein J. Org. Chem. 2012, 8, 1333. doi: 10.3762/bjoc.8.152
-
[39]
Hafenstine, G. R.; Ma, K.; Harris, A. W.; Yehezkeli, O.; Park, E. Domaille, D. W.; Cha, J. N.; Goodwin, A. P. ACS Catal. 2017, 7, 568. doi: 10.1021/acscatal.6b03213
-
[40]
Sujic, S.; Pietruszka, J.; Worgull, D. Adv. Synth. Catal. 2015, 357, 1822. doi: 10.1002/adsc.201500183
-
[41]
Akagawa, K.; Kudo, K. Org. Lett. 2011, 13, 3498. doi: 10.1021/ol2012956
-
[42]
Parmeggiani, F.; Ahmed, S. T.; Weise, N. J.; Turner, N. J. Tetrahedron 2016, 72, 7256. doi: 10.1016/j.tet.2015.12.063
-
[43]
Ahmed, S. T.; Parmeggiani, F.; Weise, N. J.; Flitsch, S. L.; Turner, N. J. Org. Lett. 2016, 18, 5468. doi: 10.1021/acs.orglett.6b02559
-
[44]
Xu, Y.-F.; Wang, M.; Feng, B.; Li, Z.-Y.; Li, Y.-H.; Li, H.-X.; Li, H. Catal. Sci. Technol. 2017, 7, 5838. doi: 10.1039/C7CY01954H
-
[45]
Kotlewska, A. J.; Rantwijk, F. V.; Sheldon, R. A.; Arends, I. W. C. E. Green Chem. 2011, 13, 2154. doi: 10.1039/c1gc15255f
-
[46]
Zhou, P.-F.; Wang, X.-P.; Yang, B.; Hollmannc, F.; Wang, Y.-H. RSC Adv. 2017, 7, 12518. doi: 10.1039/C7RA00805H
-
[1]
-

-

 下载:
下载:




 下载:
下载:






















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 58
- 文章访问数: 5001
- HTML全文浏览量: 922
