图式 1.
支链型氟表面活性剂的合成路线
Scheme 1.
Synthetic route of the branched fluorosurfactants

表面活性剂是指一种活跃在表面和界面上, 具有极高的降低表、界面张力能力和效率的一种物质, 在一定浓度以上的溶液中能形成分子有序组合体, 从而具有一系列的应用功能.表面活性剂分子通常由分布在分子两端的长链亲油基团与极性的亲水基团共同组成.表面活性剂这种特殊的“两亲”特性使其表现出发泡、乳化、分散、抗菌、润湿、增溶、洗涤等特性.碳氢表面活性剂碳氢链上的氢原子全部或部分被氟原子取代, 形成以碳氟链为疏水基的表面活性剂叫氟碳表面活性剂.氟碳表面活性剂具有比碳氢表面活性剂更高的表面活性之外, 还具有高化学稳定性和热稳定性.
氟碳表面活性剂由于其优异的性能, 在消防、生物医学、日化、纺织等领域均有应用[1~4], 其中, 最为知名、应用最广的主要为不同疏水链长的全氟辛烷磺酸(PFOS, Perfluorooctane sulfonte)、不同疏水链长的全氟辛酸(PFOA, Perfluorooctanoic acid)以及它们的衍生物等[5].然而, 由于长链氟碳烷基的超强稳定性, PFOS/ PFOA类氟碳表面活性剂具有生物毒性、生物累积性、环境迁移性, 已被斯德哥尔摩公约列为持久有机污染物(POPs, Persistent organic pollutants)[6, 7]. 2006年12月12日欧洲议会通过决议, 正式发布了对PFOS的禁令, 即《关于限制全氟辛烷磺酸使用及销售的指令》(2006/122/ EEC)[8].美国环境保护署(EPA)颁布了2010/2015 PFOA Stewardship计划, 已全面消除使用含有PFOA及高同系物全氟羧酸的产品[9].自此, 世界各国均已紧张地开展了PFOS/PFOA替代品的研制工作.目前PFOS/PFOA替代品的研发思路主要分为四类:降低全氟链的长度、引入弱位点、构造新型氟碳表面活性剂结构(双子或杂双子结构)、氟碳链支链化.
现行法规认为碳氟链小于7个碳的全氟烷基羧酸类(PFCAs, Perfluoroalkylcarboxylic acids)和小于6个碳的全氟烷基磺酸盐类(PFSAs, Perfluoroalkane sulfonates)不具生物累积性, 且短碳氟链的PFCA和PFSA的毒性也低很多[10], 基于C6基的含氟助剂不含有C8成分, 理论上目前属于非POPs产品.但是其环境安全性还存在着较大的争议, 具有类似化学结构的C6同样具有超乎寻常的难分解性, 与长链类似物相比, 具有较高的水溶性、更高的饱和蒸气压、低吸附性的短链替代物更易在环境中迁移[11, 12].尽管如此, 降低氟碳链长度仍是PFOS/PFOA替代品的一个研发方向.
在氟碳直链中引入醚键、亚甲基或类似基团对氟碳链结构进行改性, 也是PFOA替代品研发的一个重要方向.醚键或亚甲基基团在碳氟链上的存在, 丰富了碳氟链的结构, 且与碳氟链刚性结构不同, 增加分子的柔顺性同时具有了潜在可降解性.此类氟碳结构通常以偏氟乙烯(VDF)和3, 3, 3-三氟丙烯(TFP)为原料进行调聚得到[13, 14].氟烯烃调聚法通常会产生不同链长的混合物, 需要严格控制反应条件.
双子(Gemini)与杂双子(Hetero-gemini)表面活性剂是指含有两个疏水基团(有时三个)和两个亲水基团, 在靠近亲水基团处通过连接基连接得到的表面活性剂, 在20世纪90年代分别由Zana[15]和Jaeger等[16]首次合成.由于双子表面活性剂分子中两个亲水头基是靠化学键连接的, 烷基链间容易产生更强的疏水相互作用, 亲水头基间的斥力由于化学键的作用而大大减弱, 能够更加紧密地排列.此类表面活性剂在水介质中的界面性质比传统表面活性剂要高一至几个数量级.近年来, 采用“短链+多链”的双子策略进行PFOS/PFOA替代品研究引起了广泛兴趣[17~19].但是, 该类结构通常在制备过程中合成步骤多且复杂, 离应用尚有一定距离.
研究表明, 在普通的碳氢表面活性剂上构造超支化的结构后, 碳氢表面活性剂会具有接近氟碳表面活性剂的低表面能[20].此外, Dmowski等[21]合成了结构为CF3CF2CF2C(CF3)2CH2CH2COONa的氟碳表面活性剂, 并将其表面活性与全氟辛酸钠进行比较, 结果发现, CF3CF2CF2C(CF3)2CH2CH2COONa的表面活性优于全氟辛酸钠.因此, 氟碳链中引入支链是合成PFOA替代品的有效策略.支链化氟碳链的原料主要是六氟丙烯通过齐聚法得到的低聚物, 以二聚体和三聚体为主.六氟丙烯是仅次于四氟乙烯产量的简单氟化工原料, 齐聚法反应条件相对温和, 适于工业生产.
从以上对比可以发现, 以六氟丙烯齐聚物(HFPD)为原料制备氟碳表面活性剂, 产物表面活性高, 对环境危害性低, 原料便宜易得, 是PFOS/PFOA替代品研发的一个十分有意义的方向.针对目前PFOS/PFOA替代品研发现状, 我们课题组以HFPD为原料, 基于氟烯烃双键反应活性位点合成了一系列性能优于全氟辛酸钠的支链型氟碳表面活性剂[22~25].但是目前已合成的化合物结构仍较为单一, 如疏水尾端与亲水头基之间的连接基均含有苯环、同一种亲水基团链长与大小均相同.因此, 本文在前期工作基础上, 继续基于氟烯烃双键反应活性位点, 设计、合成了一系列连接基团为烷基链的氟碳表面活性剂.通过对比连接基团长短、亲水基团种类和亲水基团大小对表面活性的影响, 进一步阐述该类氟表结构和性能之间的关系, 以期为今后氟表面活性剂的开发和应用等研究提供一定的实验和理论支撑.
通常, 烯烃易于发生亲电加成反应, 而由于氟原子的强吸电性, 全氟烯烃却更易发生亲核加成反应.以全氟-2-甲基-2-戊烯的碳碳双键为活性位点进行氟碳表面活性剂的合成时, 其反应的第一步首先是碳碳双键受到阴离子进攻形成碳负离子, 其可与活泼卤代物进行SN2取代反应, 形成碳碳键(Scheme 1).本文选取卤代物为溴乙酸乙酯, 分别通过亲核取代反应与酯交换反应生成含氟中间体2, 进而经季铵化反应得到化合物3与4, 化合物4经强碱型离子交换树脂水解生成内盐5.双氧水氧化中间体2可得到化合物6.为了进一步探讨化合物结构中酰胺键上的羰基与亲水基团的大小对表面活性的影响, 将中间体1还原为醇, 醇羟基转化为易于离去的三氟甲磺酸酯, 再与胺发生取代反应, 然后经碘乙烷季铵化得到化合物10.由于化合物1未能与N-甲基哌嗪发生酯交换, 因此先将其转化为酰氯再与胺反应, 最后经季铵化得到化合物13.
表面张力由Krüss K100表面张力仪, 用Wilhemy板法进行测定.测定时先将Scheme 1中合成的氟表面活性剂配制为不同浓度的溶液, 按照从低浓度到高浓度的顺序进行测试, 每种浓度均测定三次后取平均值, 测定时溶液的温度为25 ℃.将测得的表面张力值作为纵坐标, 取浓度的常用对数函数值作为横坐标, 得到表面张力随浓度变化的曲线.通过Origin软件分别对接近临界胶束浓度之前及之后的曲线进行线性拟合, 读取两条拟合直线的交点确定不同化合物的临界胶束浓度(CMC)与该浓度时的表面张力值(γCMC), 结果如表 1所示.
 下载:
导出CSV
下载:
导出CSV
| Compd. | CMC/(mol•L-1) | γCMC (mN•m-1) |
| 3a | 2.52×10-2 | 22.91 |
| 3b | 8.50×10-3 | 33.73 |
| 3c | 1.64×10-2 | 27.09 |
| 5a | 1.41×10-2 | 21.50 |
| 5b | 1.21×10-2 | 24.88 |
| 6a | 1.73×10-2 | 19.93 |
| 6b | 1.11×10-2 | 20.11 |
| 10 | 9.89×10-4 | 36.32 |
| 13 | 1.08×10-2 | 29.08 |
| M1 | 1.89×10-4 | 20.24 |
| M2 | 4.28×10-5 | 23.79 |
| M3 | 1.74×10-5 | 24.86 |
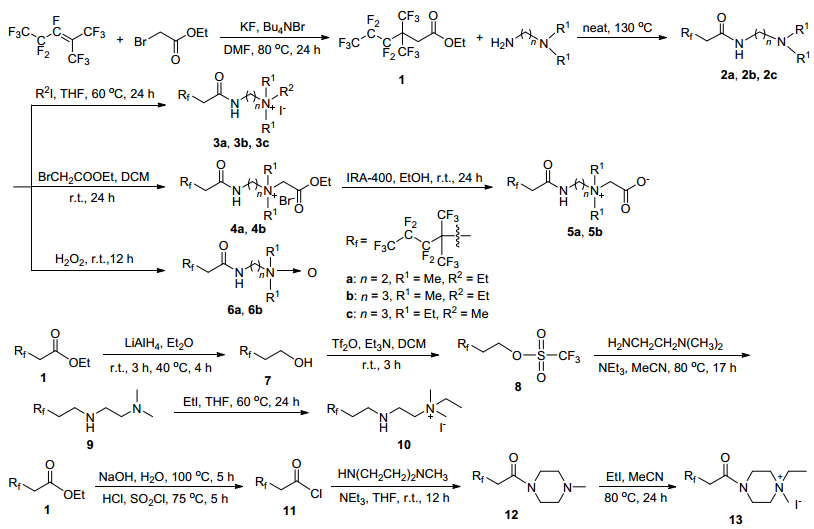
具有不同亲水基团的支链氟碳表面活性剂的表面活性对比结果如图 1所示.从图 1A可以看出, 所合成的支链型氟碳表面活性剂均表现出优异的表面活性, 氧化铵两性型氟碳表面活性剂具有最高的降低表面张力的能力, 甜菜碱两性型氟碳表面活性剂次之, 这两种两性型表面活性剂降低表面张力的效率也要高于季铵盐阳离子表面活性剂. 图 1B为在连接基团加上一个亚甲基后表面张力随浓度变化的曲线, 从中可以看出, 三种类型氟碳表面活性剂降低表面张力的能力与图 2A中所示一致, 且在连接基团增加一个亚甲基后, 其降低表面张力的效率均有所提高, 此时季铵盐阳离子表面活性剂降低表面张力的效率最好.化合物3a、5a、6a和6b的表面活性均要高于全氟辛酸钠(临界胶束浓度为3.1×10-2 mol/L, 临界胶束浓度下的表面张力值为24.7 mN/m[23]).
(A) 2-Carbon atoms in linkage; (B) 3-carbon atoms in linkage
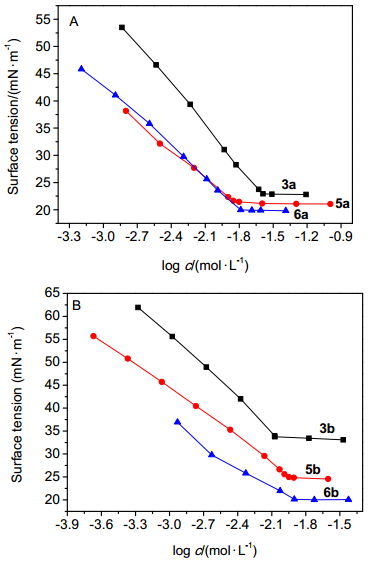
化合物3b、3c和13均为季铵盐阳离子型氟碳表面活性剂, 亲水基团的体积依次变大.化合物13的亲水基含有六元环, 刚性要比化合物3b、3c强, 它们的表面张力随浓度变化的曲线如图 2所示.从图中可以看出, 当该种支链氟碳表面活性剂的亲水基团为季铵盐时, 其降低表面张力的能力随着亲水基团体积的增大先提高再降低, 而其降低表面张力的效率是先降低再升高.由于亲水基团的大小与刚性强弱改变了气液界面及溶液内部分子间的相互作用力, 表面活性相应发生变化, 因此改变亲水基团大小与刚性可以作为调节该类支链型氟碳表面活性剂表面活性的一种有效策略.
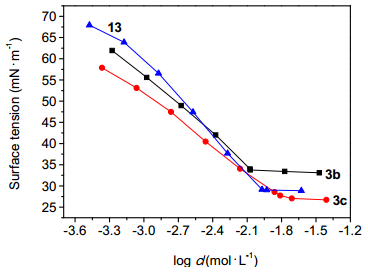
当亲水基团为氧化铵时(图 3A), 增加一个亚甲基后, 临界胶束从1.73×10-2 mol/L降低至1.11×10-2 mol/L, 而临界胶束浓度时的表面张力从19.93 mN/m增加至20.11 mN/m, 表面活性基本无变化.当亲水基团为甜菜碱时(图 3B), 化合物5a和5b的表面活性变化较为明显, 因此, 连接基长短对表面活性的影响因亲水基团种类而异.此外, 我们课题组在前期研究中发现, 当连接基团增加苯环, 刚性变强后, 表面张力和临界胶束浓度均有明显下降[25].因此, 改变连接基团长短与刚性均可以作为调节该类氟碳表面活性剂表面活性的一种手段.
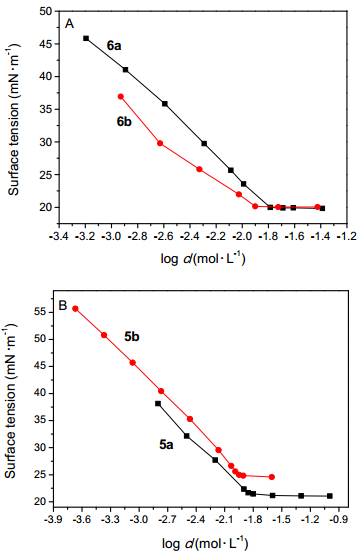
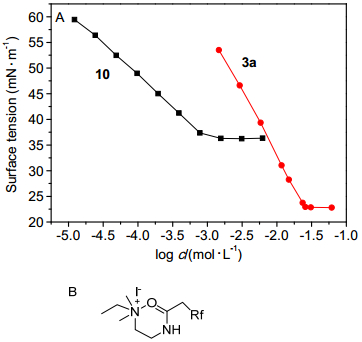
为了比较分子中的极性基团对表面活性的影响, 我们设计合成了化合物10, 化合物10比化合物3a少一个羰基, 其余的结构均相同, 均为季铵盐阳离子型氟碳表面活性剂. 图 4A给出了化合物3a和10的表面张力随浓度的变化曲线, 从图 4A中可以看出, 两者表面活性相差很大, 化合物3a的临界胶束浓度较高, 而临界胶束浓度时的表面张力更低.羰基的存在与否使得两个化合物在水溶液内部的聚集与气液界面上的排布均发生明显变化. Stähler等[26]在对一类可聚合表面活性剂的表面性能进行研究时指出, 表面活性剂中的羰基电子云密度较大, 具有供电子效应, 而季铵盐阳离子型表面活性剂中正电荷的氮原子具有吸电子效应, 因此会产生分子内相互作用, 可能形成如图 4B所示的六元环结构.此时, 化合物3a分子的刚性更强, 分子在气液界面上吸附时, 疏水尾端摆动减小, 即吸附更为牢固, 因此降低表面张力的能力更强.同时, 羰基的存在使得分子的亲水性更高, 溶解度较大, 因此降低表面张力的效率降低.由此可以看出, 极性基团修饰也是调节该类结构氟碳表面活性剂表面性能十分重要的策略.
工业应用中, 通常将氟碳表面活性剂与碳氢表面活性剂进行复配后使用, 一方面可以降低应用成本, 另一方面通过复配可以赋予体系具有单独氟碳表面活性剂所不具有的其它性能.本文所合成的化合物中, 化合物6a表面活性优异, 具有一定的应用前景, 故选用该化合物进行复配应用.碳氢表面活性剂的种类繁多, 其中烷基糖苷(Alkyl Polyglycoside, 简称APG)是单苷、二苷、三苷和多苷的混合物, 是新型高效、无毒、可生物降解的非离子表面活性剂, 具有十分优异的性能, 对人体刺激性小, 能迅速生物降解, 因此, 选择APG为碳氢表面活性剂进行复配.将化合物6a与APG分别按质量比为1:1、1:5、1:10进行复配, 逐渐稀释浓度, 静置过夜后, 测量不同浓度下的表面张力, 复配体系的表面张力随浓度变化的曲线如图 5所示.由图 5可知, 随着总浓度的逐渐增大, 水溶液的表面张力值迅速降低, 继续增大浓度到一定值后, 表面张力值趋于稳定, 直至达到最低表面张力时, 表面张力不再随浓度的增加而发生变化.
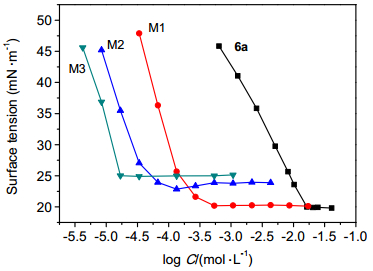
6a: m(6a):m(APG)=1:0; M1: m(6a):m(APG)=1:1; M2: m(6a):m(APG)=1:5; M3: m(6a):m(APG)=1:10
化合物6a与APG0810按不同质量比复配后, 混合体系的临界胶束浓度比单独化合物6a的水溶液的临界胶束浓度小很多, 不同体系浓度低于临界胶束浓度时, 表面张力降低的斜率基本不变, 表明复配体系降低表面张力的效率与单独化合物6a体系相当.随着m(APG0810):m(6a)增大, 临界胶束浓度逐渐降低, 而临界胶束浓度时的表面张力逐渐增大.当m(APG0810):m(6a)=1:1时, 其最低表面张力为20.24 mN/m, 和单一化合物6a水溶液的最低表面张力非常接近, 临界胶束浓度却为单独体系的10-2左右.意味着化合物6a与APG复配后, 可以在基本不改变表面张力的情况下, 化合物6a使用量降低约100倍.
本文以全氟-2-甲基-2-戊烯为原料, 基于其烯烃双键活性位点设计、合成了阳离子和两性支链型氟碳表面活性剂, 对其表面张力、临界胶束浓度进行了测试.通过合理的设计结构, 较为系统地讨论了该支链型氟碳表面活性剂的亲水基团种类、亲水基团大小、连接基团长短、结构中的酰胺键对表面活性的影响及化合物6a与APG碳氢表面活性剂的复配性能.
结果表明亲水基团为氧化铵时, 该类氟碳表面活性剂表面活性最高.亲水基团大小与连接基团长短均对表面活性有显著的影响, 当亲水基团与连接基团的刚性变强时, 更有利于表面活性的提高, 同理, 极性基团的修饰也会增强分子内部的相互作用, 提高分子的刚性, 进而提高表面活性.基于此, 在进行新型氟碳表面活性剂的设计、合成中, 应当着重考虑如何提高分子的刚性, 从而使得分子在水溶液内部以及气液界面上更为紧密、稳定, 从而表现出更好的性能.此外, 化合物3a、5a、6a和6b的表面活性要高于全氟辛酸钠, 均为潜在的替代物.化合物6a的复配性能优异, 十分具有应用价值.
核磁共振氢谱和氟谱由Varian Mercury-vx300测定, 核磁共振碳谱由Varian Mercury-400测定, 红外光谱由Nicolet 380FT-IR测定. EI低分辨质谱由Agilent 5973N质谱仪测定, EI高分辨质谱由Waters Micromass GTC-CA176质谱仪测定, ESI低分辨由Agilent 1200质谱仪测定, ESI高分辨由Bruker APeXIII 7.0 TESLAEI质谱仪测定.熔点由SGW-X4显微熔点仪测定.全氟-2-甲基-2-戊烯购于宿迁凯尔氟有限公司, 纯度为99%.其他所有试剂均为分析纯, 使用前未经进一步纯化, 需要除水的溶剂按照标准操作除水后加入4 Å分子筛干燥备用.
300 mL封管中加入KF (7.08 g, 0.12 mol, 1.2 equiv.)、全氟-2-甲基-2-戊烯(30.00 g, 0.10 mol, 1.0 equiv.)、溴乙酸乙酯(18.37 g, 0.11 mol, 1.1 equiv.)、四丁基溴化铵(0.50 g, 1.5 mmol, 0.015 equiv.)和100 mL无水N, N-二甲基甲酰胺(DMF), 密封后置于80 ℃油浴锅中搅拌反应24 h.冷却至室温后将反应体系倒入乙醚中, 乙醚液依次用去离子水和饱和食盐水洗涤, 再经无水硫酸钠干燥, 过滤, 旋干溶剂.柱层析[V(乙酸乙酯):V(石油醚)=1:20]分离提纯, 得无色液体产物1[27] 31.6 g, 产率为78%. 1H NMR (300 MHz, CDCl3)δ: 4.30~4.06 (m, 2H), 3.12 (s, 2H), 1.24 (t, J=7.1 Hz, 3H); 19F NMR (282 MHz, CDCl3) δ: -63.24 (p, J=10.8 Hz, 6F), -80.64 (t, J=13.3 Hz, 3F), -106.99~-108.13 (m, 2F), -123.09 (tt, J=21.2, 10.5 Hz, 2F); LRMS (EI) m/z (%): 361.1 (100), 406.1 (2.1).
25 mL封管中加入化合物1 (2.03 g, 5.0 mmol, 1.0 equiv.)和5 mL N, N-二甲基乙二胺(N, N-二甲基丙二胺或N, N-二乙基丙二胺), 密封后置于130 ℃油浴锅搅拌反应34 h.冷却至室温, 旋干浓缩, 柱层析[V(CH2CH2):V(MeOH):V(Et3N)=20:1:trace]分离提纯.
N, N-二甲基胺乙基-2-(全氟-1, 1-二甲基-丁基)乙酰胺(2a):无色油状物, 收率67%. 1H NMR (300 MHz, CDCl3) δ: 7.10 (s, 1H), 3.36 (dd, J=10.9, 5.2 Hz, 2H), 3.00 (s, 2H), 2.51 (t, J=5.7 Hz, 2H), 2.29 (s, 6H); 19F NMR (282 MHz, CDCl3)δ: -63.51 (p, J=11.0 Hz, 6F), -80.39~-80.64 (m, 3F), -107.61~-108.36 (m, 2F), -123.51 (dd, J=20.9, 10.4 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 163.5, 57.3, 44.6, 36.8, 33.4; IR (KBr) v: 3322, 1671, 1559, 1340, 1220, 1111, 731, 702 cm-1; LRMS (ESI) m/z: 449.0 ([M+H]+); HRMS (ESI) calcd for C12H14ON2F13[M+H]+ 449.0893, found 449.0895.
N, N-二甲基胺丙基-2-(全氟-1, 1-二甲基-丁基)乙酰胺(2b):无色油状物, 收率66%. 1H NMR (300 MHz, CDCl3) δ: 8.13 (s, 1H), 3.35 (dd, J=11.2, 5.3 Hz, 2H), 2.92 (s, 2H), 2.47~2.34 (m, 2H), 2.22 (s, 6H), 1.64 (dt, J=11.8, 5.9 Hz, 2H); 19F NMR (282 MHz, CDCl3) δ: -63.56 (dt, J=23.0, 11.5 Hz, 6F), -80.46 (t, J=14.0 Hz, 3F), -105.42~-110.30 (m, 2F), -123.71 (ddd, J=127.7, 69.2, 58.3 Hz, 2F); 13C NMR (101 MHz, CDCl3)δ: 163.0, 59.3, 45.3, 41.0, 33.8, 24.7; IR (KBr) v: 3319, 1671, 1559, 1340, 1220, 1112, 731, 702 cm-1; LRMS (ESI) m/z: 463.1 ([M+H]+); HRMS (ESI) calcd for C13H16ON2F13[M+H]+ 463.1050, found 463.1047.
N, N-二乙基胺丙基-2-(全氟-1, 1-二甲基-丁基)乙酰胺(2c):无色油状物, 收率68%. 1H NMR (300 MHz, CDCl3) δ: 8.30 (s, 1H), 3.35 (dd, J=10.8, 5.1 Hz, 1H), 2.91 (s, 2H), 2.73~2.38 (m, 6H), 1.76~1.38 (m, 2H), 1.02 (t, J=7.2 Hz, 6H); 19F NMR (282 MHz, CDCl3) δ:-63.40 (p, J=11.4 Hz, 6F), -80.47 (t, J=13.4 Hz, 3F), -107.68~-108.02 (m, 2F), -123.18~-123.46 (m, 2F); 13C NMR (101 MHz, CDCl3) δ: 162.9, 53.0, 46.5, 40.9, 33.7, 24.6, 11.2; IR (KBr) v: 3320, 1670, 1559, 1340, 1220, 1111, 731, 702 cm-1; LRMS (ESI) m/z: 491.1 ([M+H]+); HRMS (ESI) calcd for C15H20ON2F13[M+H]+ 491.1363; found 491.1356.
30 mL封管中加入2.0 mmol化合物2a~2c, 氩气置换三次, 加入10 mL无水四氢呋喃, 5 mmol碘乙烷(2a和2b反应体系)或碘甲烷(2c反应体系). 60 ℃搅拌反应24 h, 冷却至室温后过滤, 固体用乙酸乙酯洗涤, 得目标化合物.
N, N-二甲基-N-乙基-2-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]乙基碘化铵(3a):白色固体, 收率72%. m.p. 213~216 ℃; 1H NMR (300 MHz, CD3OD) δ: 3.64 (t, J=7.1 Hz, 2H), 3.47 (q, J=7.3 Hz, 2H), 3.38 (t, 2H), 3.25 (s, 2H), 3.13 (s, 6H), 1.40 (t, J=7.3 Hz, 3H); 19F NMR (282 MHz, CD3OD) δ: -67.37~-68.45 (m, 6F), -85.14~-85.99 (m, 3F), -111.28~-112.85 (m, 2F), -127.20~-127.88 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.3, 61.9, 61.4, 51.3, 34.6, 33.5, 8.6; IR (KBr) v: 3323, 2997, 1664, 1559, 1264, 1210, 986, 734 cm-1; LRMS (ESI) m/z: 477.1 ([M-I]+); HRMS (ESI) calcd for C14H18ON2F13 [M-I]+ 477.1206; found 477.1198.
N, N-二甲基-N-乙基-3-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]丙基碘化铵(3b):白色固体, 收率76%. m.p. 196~199 ℃; 1H NMR (300 MHz, CD3OD)δ: 3.49~3.30 (m, 6H), 3.24 (s, 2H), 3.08 (s, 6H), 2.11~1.82 (m, 2H), 1.37 (t, J=7.0 Hz, 3H); 19F NMR (282 MHz, CD3OD) δ: -64.32~-64.56 (m, 6F), -82.15 (t, J=13.5 Hz, 3F), -108.29~-108.71 (m, 2F), -123.88~-124.19 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.3, 62.6, 61.1, 50.9, 37.6, 33.6, 23.7, 8.4; IR (KBr) v: 3335, 3006, 1671, 1557, 1258, 1210, 733, 701 cm-1; LRMS (ESI) m/z: 491.2 ([M-I]+); HRMS (ESI) calcd for C15H20ON2F13[M-I]+ 491.1363, found 491.1359.
N-甲基-N, N-二乙基-3-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]丙基碘化铵(3c): 为白色固体, 收率78%. m.p. 175~177 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.61 (s, 1H), 3.35 (dd, J=11.9, 5.1 Hz, 2H), 3.31~3.11 (m, 8H), 3.08 (s, 3H), 2.15~1.96 (m, 2H), 1.28 (t, J=7.3 Hz, 6H); 19F NMR (376 MHz, CDCl3) δ: -62.86 (p, J=11.2 Hz, 6F), -79.06~-80.66 (m, 3F), -107.32 (dt, J=23.7, 11.8 Hz, 2F), -121.97~-123.54 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.3, 59.7, 57.8, 47.9, 37.7, 33.7, 23.4, 8.1; IR (KBr) v: 3343, 3004, 1676, 1552, 1257, 1208, 989, 733 cm-1; LRMS (ESI) m/z: 505.1 ([M-I]+); HRMS (ESI) calcd for C16H22ON2F13[M-I]+ 505.1519, found 505.1513.
50 mL圆底烧瓶中加入1.0 mmol化合物2a或2b, 0.26 mL溴乙酸乙酯(2.3 mmol, 2.3 equiv)和10 mL二氯甲烷, 室温下搅拌24 h, 直接旋干溶剂, 甲醇与乙酸乙酯体系重结晶得目标化合物.
N, N-二甲基-N-乙氧羰甲基-2-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]乙基溴化铵(4a):白色固体, 收率83%. m.p. 79~81 ℃; 1H NMR (300 MHz, CD3OD)δ: 4.44 (s, 2H), 4.31 (dd, J=14.0, 7.2 Hz, 2H), 3.78~3.53 (m, 4H), 3.35 (s, 6H), 3.23 (s, 2H), 1.33 (t, J=6.9 Hz, 3H); 19F NMR (282 MHz, CD3OD) δ: -64.37~-64.68 (m, 6F), -82.14 (t, J=13.6 Hz, 3F), -108.31~-108.84 (m, 2F), -123.98~-124.35 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.4, 165.7, 63.7, 63.6, 62.6, 52.7, 34.6, 33.3, 14.2; IR (KBr) v: 3326, 3207, 1750, 1686, 1276, 1219, 734, 659 cm-1; LRMS (ESI) m/z : 535.1 ([M-Br]+); HRMS (ESI) calcd for C16H20O3N2F13[M-Br]+ 535.1261, found 535.1273.
N, N-二甲基-N-乙氧羰甲基-3-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]丙基溴化铵(4b):白色固体, 收率84%. m.p. 88~90 ℃; 1H NMR (300 MHz, CD3OD)δ: 4.50~4.11 (m, 4H), 3.72~3.53 (m, 2H), 3.36~3.30 (m, 8H), 3.24 (s, 2H), 2.15~1.88 (m, 2H), 1.34 (t, J=7.1 Hz, 3H); 19F NMR (282 MHz, CD3OD) δ: -64.14~-64.67 (m, 6F), -82.13 (t, J=13.6 Hz, 3F), -108.29~-108.72 (m, 2F), -123.77~-124.24 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.3, 165.8, 64.5, 63.7, 62.2, 52.4, 37.5, 33.61, 23.8, 14.2; IR (KBr) v: 3479, 3285, 1755, 1686, 1265, 1208, 1110, 733 cm-1; LRMS (ESI) m/z: 549.1 ([M-Br]+); HRMS (ESI) calcd for C17H22O3N2F13[M-Br]+ 549.1417, found 549.1415.
50 mL圆底烧瓶中加入1.0 mmol化合物4a或4b, 10.0 g IRA-400强碱型离子交换树脂和15 mL无水乙醇, 室温下搅拌24 h.反应体系过滤, 收集滤液, 旋干, 丙酮和二氯甲烷体系重结晶得目标化合物.
N, N-二甲基-N-羧甲基-2-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]乙基铵内盐(5a):白色固体, 收率79%. m.p. 89~91 ℃; 1H NMR (300 MHz, CD3OD) δ: 3.86 (s, 2H), 3.81~3.68 (m, 2H), 3.68~3.57 (m, 2H), 3.28 (s, 6H), 3.22 (s, 2H); 19F NMR (282 MHz, CD3OD) δ: -64.49 (p, J=11.1 Hz, 6F), -82.17 (t, J=13.6 Hz, 3F), -108.65 (s, 2F), -124.19 (d, J=10.9 Hz, 2F); 13C NMR (101 MHz, CD3OD) δ: 168.5, 166.3, 65.2, 62.5, 52.2, 34.8, 33.4; IR (KBr) v: 3501, 3295, 1662, 1382, 1215, 1126, 991, 734 cm-1; LRMS (ESI) m/z: 507.0 ([M+H]+); HRMS (ESI) calcd for C14H16O3N2F13[M+H]+ 507.1948, found 507.0945.
N, N-二甲基-N-羧甲基-3-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]丙基铵内盐(5b):白色固体, 收率81%. m.p. 190~193 ℃; 1H NMR (300 MHz, CD3OD) δ: 3.79 (s, 2H), 3.70~3.46 (m, 2H), 3.38~3.24 (m, 4H), 3.22 (s, 6H), 2.12~1.67 (m, 2H); 19F NMR (282 MHz, CD3OD) δ: -64.30~-64.66 (m, 6F), -82.16 (t, J=13.5 Hz, 3F), -107.69~-109.38 (m, 2F), -123.52~-124.94 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 168.6, 166.2, 64.9, 63.1, 51.9, 37.6, 33.5, 23.8; IR (KBr) v: 3226, 3055, 1693, 1628, 1259, 1214, 1125, 727 cm-1; LRMS (ESI) m/z: 521.1 ([M+H]+); HRMS (ESI) calcd for C15H18O3N2F13[M+H]+ 521.1104, found 521.1100.
250 mL圆底烧瓶中加入10.0 mmol化合物2a或2b和100 mL 30%双氧水, 反应体系室温搅拌过夜后变为均匀一相.二氯甲烷萃取两次, 水相冻干后得到目标化合物.
N, N-二甲基-2-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]乙基氧化胺(6a):透明膏状, 收率73%.1H NMR (400 MHz, D2O) δ: 3.71 (t, J=6.7 Hz, 2H), 3.48 (t, J=6.6 Hz, 2H), 3.29 (s, 2H), 3.25 (s, 6H); 19F NMR (376 MHz, D2O) δ: -62.80~-63.48 (m, 6F), -80.30 (t, J=13.4 Hz, 3F), -107.70 (s, 2F), -122.72~-123.33 (m, 2F); 13C NMR (101 MHz, D2O) δ: 166.9, 67.0, 57.6, 34.2, 32.4; IR (KBr) v: 3445, 3044, 1669, 1596, 1461, 1262, 1206, 704 cm-1; LRMS (ESI) m/z: 465.1 ([M+H]+); HRMS (ESI) calcd for C12H14O2N2F13[M+H]+ 465.0842, found 465.0838.
N, N-二甲基-3-[2-(全氟-1, 1-二甲基-丁基)乙酰胺基]丙基氧化胺(6b): 透明膏状, 收率75%.1H NMR (300 MHz, CD3OD) δ: 3.39~3.27 (m, 6H), 3.23~3.14 (m, 6H), 2.14~1.94 (m, 2H); 19F NMR (282 MHz, CD3OD) δ: -64.29~-64.70 (m, 6F), -82.16 (t, J=13.6 Hz, 3F), -108.37~-108.81 (m, 2F), -123.88~-124.42 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 166.0, 69.1, 58.4, 38.0, 33.6, 24.4; IR (KBr) v: 3236, 3060, 1697, 1569, 1260, 1216, 731, 662 cm-1; LRMS (ESI) m/z: 479.1 ([M+H]+); HRMS (ESI) calcd for C13H16O2N2F13[M+H]+ 479.0999, found 479.1000.
250 mL三口烧瓶中加入2.4 g LiAlH4 (0.63 mol), 氩气置换三次后加入40 mL无水乙醚, 将21.4 g化合物1 (0.053 mol)溶于60 mL无水乙醚中, 0 ℃下通过恒压滴液漏斗逐滴加入反应瓶中.反应体系在室温下搅拌3 h, 然后升温至40 ℃搅拌反应4 h.冷却至室温后用十水硫酸钠淬灭, 硅藻土过滤, 滤液依次用去离子水和饱和食盐水洗涤后再经无水硫酸钠干燥, 过滤.减压蒸馏, 收集70~73 ℃/4.67 kPa馏分, 得到12.9 g无色液体, 收率67%. 1H NMR (300 MHz, Acetone-d6) δ: 4.39~4.16 (m, 1H), 3.86 (dd, J=13.4, 6.7 Hz, 2H), 2.51 (t, J=7.6 Hz, 2H); 19F NMR (282 MHz, Acetone-d6) δ: -65.24 (p, J=11.4 Hz, 6F), -81.82 (t, J=13.8 Hz, 3F), -108.79 (tt, J=24.6, 12.3 Hz, 2F), -123.88~-124.77 (m, 2F); LRMS (EI) m/z(%): 31.1 (100), 363.1 (2.5).
50 mL圆底烧瓶中加入0.38 mL三乙胺(3.2 mmol, 1.6 equiv.)和0.728 g化合物7 (2 mmol, 1.0 equiv.), 0 ℃下溶于10 mL二氯甲烷, 接着逐滴加入0.38 mL三氟甲磺酸酐(2.24 mmol, 1.12 equiv.).反应体系在室温下搅拌3 h, 有机相分别经去离子水、饱和碳酸氢钠水溶液和饱和食盐水洗涤, 再经无水硫酸钠干燥, 旋干溶剂.柱层析[V(乙酸乙酯):V(石油醚)=1:20]分离提纯, 得无色液体产物0.84 g, 收率85%. 1H NMR (400 MHz, CDCl3) δ: 4.74 (t, J=7.8 Hz, 2H), 2.74 (t, J=7.8 Hz, 2H); 19F NMR (282 MHz, CDCl3) δ:-64.54 (p, J=11.3 Hz, 6F), -75.28 (s, 3F), -80.71 (t, J=13.5 Hz, 3F), -108.32 (tt, J=23.6, 11.6 Hz, 2F), -123.98 (dq, J=22.1, 11.0 Hz, 2F); LRMS (EI) m/z(%): 69 (100), 133 (5.76), 363 (2.77).
50 mL圆底烧瓶中加入0.94 mL N, N-二甲基乙二胺(10 mmol, 5 equiv.)和0.4 mL三乙胺(3.0 mmol, 1.5 equiv.), 溶于10 mL乙腈中.将0.99 g化合物8 (2.0 mmol, 1 equiv.)溶于5 mL乙腈, 0 ℃下加入反应瓶中, 室温反应17 h.旋干乙腈, 加入1 mol•L-1 NaOH水溶液调节pH至8~9, 二氯甲烷萃取.有机相经去离子水和饱和食盐水洗涤后用无水硫酸钠干燥, 过滤, 旋干溶剂, 经柱层析[V(CH2Cl2):V(MeOH):V(Et3N)=20:1:trace]分离提纯, 得无色油状物0.78 g, 收率90%. 1H NMR (400 MHz, CDCl3) δ: 2.89 (t, 2H), 2.68 (t, J=5.9 Hz, 2H), 2.39 (t, J=5.9 Hz, 2H), 2.33 (t, 2H), 2.20 (s, 6H); 19F NMR (376 MHz, CDCl3)δ: -63.94 (p, J=11.4 Hz, 6F), -80.29 (t, J=13.6 Hz, 3F), -107.74 (tt, J=24.3, 12.2 Hz, 2F), -123.11~-123.41 (m, 2F); 13C NMR (101 MHz, CDCl3) δ: 59.1, 47.1, 45.6, 44.3, 28.4; IR (KBr) v: 3307, 2949, 2823, 1459, 1327, 1217, 1042, 734, 529 cm-1; LRMS (ESI) m/z: 435.1 ([M+H]+); HRMS (ESI) calcd for C12H16N2F13[M+H]+ 435.1100, found 435.1097.
30 mL封管中加入0.87 g化合物9 (2.0 mmol, 1 equiv.), 氩气置换三次, 加入10 mL无水四氢呋喃, 0.80 mL碘乙烷(10 mmol, 5 equiv.). 60 ℃搅拌反应48 h后冷却至室温, 旋干溶剂, 粗产物经乙酸乙酯和正己烷混合溶剂重结晶, 得0.93 g白色固体, 收率79%. m.p. 134~137 ℃; 1H NMR (400 MHz, Acetone-d6) δ: 9.98 (s, 1H), 4.23 (t, 2H), 3.81 (dt, J=14.4, 7.7 Hz, 4H), 3.48 (d, J=10.3 Hz, 8H), 3.24 (t, 2H), 1.56 (t, J=7.2 Hz, 3H); 19F NMR (376 MHz, Acetone-d6) δ: -63.96 (p, J=11.5 Hz, 6F), -80.86 (t, J=13.6 Hz, 3F), -107.85~-108.29 (m, 2F), -123.11 (tt, J=23.1, 11.6 Hz, 2F); 13C NMR (101 MHz, Acetone-d6) δ: 61.4, 57.1, 51.6, 43.3, 41.2, 23.7, 8.6; IR (KBr) v: 3447, 2676, 1701, 1465, 1250, 1031, 735, 478 cm-1; LRMS (ESI) m/z: 463.1 ([M-I]+); HRMS (ESI) calcd for C14H20N2F13[M-I]+ 463.1413, found 463.1416.
100 mL三口瓶中加入6.0 g NaOH (150.0 mmol, 2 equiv.)和18 mL去离子水, 升温至100 ℃回流搅拌, 经恒压滴液漏斗逐滴加入30.46 g化合物1 (75.0 mmol, 1 equiv.). 3 h后反应体系变为一相, 继续反应1 h.冷却至室温, 滴入35% HCl酸化至pH=3, 有机相经饱和食盐水洗涤三次, 二氯甲烷萃取, 无水硫酸钠干燥, 旋干溶剂.粗产物中直接加入17.85 g氯化亚砜(150.0 mmol, 2 equiv.), 75 ℃回流搅拌5 h, 旋除过量氯化亚砜.粗产物减压蒸馏, 收集53~55 ℃/3.20 kPa馏分, 得23.9 g无色液体, 收率81%. 1H NMR (300 MHz, CDCl3) δ: 3.76 (s, 2H); 19F NMR (282 MHz, CDCl3) δ: -64.18 (p, J=10.9 Hz, 6F), -80.98 (t, J=13.2 Hz, 3F), -107.85 (tt, J=22.5, 11.1 Hz, 2F), -123.11~-124.02 (m, 2F); LRMS (EI) m/z (%): 69 (100), 361 (23.12), 377 (1.13).
50 mL圆底烧瓶氩气置换三次, 加入1.18 mL N-甲基哌嗪(9.75 mmol, 1.5 equiv.)和1.36 mL三乙胺(9.75 mmol, 1.5 equiv.).溶于10 mL无水四氢呋喃.将2.57 g化合物11 (6.5 mmol, 1.0 equiv.)溶于10 mL无水四氢呋喃, 并通过恒压滴液漏斗逐滴加入至反应瓶中.室温搅拌过夜, 反应体系分别经去离子水和饱和食盐水洗涤, 无水硫酸钠干燥.旋干溶剂, 粗产物经柱层析[V(CH2Cl2):V(MeOH):V(Et3N)=10:1:trace]分离提纯, 得到2.73 g白色固体, 收率90%. m.p. 74~76 ℃; 1H NMR (300 MHz, CDCl3) δ: 3.83~3.60 (m, 2H), 3.58~3.38 (m, 2H), 3.18 (s, 2H), 2.52~2.34 (m, 4H), 2.31 (s, 3H); 19F NMR (282 MHz, CDCl3)δ: -62.97 (p, J=11.2 Hz, 6F), -80.49 (t, J=13.4 Hz, 3F), -107.74 (s, 2F), -123.16 (d, J=10.8 Hz, 2F); 13C NMR (101 MHz, CDCl3) δ: 162.4, 54.9, 54.5, 46.7, 45.9, 42.3, 28.2; IR (KBr) v: 2958, 1661, 1464, 1221, 1108, 1001, 892, 734 cm-1; LRMS (ESI) m/z: 483.0 ([M+Na]+); HRMS (ESI) calcd for C13H14ON2F13[M+H]+ 461.0893, found 461.0887.
50 mL封管中加入1.38 g化合物12 (3.0 mmol, 1 equiv.), 2.4 mL碘乙烷(15.0 mmol, 5 equiv.)和20 mL MeCN, 回流搅拌24 h.冷却至室温后过滤, 滤饼用乙酸乙酯洗涤, 干燥得1.16 g白色固体, 收率69%. m.p.>250 ℃; 1H NMR (300 MHz, CD3OD) δ: 4.31~4.02 (m, 2H), 4.02~3.71 (m, 2H), 3.71~3.38 (m, 8H), 3.22 (s, 3H), 1.42 (t, J=7.1 Hz, 3H); 19F NMR (282 MHz, CD3OD) δ: -66.39~-67.24 (m, 6F), -84.94 (t, J=13.5 Hz, 3F), -109.8~-111.92 (m, 2F), -125.74~ -127.36 (m, 2F); 13C NMR (101 MHz, CD3OD) δ: 164.7, 61.2, 60.3, 60.0, 46.9, 41.4, 37.0, 30.2, 7.8; IR (KBr) v: 3445, 2999, 1663, 1463, 1266, 1155, 1112, 981, 729 cm-1; LRMS (ESI) m/z: 489.1 ([M-I]+); HRMS (ESI) calcd for C15H18ON2F13[M-I]+ 489.1206, found 489.1200.
辅助材料(Supporting Information) 所有的中间体和最终产物的1H NMR, 19F NMR (13C NMR), IR, MS, HRMS谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Kissa, E. Fluorinated Surfactants: Synthesis, Properties, Applica- tions, M. Dekker, New York, 1994.
Rao, N. S.; Baker, B. E. Textile Finishes and Fluorosurfactants, In Organofluorine Chemistry, Springer, Boston, 1994, p. 321.
Renner, R. Environ. Sci. Technol. 2006, 40, 12. doi: 10.1021/es062612a
Porter, M. R. Handbook of Surfactants, Springer, Glasgow, 2013.
Kovarova, J.; Svobodova, Z. Neuroendocrinol. Lett. 2008, 29, 599. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM18987583
Giesy, J. P.; Kannan, K. Environ. Sci. Technol. 2002, 36, 146. doi: 10.1021/es010947g
Conder, J. M.; Hoke, R. A.; Wolf, W. d.; Russell, M. H.; Buck, R. C. Environ. Sci. Technol. 2008, 42, 995. doi: 10.1021/es070895g
司戈, 消防技术与产品信息, 2010, 12, 75.Si, G. Fire Tech. Prod. Inform. 2010, 12, 75 (in Chinese).
U. S. Environmental Protection Agency, http://www.epa.gov/oppt/pfoa/pubs/stewardship/index.html.
Krafft, M. P.; Riess, J. G. Curr. Opin. Colloid Interface Sci. 2015, 20, 192. doi: 10.1016/j.cocis.2015.07.004
Venkatesan, A. K.; Halden, R. U. Environ. Res. 2014, 132, 321. doi: 10.1016/j.envres.2014.04.024
Vierke, L.; Möller, A.; Klitzke, S. Environ. Pollut. 2014, 186, 7. doi: 10.1016/j.envpol.2013.11.011
Guerra, K. H. M. A.; Jurgens, M.; Guerra, M. A. US 0276103, 2007[Chem. Abstr. 2007, 148, 35460].
Boutevin, G.; Tiffes, D.; Loubat, C.; Boutevin, B.; Ameduri, B. J. Fluorine Chem. 2012, 134, 77. doi: 10.1016/j.jfluchem.2011.06.019
Zana, R.; Benrraou, M.; Rueff, R. Langmuir 1991, 7, 1072. doi: 10.1021/la00054a008
Jaeger, D. A.; Li, B.; Clark, T. Langmuir 1996, 12, 4314. doi: 10.1021/la960012r
Schuster, T.; Krumpfer, J. W.; Schellenberger, S.; Friedrich, R.; Klapper, M.; Müllen, K. J. Colloid Interface Sci. 2014, 428, 276. doi: 10.1016/j.jcis.2014.04.051
Wang, X. Y.; Dai C. J.; Liu, X. Y.; Chen, T.; Wang, J. P. Key Eng. Mater. 2015, 671, 210. doi: 10.4028/www.scientific.net/KEM.671
Fisicaro, E.; Contardi, L.; Compari, C.; Bacciottini, F.; Pongiluppi, E.; Viscardi, G.; Barbero, N.; Quagliotto, P.; Różycka-Roszak, B. Colloids Surf., A 2016, 507, 236. doi: 10.1016/j.colsurfa.2016.07.051
Sagisaka, M.; Narumi, T.; Niwase, M.; Narita, S.; Ohata, A.; James, C.; Yoshizawa, A.; Taffin de Givenchy, E.; Guittard, F.; Alexander, S.; Eastoe, J. Langmuir 2014, 30, 6057. doi: 10.1021/la501328s
Dmowski, W.; Plenkiewicz, H.; Piasecka-Maciejewska, K.; Prescher, D.; Schulze, J. Endler, I. J. Fluorine Chem. 1990, 48, 77. doi: 10.1016/S0022-1139(00)82603-2
Sha, M.; Pan, R. M.; Zhan, L. W.; Xing, P.; Jiang, B. Chin. J. Chem. 2014, 32, 995. doi: 10.1002/cjoc.201400377
Sha, M.; Pan, R. M.; Xing, P.; Jiang, B. J. Fluorine Chem. 2015, 169, 61. doi: 10.1016/j.jfluchem.2014.11.005
沙敏, 张丁, 潘仁明, 邢萍, 姜标, 化学学报, 2015, 73, 395. http://www.cnki.com.cn/Article/CJFDTotal-HXXB201505003.htmSha, M.; Zhang, D.; Pan, R. M.; Xing, P.; Jiang, B. Acta Chim. Sinica 2015, 73, 395 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HXXB201505003.htm
Lin, C.; Pan, R. M.; Xing, P.; Jiang, B. Chin. Chem. Lett. 2018, 29, 1613.
Stähler, K.; Selb, J.; Barthelemy, P.; Pucci, B.; Candau, F. Langmuir 1998, 14, 4765 doi: 10.1021/la980245d
Tomota, H. JP 07145119, 1995[Chem. Abstr. 1995, 123, 285205].

图 1 亲水基团种类对表面活性的影响
Figure 1 Effect of the species of the headgroups on the surface activities
(A) 2-Carbon atoms in linkage; (B) 3-carbon atoms in linkage

图 2 亲水基团大小对表面活性的影响
Figure 2 Effect of the size of the headgroups on the surface activities

图 3 连接基团长短对表面活性的影响(A)亲水基为氧化铵(B)亲水基为甜菜碱
Figure 3 Effect of the linkage on the surface activities (A) ammonium oxide as hydrophilic group (B) betaine as hydrophilic group

图 4 (A) 极性基团对表面活性的影响和(B)表面活性剂在水中可能的结构
Figure 4 (A) Effect of the polar groups on the surface activities and (B) possible conformation for surfactant in aqueous solution

图 5 化合物6a与APG的复配性能
Figure 5 Combined properties of compound 6a with APG
6a: m(6a):m(APG)=1:0; M1: m(6a):m(APG)=1:1; M2: m(6a):m(APG)=1:5; M3: m(6a):m(APG)=1:10
表 1 支链型氟表面活性剂的表面张力与临界胶束浓度
Table 1. Surface tensions and critical micelle concentrations of branched fluorinated surfactants
| Compd. | CMC/(mol•L-1) | γCMC (mN•m-1) |
| 3a | 2.52×10-2 | 22.91 |
| 3b | 8.50×10-3 | 33.73 |
| 3c | 1.64×10-2 | 27.09 |
| 5a | 1.41×10-2 | 21.50 |
| 5b | 1.21×10-2 | 24.88 |
| 6a | 1.73×10-2 | 19.93 |
| 6b | 1.11×10-2 | 20.11 |
| 10 | 9.89×10-4 | 36.32 |
| 13 | 1.08×10-2 | 29.08 |
| M1 | 1.89×10-4 | 20.24 |
| M2 | 4.28×10-5 | 23.79 |
| M3 | 1.74×10-5 | 24.86 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
