在光动力治疗(photodynamic therapy, 简称PDT)抗癌药物(光敏剂)的筛选过程中, 叶绿素二氢卟吩已经在诸多方面凸显出独到的研究优势.除了自身所具有的非对称性分子架构和良好的生物利用度以外, 其周环结构的化学修饰还可以有效地改善大环分子的光物理和光生物性质, 也是设计理想PDT光敏剂的重要合成策略和切实可行的实施路径[1 4 .许多叶绿素降解产物的定量构效关系(QSAR)研究表明, 其周环连带结构的几何形状、电荷密度以及相对位置均对PDT活性产生深刻的影响, 也是决定大环分子光动力疗效的至关重要的因素[5 9 .基于杂环分子的非碳原子参杂的环状结构、n、p电子交织的π键离域体系和非均匀的电荷分布等基本特征, 我们在叶绿素二氢卟吩碳架上设计构建不同的杂环单元, 进而观察和探索该类大环分子的结构转换与基本属性变化之间的相互关联, 充实和拓展叶绿素光敏剂在光动力医学领域中的研究和应用.本工作选择焦脱镁叶绿酸-a甲酯(MPPa, 1a )为起始原料, 通过环上取代基团的化学转换, 在二氢卟吩色基上分别构建了醛、邻位二酮、烯腈和烯酮等活性受电子官能结构, 通过这些新形成的高活性取代结构和环上原有的碳碳或者碳氧双键与不同的多电子体系实施关环, 合成出一系列未见报道的含有多种杂环结构的焦脱镁叶绿酸衍生物.
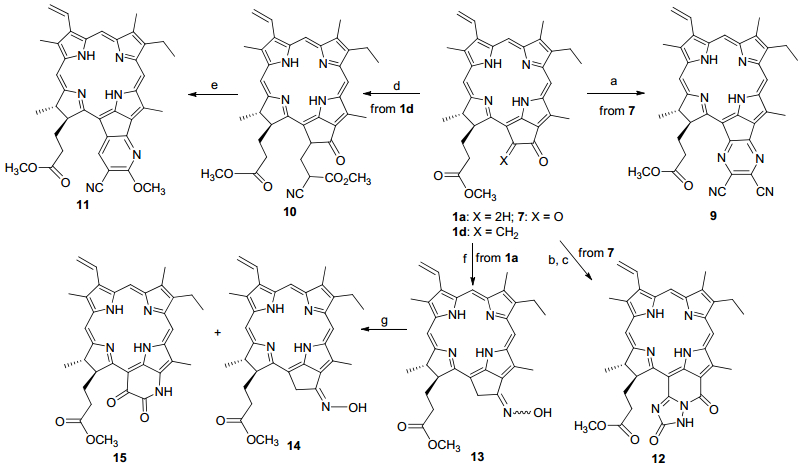
化合物1b 与丙二腈的knoevenagel反应高产率地给出3-β , β -二氰乙烯基焦脱镁叶绿酸(2 ), 将其溶解于乙酸中并与醋酸铵和二甲基环己二酮共回流, 则以46%的产率生成3-位氢化喹啉酮取代的二氢卟吩(3 ).通过相同的缩合反应得到在外接环上连有二氰亚甲基的二氢卟吩(4 , 66%), 随后与氨基酸和芳醛所形成的亚胺类偶极体进行1, 3-偶极环加成反应, 以理想的产率得到一对难以分离的具有螺环结构的差向异构产物5 .利用焦脱镁叶绿酸-b (1c )中7-位上原有的甲酰基与甲萘胺和二甲基环己二酮进行三组分缩合, 以31%的产率分离出连有苯并吖啶环系结构的叶绿素衍生物6 .在碱性条件下, 起始原料1a 经历氢氧化锂促进的空气氧化可以转化成132 -氧代焦脱镁叶绿酸(7 ).在三乙胺存在下, N -溴代丁二酰亚胺(NBS)对苯甲醛肟实施溴代和碱催化脱卤化氢生成1, 3-偶极体氧化芳甲腈, 不经分离直接与7 的3-位碳碳双键进行1, 3-偶环环加成反应, 以68%的产率得到C(3)-异噁唑啉基取代的脱镁叶绿酸衍生物8 (Scheme 1
图式 1
图式 2
在外接环上稠并杂环结构可以显著地改变大环分子的光化学和光物理性质[10 , 而五元外接环酮则是稠并各种杂环结构的有效切入位点.利用化合物7 中外接环上的α -二酮结构, 在乙酸中与顺-1, 2-二氰基-1, 2-二氨基乙烯进行酮胺缩合, 结果以15%的产率从纷杂的混合物中分离出吡嗪并焦脱镁叶绿酸(9 ).二氢卟吩烯酮1d 与氰乙酸甲酯在外接环上顺利地发生了迈克尔加成, 所生成的加成产物(10 , 78%)在醋酸铵/醋酸溶液中回流反应3 h, 以54%的产率环合成吡啶并焦脱镁叶绿酸(11 ).除了在原有外连结构上稠并含氮杂环以外, 我们还尝试通过碳架的改变来构建新的外接环系结构.用含有氢氧化钾的甲醇溶液处理二氢卟吩二酮7 , 几乎定量地转化成另一叶绿素降解产物红紫素-18, 然后直接与氨基硫脲在70 ℃下搅拌反应3 h, 以12%的产率生成1, 2, 4-三唑并红紫素-18二酰亚胺(12 ). MPPa (1a )与盐酸羟胺的肟化给出顺反异构的二氢卟吩肟(13 ), 选择N , N -二甲基甲酰胺和三氯三嗪为催化剂并对该混合物实施贝克曼重排, 除了得到期望的重排产物氮杂罗丁二氢卟吩(Azorhodinochlorin) (15 , 16%)以外, 还分离出57%的E -式酮肟14 .如果在相同条件下, 利用所得单一的E -式酮肟重复上述反应, 则没有得到相应的设计产物, 这一结果说明只有不稳定的Z -式酮肟才可发生贝克曼重排.
1.
结果与讨论
1.1
二氢卟吩色基上取代杂环的形成及其立体异构
在叶绿素色基上引进杂环单元的基本策略是首先构建亲电性双官能团结构, 或者利用周环上原有的复键基团与合适的电子给体实施环合.叶绿素色基中所存在的芳香性多氮杂轮烯结构活化了N21 -N23 轴向的不同取代基团, 其中, 原有的3-位乙烯基、3-和7-位甲酰基以及桥连于131 -和15-位的五元外接环是最重要的三个活性反应区域.通过结构转换, 可以顺利地在这些位点上形成包括烯酮(如1d )、烯腈(如2 和4 )、邻位二酮(如7 )和1, 4-二羰基(如10 )在内的双官能结构.
3-位甲酰基和131 -位酮羰基与丙二腈的Knoevenagel反应在二氢卟吩周环上建立了高反应活性的β , β -二氰乙烯基结构, 二氢卟吩2 与5, 5-二甲基-1, 3-环己二酮的迈克尔加成开始于酮羰基的烯胺化, 所形成的双甲酮烯胺首先进攻3a -位烯键碳原子生成双亚胺中间体A , 然后再异构成烯胺B , 通过分子内环化而转换成3-位多氢化喹啉取代的二氢卟吩3 ; MPPb (1c )、1-萘胺和双甲酮的多组分反应首先在7-位上发生羟醛缩合, 所形成的烯酮中间体C 中C-子环上的取代酮基再与1-萘胺的相互作用而给出亚胺中间体D , 随后的分子内Povarov反应环合成氢化苯并吖啶酮取代的中间体E , 然后再经过脱氢芳化形成二氢卟吩6 (Scheme 3
图式 3
虽然化合物3 和6 在3-位和7-位上所引进的多环氢化喹啉和氢化吖啶体系具有对称的平面结构, 但由于所占空间较大, 二者均不能围绕与色基相连的碳碳单键自由旋转而表现出阻旋异构特征, 其氢谱中一些成对的化学位移说明二氢卟吩3 和6 分别是由一对阻旋异构体所组成.
3-位乙烯基和131 -位β , β -二氰亚甲基都可以作为亲偶极体的受电子双键与偶极体发生1, 3-偶极环加成反应, 而且后者显示出了更高的反应活性.由苯甲醛肟溴化和脱溴化氢或者由脯氨酸与芳醛缩合再脱羧所制成的氧化苯甲腈a 和甲亚胺类1, 3-偶极体b (Scheme 4 1 -位上形成新的手性中心, 从而生成一对难以相互分离的差向异构体.除了电子效应以外, 反应过程的立体环境也是决定环合取向的重要影响因素, 因此, 两个偶极体所连接的芳环都选择远离大环色基的方向接近碳碳双键, 所形成的两个差向异构产物的核磁共振谱图也极其相似.在氧化苯甲腈a 与1a 进行偶极环加成时, 偶极体中的苯基取向选择远离大环色基, 以保证其环合过渡态的内能处于最低水平.
图式 4
出于同样缘由, 甲亚胺类1, 3-偶极体b 在接近131 -位二氰亚甲基的过程中, 其芳基也是保持外延的方向(exo-form)以规避更大的空间位阻.如果芳基的选择以内向(endo-form)的方式接近131 -位二氰亚甲基, 则甲亚胺偶极体b 与二氢卟吩4 的外接环形成更大的重叠面积, 再加上17-位取代结构对其也将产生范德华排斥作用, 从而导致环合过程需求更高的反应活化能.
1.2
二氢卟吩外接环上并合杂环的形成及其电子光谱
吡嗪并焦脱镁叶绿酸9 和三唑酮并红紫素-18二亚酰胺12 都是通过分子间双位点胺羰缩合环合而成, 而吡啶并焦脱镁叶绿酸11 的形成则是经历了分子内单一位点的氨羰作用.
相比之下, 焦脱镁叶绿酸酮肟13 的重排过程则较为复杂(Scheme 5 N , N -二甲基甲酰胺(DMF)和三氯三嗪的催化作用催化的反应启动于二者的SN2 Ar亲核取代反应[11 , 随后, 二氢卟吩13 的肟羟基与氯代亚胺盐发生亲核加成, 所生成O -取代肟中间体F 中处于反位的大环色基进行迁移, 进而形成吡啶并二氢卟吩碳正离子G , 与水分子作用后异构成贝克曼重排产物H ; 其151 -位的空气氧化从二氢卟吩酰亚胺烯醇异构体I 开始, 其烯醇式羟基与氯代亚胺盐的亲核加成给出烯醇醚J , 然后再通过电子转移与单线态氧反应并形成过氧负离子K 和氯代亚胺盐, 二者连结而成的过氧化物L 重新异构成烯醇中间体M , 再分别脱去一分子氯化氢和一分子DMF, 最后生成重排氧化产物15 .其间, E -式肟羟基同样可以生成O -取代肟中间体F' , 但并没有发现烷基迁移的重排产物J' , 反应结束后只是分离出单一的E -式二氢卟吩肟14 .
图式 5
叶绿素外接环的结构转换与其电子光谱的吸收变化密切相关, 稠并的芳香性环系可以有效地扩展色基中π-电子体系的离域范围, 同时也导致大环平面发生了相应的扭曲.外接环的刚性拉伸作用改善了四吡咯分子的共平面程度, 从而大幅度地延长了大环分子的最大可见光吸收波长(Qy谱带).当五元外接环并入芳香性的吡啶和吡嗪环之后, 所形成化合物9 和11 的最大可见光吸收谱带分别出现在754和757 nm处, 与焦脱镁叶绿酸1a 相比[12a , 其Qy吸收峰均形成了非常强烈的红移(9 , ΔQy=56 nm; 11 , ΔQy=59 nm).虽然并有咪唑环的红紫素-18二酰亚胺12 的外接环中不存在芳香性体系, 但与二氢卟吩二酮7 相比[12b , 其最大可见光的吸收谱带仍然向长波方向大幅推移(38 nm).相对而言.由于非芳香性的氮杂罗丁二氢卟吩15 的外接环中只有一个碳氧双键在15-位上直接与色基相连, 因此, 其最大可见光吸收波长仅比反应前体MPPa 1a 的Qy值仅仅延长了5 nm(图 1
图 1
2.
结论
通过对焦脱镁叶绿酸的3-位乙烯基和外接环酮的化学转换, 在周环上形成了醛、邻位二酮、烯腈和烯酮等活性受电子官能基团, 进而与含有杂原子的富电子体系进行环合, 在叶绿素二氢卟吩碳架上以取代和并合的两种方式构建了不同的杂环结构.杂环取代对分子的最大可见光吸收影响相对较小, 改变和重构环上连带成分和外接环碳架可以有效地改善它们的电子光谱, 其稠并芳香性杂环的同系物的最大可见光吸收均可延长50 nm以上.以经典的杂环化反应作为新的合成策略, 对叶绿素大环色基实施化学修饰, 为进一步完善其光物理、光生物等诸多性质和合成具有应用前景的新型四吡咯大环分子提供了有效的切入途径.
3.
实验部分
3.1
仪器与试剂
元素分析用Perkin-Elmer 2400型元素分析仪测定; IR用Perkin-Elmer 1730型红外分光光度仪测定(KBr压片); UV-Vis用UV-160A型紫外分光光度计测定; 1 H NMR用Brucker ARX-300型核磁共振仪测定, 内标为TMS.所用试剂均为分析纯或化学纯.焦脱镁叶绿酸-a甲酯(1a )按文献[12a 1b )按文献[12b 1c )按文献[12c 2 -亚甲基焦脱镁叶绿酸-a (1d )按文献[4b
3.2
3-[4'-(1', 4', 5', 6', 7', 8'-六氢-2'-氨基-3'-氰基-5'-氧代-7', 7'-二甲基喹啉基)]-3-去乙烯基焦脱镁叶绿酸-a甲酯(3)的合成
将122 mg (0.222 mmol)焦脱镁叶绿酸-d甲酯(1b )溶解于10 mL四氢呋喃中, 加入0.3 mL三乙胺后, 再缓慢滴加溶解于5 mL四氢呋喃中的20 mg (0.303 mmol)丙二腈, 室温避光搅拌25 min反应结束.搅拌下向反应体系先后加入20 mL水和20 mL二氯甲烷, 分出有机相, 用二氯甲烷萃取水相(20 mL×3), 合并有机相后再水洗一次, 无水硫酸钠干燥, 减压除尽溶剂, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1]得到105 mg (0.175 mmol)绿色固体2 .产率79%.其物理常数与分析数据与文献[13a 2 溶解于5 mL乙酸, 搅拌下加入碾碎的37 mg (0.264 mmol) 5, 5-二甲基-1, 3环己二酮和35 mg乙酸铵, 避光条件下110 ℃搅拌回流2 h.冷却后先后向反应体系加入20 mL水和20 mL二氯甲烷, 分出有机层后用二氯甲烷萃取水层(15 mL×3), 合并有机层, 再分别用15 mL饱和碳酸氢钠溶液和15 mL水洗涤, 经无水硫酸钠干燥后减压浓缩, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=3:1]得到59 mg (0.081 mmol)墨绿色固体3 , 产率46%. m.p. 245~247 ℃; UV-vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 407 (1.23×105 ), 475 (1.64×103 ), 546 (1.32×103 ), 619 (6.25×103 ), 665 (4.06×104 ); 1 H NMR (CDCl3 ) δ : -1.86 (br s, 1H, NH), 0.35 (br s, 1H, NH), 1.07 (s, 3H, 7'-CH3 ), 1.28 (1.25) (s, 3H, 7'-CH3 ), 1.58 (t, J =7.0 Hz, 3H, 8-CH3 ), 1.79 (1.75) (d, J =7.2 Hz, 3H, 18-CH3 ), 1.88~2.05 (m, 1H, 17a+17b-H), 2.15~2.41 (m, 3H, 17a+17b+6'+8'-H), 2.45~2.71 (m, 3H, 17a+17b+6'+8'-H), 3.40~3.58 (m, 3H, 8a+4'-H), 3.05, 3.49, 3.51, 3.59 (3.57) (each s, each 3H, 18H, CH3 +OCH3 ), 3.56 (t, J =7.6 Hz, 2H, 8a-H), 4.07 (d, J =5.4 Hz, 2H, 3a-H), 4.40~4.48 (m, 1H, 18-H), 4.20~4.50 (m, 1H, 17-H), 4.89 (br s, 1H, 2'-NH), 4.91 (br s, 1H, 2'-NH), 5.07 (d, J =19.8 Hz, 1H, 132 -H), 5.24 (5.21) (d, J =19.8 Hz, 1H, 132 -H), 5.54 (br s, 1H, 1'-NH), 8.56, 8.98, 9.24 (each s, each 1H, meso -H); IR (KBr) ν : 3428 (N—H), 2958 (C—H), 1735~1694 (C=O), 1662 (C=C), 1522 (chlorin skeleton), 1492, 1440, 1172, 1508, 912 cm-1 ; MS (ESI) m /z : d 738.3 (M+H+ ). Anal. calcd for C44 H47 N7 O4 : C 71.62, H 6.42, N 13.29; found C 71.50, H 6.21, N 13.37.
3.3
131 (R /S )-2'-对甲氧基-3', 3'-二氰基-1', 5'-亚丙基吡咯烷螺[4', 131 ]-131 -去氧焦脱镁叶绿酸-a甲酯(5)的合成
将152 mg (0.277 mmol)焦脱镁叶绿酸-a甲酯(1a )溶解于20 mL无水乙醇中, 再先后加入73 mg (1.105 mmol)丙二腈和0.5 mL三乙胺, 氮气保护下避光回流反应3 h, 搅拌下向反应体系先后加入20 mL水和20 mL二氯甲烷, 分出有机相, 用二氯甲烷萃取水相(20 mL×3), 合并有机相后再水洗一次, 无水硫酸钠干燥, 剩余物用硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1], 得109 mg (0.183 mmol)绿色固体4 , 产率66%.其物理常数与分析数据与文献[13b V (石油醚):V (乙酸乙酯)=5:1], 得93 mg (0.118 mmol)绿色固体5 , 产率63%. m.p. 207~209 ℃; UV-Vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 359 (2.23×104 ), 401 (1.19×105 ), 470 (3.13×104 ), 501 (1.28×104 ), 557 (2.56×104 ), 655 (3.89×104 ); 1 H NMR (CDCl3 ) δ : -3.24 (br s, 1H, NH), -1.31 (br s, 1H, NH), 1.07 (s, 3H, 7'-CH3 ), 1.28 (1.25) (s, 3H, 7'-CH3 ), 1.79 (t, J =7.0 Hz, 3H, 8-CH3 ), 1.86 (1.72) (d, J =7.2 Hz, 3H, 18-CH3 ), 2.13~2.37 (m, 1H, 17a+17b+6'+7'-H), 2.79~2.87 (m, 3H, 17a+17b+6'+7'-H), 2.40~2.757 (m, 4H, 17a+17b+6'+7'-H), 3.04~3.12 (m, 1H, 8'-H), 3.33~3.42 (m, 1H, 8'-H), 3.37 (3.25), 3.41, 3.53, 3.62 (3.59), 4.02 (each s, each 3H, 18H, CH3 +OCH3 ), 3.87 (t, J =7.6 Hz, 2H, 8a-H), 4.07 (d, J =5.4 Hz, 2H, 3a-H), 4.41 (4.22) (d, J =8.6 Hz, 1H, 17-H), 4.58 (4.49) (q, J =7.6 Hz, 1H, 18-H), 4.82 (4.67) (d, J =19.8 Hz, 1H, 132-H), 4.98 (4.93) (d, J =19.8 Hz, 1H, 132-H), 5.14 (t, J =6.7 Hz, 1H, 5'-H), 5.72 (5.65) (s, 1H, 2'-H), 6.17 (dd, J =11.6, 1.2 Hz, cis -3b-H), 6.34 (dd, J =17.8, 1.2 Hz, trans -3b-H), 6.45 (6.30) (d, J =8.7 Hz, 2H, Ar-H), 7.32 (7.30) (d, J =8.7 Hz, 2H, Ar-H), 8.21 (dd, J =17.8, 11.6 Hz, 1H, 3a-H), 8.86, 9.77 (9.76), 9.85 (each s, each 1H, meso -H); IR (KBr) ν : 3458 (N—H), 2959 (C—H), 1737, 1710 (C=O), 1662 (C=C), 1564 (chlorin skeleton), 1443, 1241, 1172, 911 cm-1 ; MS (ESI) m /z : 786.3 (M+H+ ). Anal. calcd for C49 H51 N7 O3 : C 74.88, H 6.54, N 12.47; found C 74.96, H 6.31, N 12.59.
3.4
7-[10'-(5', 6', 7', 8-四氢-5-氧代苯并[c ]吖啶基)]-3-去乙烯基焦脱镁叶绿酸-a甲酯(6)的合成
将135 mg (0.240 mmol)焦脱镁叶绿酸-b甲酯1c 溶解于10 mL无水乙醇中, 搅拌下加入52 mg (0.363 mmol) 1-萘胺, 回流搅拌15 min后, 再一次性加入溶解于5 mL无水乙醇中的51 mg (0.364 mmol) 5, 5-二甲基- 1, 3环己二酮, 搅拌回流反应6 h, 冷却后加入20 mL水和20 mL二氯甲烷, 分出有机层, 水层用二氯甲烷萃取水层(15 mL×3), 合并有机层, 再分别用20 mL饱和碳酸氢钠溶液和20 mL水洗涤, 经无水硫酸钠干燥后减压浓缩, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1]得到60 mg (0.074 mmol)墨绿色固体6 , 产率31%. m.p. 227~230 ℃; UV-vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 409 (1.69, 1.17×105 ), 504 (1.11×104 ), 5.36 (8.88×103 ), 601 (7.69×103 ), 655 (4.85×104 ) nm; 1 H NMR (CDCl3 ) δ : -1.47 (-1.50) (br s, 1H, NH), 0.59 (br s, 1H, NH), 1.08 (1.06) (s, 3H, 7'-CH3 ), 1.18 (1.17) (s, 3H, 7'-CH3 ), 1.69 (t, J =7.6 Hz, 1H, 8-CH3 ), 1.74 (1.71) (d, J =7.2 Hz, 3H, 18-CH3 ), 2.02~2.35 (m, 5H, 6'+8'+17a+17b-H), 2.40~2.74 (m, 2H, 6'+8'+17a+7b-H), 2.85~2.97 (m, 1H, 6'+8'-H), 3.20, 3.38, 3.52, 3.57 (each s, each 3H, CH3 +OCH3 ), 3.64~3.76 (m, 2H, 8a-H), 4.12~4.22 (m, 1H, 17-H), 4.40 (q, J =7.2 Hz, 1H, 18-H), 4.98 (4.90) (d, J =19.8 Hz, 1H, 132-H), 5.12 (5.08) (d, J =19.8 Hz, 1H, 132-H), 6.16 (d, J =11.5 Hz, 1H, cis -3b-H), 6.28 (d, J =17.8 Hz, 1H, trans -3b-H), 7.08~7.17 (m, 2H, Ar-H), 7.30~7.64 (m, 3H, Ar-H), 7.93~8.04 (m, 2H, 3a+Ar-H), 8.49, 9.22, 10.02 (10.01) (each s, each 1H, meso -H); IR (KBr) ν : 3437 (N—H), 2959 (C—H), 1738~1696 (C=O), 1623 (C=N), 1562 (chlorin skeleton), 1464, 1371, 1028, 986 cm-1 ; MS (ESI) m /z : 808.2 (M+H+ ). Anal. calcd for C52 H49 N5 O4 : C 77.30, H 6.11, N 8.67; found C 77.48, H 6.16, N 8.79.
3.5
3-(5'(R /S )-3'-苯基-4'5'-二氢-5'-异噁唑基)-132 -氧代-焦脱镁叶绿酸-a甲酯(8)的合成
将267 mg (0.487 mmol)焦脱镁叶绿酸-a甲酯1 、溶解于10 mL四氢呋喃中, 再加入15 mL甲醇稀释, 剧烈搅拌下加入含有300 mg氢氧化锂的水(5 mL), 开口剧烈搅拌3 h, 用乙酸将反应液调节pH至3, 搅拌下向反应体系先后加入20 mL水和20 mL二氯甲烷, 分出有机相, 用二氯甲烷萃取水相(20 mL×3), 合并有机相后再水洗一次, 无水硫酸钠干燥, 减压除尽溶剂, 将剩余物溶解于50 mL 5%的硫酸甲醇溶液, 氮气保护下搅拌过夜, 加入25 mL水和25 mL二氯甲烷, 分出水相, 先后用碳酸钠饱和溶液洗涤有机相, 无水硫酸钠干燥, 减压浓缩, 剩余物用硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1], 得93 mg (0.166 mmol)黄色固体7 , 产率34%.其物理常数与分析数据与文献[12b 7 后再滴加溶解于1 mL二氯甲烷的三乙胺(35 mg, 0.350 mmol), 室温搅拌反应24 h, 冷却后加入20 mL水和20 mL二氯甲烷, 分出有机层, 水层用二氯甲烷萃取水层(15 mL×3), 合并有机层, 再分别用20 mL饱和碳酸氢钠溶液和20 mL水洗涤, 经无水硫酸钠干燥后减压浓缩, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1]得到77 mg (0.113 mmol)墨绿色固体8 , 产率68%. m.p. 205~207 ℃; UV-vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 386 (1.31×104 ), 415 (8.91×103 ), 5.13 (1.32×103 ), 638 (1.02×103 ), 676 (6.81×103 ) nm; 1 H NMR (CDCl3 ) δ : -0.21 (br s, 1H, NH), 0.65 (br s, 1H, NH), 1.61 (t, J =7.6 Hz, 1H, 8-CH3 ), 1.77 (1.75) (d, J =7.2 Hz, 3H, 18-CH3 ), 2.16~2.49 (m, 3H, 17a+17b-H), 2.71~2.78 (m, 1H, 17a+17b-H), 2.99 (2.98), 3.36, 3.59, 3.75 (3.74) (each s, each 3H, CH3 +OCH3 ), 3.54~3.62 (m, 2H, 8a-H), 4.00 (4.39) (dd, J =17.0, 9.8 Hz, 1H, 4'-H), 4.29~4.35 (m, 1H, 4'-H), 4.41 (4.39) (q, J =7.2 Hz, 1H, 18-H), 5.18 (d, J =8.4 Hz, 1H, 17-H), 7.04 (7.02) (t, J =9.8 Ha, 1H, 5'-H), 7.48~7.58 (m, 3H, Ph-H), 7.88~7.96 (m, 2H, Ph-H), 8.85, 9.44 (9.43), 9.95 (9.93) (each s, each 1H, meso -H); IR (KBr) ν : 3450 (N—H), 2978 (C—H), 1735, 1690 (C=O), 1633 (C=N), 1562 (chlorin skeleton), 1475, 1371, 1168, 1082, 985, 721 cm-1 ; MS (ESI) m /z : 692.4 (M+ H+ ). Anal. calcd for C41 H39 N5 O5 : C 72.23, H 5.77, N 10.27; found C 72.38, H 5.70, N 12.41.
3.6
5', 6'-二氰基吡嗪并[2', 3'-n ]焦脱镁叶绿酸-a甲酯(9)的合成
将73 mg (0.130 mmol)二氢卟吩二酮7 溶解于5 mL冰乙酸中, 滴加含有20 mg (0.185 mmol)顺-2, 3-二氨基- 2-丁烯二腈的乙酸溶液(5 mL), 室温搅拌36 h, 加入20 mL水和30 mL二氯甲烷分层, 分出有机层并水洗两次, 无水硫酸钠干燥, 减压除尽溶剂, 剩余物用硅胶柱层析[洗脱剂: V (石油醚):V (乙酸乙酯)=3:1]分离, 得12 mg (0.020 mmol)黄色固体9 , 产率15%. m.p. 228~231 ℃; UV-Vis (CHCl3 ) λ max [ε /(L•mol-1 •cm-1 )]: 416 (1.00), 570 (0.132), 675 (0.214), 700 (0.23), 754 (0.141) nm; 1 H NMR (CDCl3 ) δ : -2.06 (br s, 1H, NH), 0.25 (br s, 1H, NH), 1.65 (t, J =7.6 Hz, 3H, 8a-CH3 ), 1.91 (d, J =7.2 Hz, 3H, 18-CH3 ), 2.15~2.27 (m, 1H, 17a+17b-H), 2.32~2.46 (m, 2H, 17a+17b-H), 2.70~2.78 (m, 1H, 17a+17b-H), 3.05, 3.34, 3.50, 3.64 (each s, each 3H, CH3 +OCH3 ), 3.69 (q, J =7.6 Hz, 2H, 8b-H), 4.64 (q, J =7.3 Hz, 1H, 18-H), 5.09 (d, J =7.6 Hz, 1H, 17-H), 6.30 (dd, J =11.6, 1.0 Hz, 1H, cis -3b-H), 6.38 (dd, J =17.8, 1.0 Hz, 1H, trans -3b-H), 8.07 (dd, J =17.8, 11.6 Hz, 1H, 3a-H), 8.95, 9.20, 9.79 (each s, each 1H, meso -H); IR (KBr) ν : 3443 (N—H), 2968 (C—H), 1741~1689 (C=O), 1650 (C=C), 1562 (chlorin skeleton), 1438, 1255, 1088, 725 cm-1 ; MS (ESI) m /z : 635.4 (M+H+ ). Anal. calcd for C38 H34 N8 O2 : C 71.91, H 5.40, N 17.65; found C 71.80, H 5.33, N 17.76.
3.7
132 (R /S )-132 -(2'-甲氧甲酰基氰乙基)焦脱镁叶绿酸-a甲酯(10)的合成
在8 mL甲醇中溶解23 mg (1.000 mmol)金属钠, 然后先后加入180 mg (0.321 mmol)化合物1d 和35 mg (0.353 mmol)氰基乙酸甲酯, 氮气保护, 35 ℃下搅拌反应10 h, 先后向反应体系加入20 mL二氯甲烷和20 mL水, 分出有机层, 水层用二氯甲烷萃取(20 mL×3), 合并有机层, 水洗后用无水硫酸钠干燥, 减压除去溶剂, 剩余物经硅胶柱层析[洗脱剂: V (石油醚):V (乙酸乙酯)=5:1], 得165 mg (0.250 mmol)黑绿色产物10 , 产率78%. m.p. 198~201 ℃; UV-vis (CH2 Cl2 ) λ max [ε /(L• mol-1 •cm-1 )]: 358 (9.09×104 ), 414 (2.811.12×105 ), 508 (2.95×104 ), 539 (2.87×104 ), 610 (2.01×103 ), 667 (1.02×105 ) nm; 1 H NMR (CDCl3 ) δ : -1.62 (br s, 1H, NH), 0.43 (br s, 1H, NH), 1.72 (1.71) (t, J =7.6 Hz, 3H, 8-CH3 ), 1.97 (d, J =7.2 Hz, 3H, 18-CH3 ), 2.17~2.33 (m, 1H, 17a+17b-H), 2.55~2.75 (m, 1H, 17a+17b-H), 2.79~3.03 (m, 2H, 17a+17b-H), 3.19~3.13 (m, 1H, 1'-H), 3.40~3.34 (m, 1H, 1'-H), 3.26 (3.25), 3.30 (3.89), 3.43 (3.42), 3.60 (3.58), 3.71 (3.70) (each s, each 3H, CH3 +OCH3 ), 3.56 (q, J =7.6 Hz, 2H, 8a-H), 4.42~4.49 (m, 2H, 2'-H), 4.47 (4.43) (dd, J =9.2, 1.7 Hz, 1H, 17-H), 4.54 (4.60) (q, J =7.4 Hz, 1H, 18-H), 5.55 (5.45) (dd, J =10.0, 3.2 Hz, 1H, 132 -H), 6.21 (dd, J =11.5, 1.2 Hz, 1H, cis -3b-H), 6.31 (dd, J =17.8, 1.2 Hz, 1H, trans -3b-H), 8.01 (dd, J =17.8, 11.5 Hz, 1H, 3a-H), 8.60, 9.41 (9.39), 9.57 (9.55) (each s, each 1H, meso -H); IR (KBr) ν : 3445 (N—H), 2951 (C—H), 1729 (C=O), 1650 (C=C), 1560 (chlorin skeleton), 1438, 1255, 1082, 766 cm-1 ; MS (ESI) m /z : 660.3 (M+H+ ). Anal. calcd for C39 H41 N5 O5 : C 71.00, H 6.26, N 10.61; found C 71.18, H 6.47, N 10.87.
3.8
5'-氰基-6'-甲氧基吡啶并[2', 3'-n]焦脱镁叶绿酸-a甲酯(11)的合成
将122 mg (0.185 mmol)二氢卟吩10 溶解于20 mL冰乙酸中, 再加入760 mg醋酸铵, 120 ℃搅拌反应2 h, 加入20 mL水和40 mL二氯甲烷分层, 分出有机层并水洗两次, 无水硫酸钠干燥, 减压除尽溶剂, 用5 mL二氯甲烷重新溶解剩余物, 加入5 mL自制的重氮甲烷乙醚溶液, 搅拌反应2 min后迅速用乙酸猝灭反应, 除去溶剂后用硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=3:1], 得到64 mg (0.100 mmol)绿色固体11 .产率54%. m.p. 214~217 ℃; UV-Vis (CH2 Cl2 ) λ max [ε /(L• mol-1 •cm-1 )]: 435 (1.35×104 ), 512 (7.54×103 ), 613 (1.01×104 ), 709 (5.01×102 ), 757 (9.34×102 ) nm; 1 H NMR (CDCl3 ) δ : 0.20 (br s, 1H, NH), 0.60 (br s, 1H, NH), 1.58 (t, J =7.6 Hz, 3H, 8a-CH3 ), 1.73 (d, J =7.2 Hz, 3H, 18-CH3 ), 1.82~2.10 (m, 1H, 17a+17b-H), 2.18~2.35 (m, 1H, 17a+17b-H), 2.47~2.67 (m, 2H, 17a+17b-H), 3.03, 3.21, 3.35, 3.74, 4.22 (each s, each 3H, CH3 +OCH3 ), 3.46 (q, J =6.4 Hz, 2H, 8b-H), 4.23 (q, J =7.2 Hz, 1H, 18-H), 4.32 (d, J =10.8 Hz, 1H, 17-H), 6.09 (d, J =11.6 Hz, 1H, cis -3b-H), 6.18 (d, J =18.0 Hz, 1H, trans -3b-H), 7.80 (dd, J =17.7, 11.6 Hz, 3a-H), 8.11 (s, 1H, 4'-H), 8.19, 8.92, 8.98 (each s, each 1H, meso -H); IR (KBr) ν : 3342 (N—H), 2917 (C—H), 1733 (C=O), 1603 (C=C), 1497 (chlorin skeleton), 1438, 1365, 1196, 981 cm-1 ; MS (ESI) m /z : 639.4 (M+H+ ). Anal. calcd for C39 H38 N6 O3 : C 73.33, H 6.00, N 13.16; found C 73.19, H 6.17, N 13.24.
3.9
3'-氧代-1', 2', 4'-三唑并[1, 5-o ]红紫素-18-133 -亚胺-132 -酰亚胺甲酯(12)的合成
将148 mg (0.263 mmol)化合物7 溶解于5 mL氢氧化钾饱和的甲醇溶液, 室温下开口剧烈搅拌25 min, 用乙酸将pH值调节为2, 再加入25 mL水和30 mL二氯甲烷, 分出有机层并水洗两次, 无水硫酸钠干燥后加入5 mL自制的重氮甲烷乙醚溶液, 搅拌反应2 min后迅速用乙酸猝灭反应, 减压除尽溶剂.将剩余物在溶解于15 mL乙酸酐中并加入54 mg (0.720 mmol)盐酸氨基脲, 140 ℃搅拌反应10 h, 减压除尽乙酸酐后再用20 mL二氯甲烷溶解, 先后用水、饱和碳酸钠和水洗涤, 除去溶剂, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1], 得到20 mg (0.032 mmol)黑绿色固体12 , 产率12%. m.p. 195~198 ℃; UV-Vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 369 (7.83×103 ), 424 (1.09×104 ), 514 (2.07×103 ), 557 (2.94×103 ), 716 (4.36×103 ) nm; 1 H NMR (CDCl3 ) δ : -1.20 (br s, 1H, NH), 0.40 (br s, 1H, NH), 1.61 (d, J =7.0 Hz, 3H, 18-CH3 ), 1.63 (t, J =7.6 Hz, 3H, 8-CH3 ), 2.02~2.34 (m, 2H, 17a+17b-H), 2.38~2.50 (m, 2H, 17a+17b-H), 3.57 (q, J =7.6 Hz, 2H, 8a-H), 3.10, 3.20, 3.29, 3.54 (each s, each 3H, 18H, CH3 +OCH3 ), 4.09 (q, J =7.2 Hz, 1H, 18-H), 4.97 (d, J =8.0 Hz, 1H, 17-H), 6.04 (d, J =11.6 Hz, 1H, cis -3b-H), 6.13 (d, J =17.6 Hz, 1H, trans -3b-H), 7.81 (dd, J =17.6, 11.6 Hz, 1H, 3a-H), 8.27, 9.14, 9.35 (each s, each 1H, meso -H); IR (KBr) ν : 3450 (N—H), 2924 (C—H), 1742~1708 (C=O), 1686 (C=C), 1559 (chlorin skeleton), 1459, 1342, 1806, 1039, 969 cm-1 ; MS (ESI) m /z : 618.4 (M+H+ ). Anal. calcd for C35 H35 N7 O4 : C 68.06, H 5.71, N 15.87; found C 68.16, H 5.53, N 15.72.
3.10
131 -氮杂-132 , 133 -二氧代罗丁甲酯(15)的合成
将138 mg (0.252 mmol)化合物1a 溶解于20 mL乙醇中, 然后分别加入2 mL溶有45 mg盐酸羟胺的水和2 mL溶有100 mg氢氧化钠的水, 95 ℃下搅拌反应3 h, 用2%的盐酸溶液调节pH值为3后加入20 mL二氯甲烷, 分出有机层, 水层用二氯甲烷萃取至无色, 合并有机相, 减压浓缩至干, 将所得固体溶解于20 mL含有5%硫酸的甲醇溶液室温搅拌过夜, 将反应混合物倒入3 mL冰水中, 用二氯甲烷萃取至无色, 将有机层合并, 无水硫酸钠干燥后除尽溶剂, 剩余物用硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=4:1]得118 mg (0.209 mmol)绿色混合物13 , 产率83%.取113 mg (0.613 mmol) 2, 4, 6-三氯三嗪溶解于2 mL DMF中, 再用5 mL DMF溶解所得化合物13 , 然后将二者合并, 室温搅拌反应4 h, 先后加入25 mL水和25 mL二氯甲烷, 分出有机层并水洗两次, 无水硫酸钠干燥, 减压浓缩, 剩余物经硅胶柱层析分离[洗脱剂: V (石油醚):V (乙酸乙酯)=5:1], 得到67 mg (0.119 mmol)绿色固体14 , 产率57%, 其物理常数与分析数据与文献[13c 15 , 产率16%.15 : m.p. 211~213 ℃; UV-Vis (CH2 Cl2 ) λ max [ε /(L•mol-1 •cm-1 )]: 410 (1.0, 9.46×104 ), 509 (0.07, 1.14×104 ), 5.44 (0.1, 4.80×103 ), 638 (4.80×103 ), 673 (0.41, 3.87×104 ) nm; 1 H NMR (CDCl3 ) δ : -1.46 (br s, 1H, NH), 0.47 (br s, 1H, NH), 1.72 (t, J =7.6 Hz, 3H, 8-CH3 ), 2.46 (d, J =7.2 Hz, 3H, 18-CH3 ), 1.97~2.10 (m, 2H, 17a+17b-H), 2.22~2.33 (m, 1H, 17a+17b-H), 2.73~2.84 (m, 1H, 17a+17b-H), 2.93, 3.30, 3.51, 3.63 (each s, each 3H, CH3 +OCH3 ), 3.74 (q, J =7.6 Hz, 2H, 8a-H), 4.43 (q, J =7.2 Hz, 1H, 18-H), 5.57 (dd, J =9.1, 2.7 Hz, 1H, 17-H), 6.21 (dd, J =11.5, 1.2 Hz, 1H, cis -3b-H), 6.36 (dd, J =17.8, 1.2 Hz, 1H, trans - 3b-H), 8.09 (dd, J =17.8, 11.5 Hz, 1H, 3a-H), 8.09, 9.56, 9.58 (each s, each 1H, meso -H), 9.60 (s, 1H, CHO); IR (KBr) ν : 3167 (N—H), 2966 (C—H), 1740~1704 (C=O), 1672 (C=C), 1542 (chlorin skeleton), 1508, 1401, 1223, 1021, 980 cm-1 ; MS (ESI) m /z : 578.3 (M+H+ ). Anal. calcd for C34 H35 N5 O4 : C 70.69, H 6.11, N 12.12; found C 70.80, H 6.07, N 12.26.
辅助材料(Supporting Information) 所得新化合物的1 H NMR图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/ )上下载.


 下载:
下载:





 下载:
下载:







