CAS-Key Laboratory of Synthetic Biology, CAS Center for Excellence in Molecular Plant Sciences, Shanghai Institute of Plant Physiology and Ecology, Chinese Academy of Sciences, Shanghai 200032
b.
University of Chinese Academy of Sciences, Beijing 100049
Received Date:
31 May 2018 Revised Date:
30 July 2018 Available Online:
01 September 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21672228), the Key Deployment Projects of the Chinese Academy of Sciences (Nos. KFZD-SW-215), the "Strategic Priority Research Program" of the Chinese Academy of Sciences (No. XDPB0400) and the International Great Science Program of the Chinese Academy of Sciences (No. 153D31KYSB20170121)
Abstract:
Plant natural products and their derivatives are the important reservoirs for the development of medicines, health products and food additives. The synthetic biology technology brings new strategies to manufacture rare plant natural products with complicated structures at large scale by artificially constructing and optimizing the biosynthesis pathway of target compounds in microbial chassis cells. In this paper, the research progress on the artificial syntheses of important plant natural products such as artemisinin, ginsenosides, morphinan alkaloids, paclitaxel and vinblastine is reviewed. These examples not only demonstrate the great potentials of synthetic biology as well as its combination with synthetic chemistry applied in the artificial syntheses of plant natural compounds, but also show us the roadmap for future research and development on de novo synthesis of plant natural products. New technologies developed in synthetic biology and synthetic chemistry would further promote the unveiling of biosynthetic pathways of complex natural compounds, the discovery and characterization of key bioparts, the design and building of novel chassis cells, the strong-strong combination of biosynthesis and chemical synthesis, and thus accelerate the process to transfer the developed technologies to artificially synthesize plant natural products from laboratories to markets.
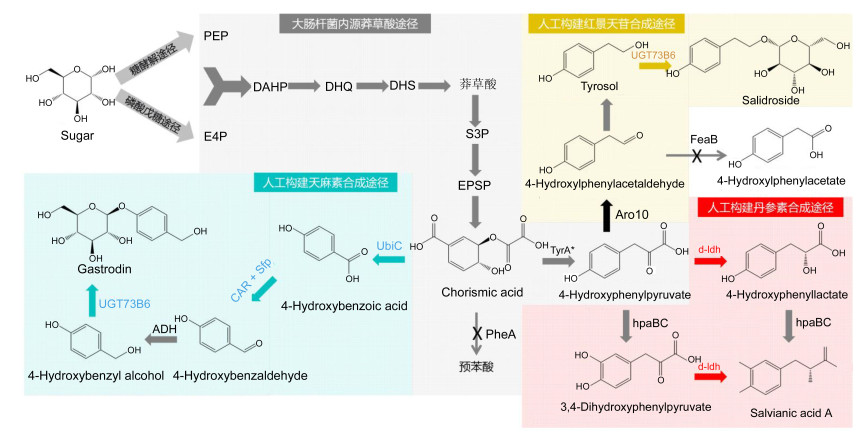
Figure 4.
Biosyntheses of the bioactive polyphenol aromatic acid compounds salvianic acid A, salidroside and gastrodin from traditional Chinese medicines
Chang, M. C. Y.; Keasling, J. D. Nat. Chem. Biol. 2006, 2, 674. doi: 10.1038/nchembio836
[2]
Paddon, C. J.; Keasling, J. D. Nat. Rev. Microbiol. 2014, 12, 355. doi: 10.1038/nrmicro3240
[3]
Morrison, K. C.; Hergenrother, P. J. Nat. Prod. Rep. 2014, 31, 6. doi: 10.1039/C3NP70063A
[4]
(a) Chen, J. C.; Chen, X. C.; Bois-Choussy, M.; Zhu, J. P. J. Am. Chem. Soc. 2006, 128, 87. (b) Cuevas, C.; Perez, M.; Martin, M. J.; Chicharro, J. L.; Fernandez-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de la Calle, F.; Garcia, J.; Polanco, C.; Rodriguez, I.; Manzanares, I. Org. Lett. 2000, 2, 2545.
(a) Shibata, S. J. Korean Med. Sci. 2001, 16, S28. (b) Liu, Z. Q. Chem. Rev. 2012, 112, 3329.
[7]
Cameron, D. E.; Bashor, C. J.; Collins, J. J. Nat. Rev. Microbiol. 2014, 12, 381. doi: 10.1038/nrmicro3239
[8]
Paddon, C. J.; Westfall, P. J.; Pitera, D. J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M. D.; Tai, A.; Main, A.; Eng, D.; Polichuk, D. R.; Teoh, K. H.; Reed, D. W.; Treynor, T.; Lenihan, J.; Fleck, M.; Bajad, S.; Dang, G.; Dengrove, D.; Diola, D.; Dorin, G.; Ellens, K. W.; Fickes, S.; Galazzo, J.; Gaucher, S. P.; Geistlinger, T.; Henry, R.; Hepp, M.; Horning, T.; Iqbal, T.; Jiang, H.; Kizer, L.; Lieu, B.; Melis, D.; Moss, N.; Regentin, R.; Secrest, S.; Tsuruta, H.; Vazquez, R.; Westblade, L. F.; Xu, L.; Yu, M.; Zhang, Y.; Zhao, L.; Lievense, J.; Covello, P. S.; Keasling, J. D.; Reiling, K. K.; Renninger, N. S.; Newman, J. D. Nature 2013, 496, 528. doi: 10.1038/nature12051
[9]
Yan, X.; Fan, Y.; Wei, W.; Wang, P. P.; Liu, Q. F.; Wei, Y. J.; Zhang, L.; Zhao, G. P.; Yue, J. M.; Zhou, Z. H. Cell Res. 2014, 24, 770. doi: 10.1038/cr.2014.28
[10]
Rodriguez, A.; Kildegaard, K. R.; Li, M. J.; Borodina, I.; Nielsen, J. Metab. Eng. 2015, 31, 181. doi: 10.1016/j.ymben.2015.08.003
[11]
Li, M.; Kildegaard, K. R.; Chen, Y.; Rodriguez, A.; Borodina, I.; Nielsen, J. Metab. Eng. 2015, 32, 1. doi: 10.1016/j.ymben.2015.08.007
[12]
Galanie, S.; Thodey, K.; Trenchard, I. J.; Filsinger, I. M.; Smolke, C. D. Science 2015, 349, 1095. doi: 10.1126/science.aac9373
[13]
Westfall, P. J.; Pitera, D. J.; Lenihan, J. R.; Eng, D.; Woolard, F. X.; Regentin, R.; Horning, T.; Tsuruta, H.; Melis, D. J.; Owens, A.; Fickes, S.; Diola, D.; Benjamin, K. R.; Keasling, J. D.; Leavell, M. D.; McPhee, D. J.; Renninger, N. S.; Newman, J. D.; Paddon, C. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E111. doi: 10.1073/pnas.1110740109
Martin, V. J.; Yoshikuni, Y.; Keasling, J. D. Biotechnol. Bioeng. 2001, 75, 497. doi: 10.1002/(ISSN)1097-0290
[16]
Martin, V. J.; Pitera, D. J.; Withers, S. T.; Newman, J. D.; Keasling, J. D. Nat. Biotechnol. 2003, 21, 796. doi: 10.1038/nbt833
[17]
Ro, D. K.; Paradise, E. M.; Ouellet, M.; Fisher, K. J.; Newman, K. L.; Ndungu, J. M.; Ho, K. A.; Eachus, R. A.; Ham, T. S.; Kirby, J.; Chang, M. C.; Withers, S. T.; Shiba, Y.; Sarpong, R.; Keasling, J. D. Nature 2006, 440, 940. doi: 10.1038/nature04640
[18]
Chang, M. C.; Eachus, R. A.; Trieu, W.; Ro, D. K.; Keasling, J. D. Nat. Chem. Biol. 2007, 3, 274. doi: 10.1038/nchembio875
[19]
Zhang, Y.; Teoh, K. H.; Reed, D. W.; Maes, L.; Goossens, A.; Olson, D. J.; Ross, A. R.; Covello, P. S. J. Biol. Chem. 2008, 283, 21501. doi: 10.1074/jbc.M803090200
[20]
Teoh, K. H.; Polichuk, D. R.; Reed, D. W.; Covello, P. S. Botany 2009, 87, 635. doi: 10.1139/B09-032
[21]
Carlsen, S.; Ajikumar, P. K.; Formenti, L. R.; Zhou, K.; Phon, T. H.; Nielsen, M. L.; Lantz, A. E.; Kielland-Brandt, M. C.; Stephanopoulos, G. Appl. Microbiol. Biotechnol. 2013, 97, 5753. doi: 10.1007/s00253-013-4877-y
[22]
(a) Wu, Y.-L.; He, Z.-L. Highlights of Total Synthesis Natural Products: Terpenes, Science Press, Beijing, 2009, pp. 41~42 (in Chinese). (吴毓林, 何子乐, 天然产物全合成荟萃——萜类, 科学出版社, 北京, 2009, pp. 41~42.) (b) Xu, X.-X.; Zhu, J.; Huang, D.-Z.; Zhou, W.-S. Acta Chim. Sinica1983, 41, 574 (in Chinese). (许杏祥, 朱杰, 黄大中, 周维善, 化学学报, 1983, 41, 574.)
[23]
(a) Ye, B.; Wu, Y. L. Tetrahedron1989, 45, 7287. (b) Ye, B.; Wu, Y. L. J. Chem. Soc., Chem. Commun. 1990, 726.
[24]
Lévesque, F.; Seeberger, P. P. H. Angew. Chem., Int. Ed. 2012, 51, 1706. doi: 10.1002/anie.v51.7
Chen, H. J.; Han, W. B.; Hao, H. D.; Wu, Y. Tetrahedron 2013, 69, 1112. doi: 10.1016/j.tet.2012.11.056
[27]
Turconi, J.; Griolet, F.; Guevel, R.; Oddon, G.; Villa, R.; Geatti, A.; Hvala, M.; Kai, R.; Göller, R.; Burgard, A. Org. Process Res. Dev. 2014, 18, 417. doi: 10.1021/op4003196
[28]
Chae, S.; Kang, K. A.; Chang, W. Y.; Kim, M. J.; Lee, S. J.; Lee, Y. S.; Kim, H. S.; Kim, D. H.; Hyun, J. W. J. Agric. Food. Chem. 2009, 57, 5777. doi: 10.1021/jf900331g
[29]
Han, J. Y.; Kim, H. J.; Kwon, Y. S.; Choi, Y. E. Plant Cell Physiol. 2011, 52, 2062. doi: 10.1093/pcp/pcr150
[30]
Han, J. Y.; Hwang, H. S.; Choi, S. W.; Kim, H. J.; Choi, Y. E. Plant Cell Physiol. 2012, 53, 1535. doi: 10.1093/pcp/pcs106
[31]
Dai, Z. B.; Wang, B. B.; Liu, Y.; Shi, M. Y.; Wang, D.; Zhang, X. N.; Liu, T.; Huang, L. Q.; Zhang, X. L. Sci. Rep. 2014, 4, 3698.
[32]
(a) Wang, P. P.; Wei, Y. J.; Fan, Y.; Liu, Q. F.; Wei, W.; Yang, C. S.; Zhang, L.; Zhao, G. P.; Yue, J. M.; Yan, X.; Zhou, Z. H. Metab. Eng. 2015, 29, 97. (b) Wei, W.; Wang, P. P.; Wei, Y. J.; Liu, Q. F.; Yang, C. S.; Zhao, G. P.; Yue, J. M.; Yan, X.; Zhou, Z. H. Mol. Plant2015, 8, 1412.
[33]
(a) Kai, G. Y.; Xu, H.; Zhou, C. C.; Liao, P.; Xiao, J. B.; Luo, X. Q.; You, L. J.; Zhang, L. Metab. Eng. 2011, 13, 319. (b) Zhou, L. M.; Zuo, Z.; Chow, M. S. S. J. Clin. Pharmacol. 2005, 45, 1345.
[34]
Zhou, Y. J. J.; Gao, W.; Rong, Q. X.; Jin, G. J.; Chu, H. Y.; Liu, W. J.; Yang, W.; Zhu, Z. W.; Li, G. H.; Zhu, G. F.; Huang, L. Q.; Zhao, Z. B. K. J. Am. Chem. Soc. 2012, 134, 3234. doi: 10.1021/ja2114486
[35]
Dai, Z. B.; Liu, Y.; Huang, L. Q.; Zhang, X. L. Biotechnol. Bioeng. 2012, 109, 2845. doi: 10.1002/bit.v109.11
[36]
Guo, J.; Zhou, Y. J. J.; Hillwigc, M. L.; Shen, Y.; Yang, L.; Wang, Y. J.; Zhang, X. A.; Liu, W. J.; Peters, R. J.; Chen, X. Y.; Zhao, Z. B. K.; Huang, L. Q. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12108. doi: 10.1073/pnas.1218061110
Bai, Y. F.; Bi, H. P.; Zhuang, Y. B.; Liu, C.; Cai, T.; Liu, X. N.; Zhang, X. L.; Liu, T.; Ma, Y. H. Sci. Rep. 2014, 4, 6640.
[39]
Bai, Y. F.; Yin, H.; Bi, H. P.; Zhuang, Y. B.; Liu, T.; Ma, Y. H. Metab. Eng. 2016, 35, 138. doi: 10.1016/j.ymben.2016.01.002
[40]
Hawkins, K. M.; Smolke, C. D. Nat. Chem. Biol. 2008, 4, 564-73. doi: 10.1038/nchembio.105
[41]
DeLoache, W. C.; Russ, Z. N.; Narcross, L.; Gonzales, A. M.; Martin, V. J.; Dueber, J. E. Nat. Chem. Biol. 2015, 11, 465. doi: 10.1038/nchembio.1816
[42]
(a) Winzer, T.; Kern, M.; King, A. J.; Larson, T. R.; Teodor, R. I.; Donninger, S. L.; Li, Y.; Dowle, A. A.; Cartwright, J.; Bates, R.; Ashford, D.; Thomas, J.; Walker, C.; Bowser, T. A.; Graham, I. A. Science2015, 349, 309. (b) Farrow, S. C.; Hagel, J. M.; Beaudoin, G. A.; Burns, D. C.; Facchini, P. J. Nat. Chem. Biol. 2015, 11, 728.
[43]
Li, Y.; Li, S.; Thodey, K.; Trenchard, I.; Cravens, A.; Smolke, C. D. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3922. doi: 10.1073/pnas.1721469115
Fossati, E.; Ekins, A.; Narcross, L.; Zhu, Y.; Falgueyret, J. P.; Beaudoin, G. A. W.; Facchini, P. J.; Martin, V. J. J. Nat. Commun. 2014, 5, 3283. doi: 10.1038/ncomms4283
[46]
Mountford, P. G. In Green Chemistry in the Pharmaceutical Industry, Eds.: Dunn, P. J.; Wells, A. S.; Williams, M. T., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 145~160.
[47]
(a) Nicolaou, K. C.; Dai, W. M.; Guy, R. K. Angew. Chem., Int. Ed. 1994, 33, 15. (b) Gueritte-Voegelein, F.; Guenard, D.; Dubois, J.; Wahl, A.; Potier, P. J. Pharm. Belg. 1994, 49, 193. (c) Guenard, D.; Guerittevoegelein, F.; Potier, P. Acc. Chem. Res. 1993, 26, 160.
(a) Chau, M.; Croteau, R. Arch. Biochem. Biophys. 2004, 427, 48. (b) Chau, M.; Jennewein, S.; Walker, K.; Croteau, R. Chem. Biol. 2004, 11, 663. (c) Jennewein, S.; Long, R. M.; Williams, R. M.; Croteau, R. Chem. Biol. 2004, 11, 379. (d) Jennewein, S.; Rithner, C. D.; Williams, R. M.; Croteau, R. B. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13595. (e) Jennewein, S.; Wildung, M. R.; Chau, M.; Walker, K.; Croteau, R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9149. (f) Schoendorf, A.; Rithner, C. D.; Williams, R. M.; Croteau, R. B. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 1501.
[53]
(a) Guerra-Bubb, J.; Croteau, R.; Williams, R. M., Nat. Pro. Rep. 2012, 29, 683. (b) McElroy C.; Jennewein S. In Biotechnology of Natural Products, Eds.: Schwab, W.; Lange, B.; Wüst, M., Springer, Cham, 2018, pp. 145~185.
[54]
(a) Walker, K.; Croteau, R. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13591. (b) Walker, K.; Croteau, R. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 583. (c) Walker, K.; Schoendorf, A.; Croteau, R. Arch. Biochem. Biophys. 2000, 374, 371.
[55]
Walker, K.; Fujisaki, S.; Long, R.; Croteau, R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12715. doi: 10.1073/pnas.192463699
[56]
Walker, K.; Long, R.; Croteau, R. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9166. doi: 10.1073/pnas.082115799
Zhou, K.; Qiao, K. J.; Edgar, S.; Stephanopoulos, G. Nat. Biotechnol. 2015, 33, 377. doi: 10.1038/nbt.3095
[59]
Biggs, B. W.; Lim, C. G.; Sagliani, K.; Shankar, S.; Stephanopoulos, G.; De Mey, M.; Ajikumar, P. K. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3209. doi: 10.1073/pnas.1515826113
Van Der Heijden, R.; Jacobs, D. I.; Snoeijer, W.; Hallard, D.; Verpoorte, R. Curr. Med. Chem. 2004, 11, 607. doi: 10.2174/0929867043455846
[62]
(a) Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; Verpoorte, R.; Oksman-Caldentey, K. M.; Martinoia, E.; Bouwmeester, H.; Goossens, A.; Memelink, J.; Werck-Reichhart, D. Nat. Commun. 2014, 5, 3606. (b) Collu, G.; Unver, N.; Peltenburg-Looman, A. M.; van der Heijden, R.; Verpoorte, R.; Memelink, J. FEBS Lett. 2001, 508, 215. (c) Irmler, S.; Schroder, G.; St-Pierre, B.; Crouch, N. P.; Hotze, M.; Schmidt, J.; Strack, D.; Matern, U.; Schroder, J. Plant J. 2000, 24 (6), 797. (d) Geu-Flores, F.; Sherden, N. H.; Courdavault, V.; Burlat, V.; Glenn, W. S.; Wu, C.; Nims, E.; Cui, Y.; O'Connor, S. E. Nature2012, 492, 138. (e) Simkin, A. J.; Miettinen, K.; Claudel, P.; Burlat, V.; Guirimand, G.; Courdavault, V.; Papon, N.; Meyer, S.; Godet, S.; St-Pierre, B.; Giglioli-Guivarc'h, N.; Fischer, M. J.; Memelink, J.; Clastre, M. Phytochemistry2013, 85, 36.
[63]
Brown, S.; Clastre, M.; Courdavault, V.; O'Connor, S. E. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3205. doi: 10.1073/pnas.1423555112
[64]
Qu, Y.; Easson, M.; Simionescu, R.; Hajicek, J.; Thamm, A. M. K.; Salim, V.; De Luca, V. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3180. doi: 10.1073/pnas.1719979115
[65]
Caputi, L.; Franke, J.; Farrow, S. C.; Chung, K.; Payne, R. M. E.; Nguyen, T. D.; Dang, T. T.; Soares Teto Carqueijeiro, I.; Kou-dounas, K.; Duge de Bernonville, T.; Ameyaw, B.; Jones, D. M.; Vieira, I. J. C.; Courdavault, V.; O'Connor, S. E. Science 2018.
[66]
(a) Liscombe, D. K.; O'Connor, S. E. Phytochemistry2011, 72, 1969. (b) Qu, Y.; Easson, M. L.; Froese, J.; Simionescu, R.; Hudlicky, T.; De Luca, V. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6224.
[67]
Sottomayor, M.; Lopez-Serrano, M.; DiCosmo, F.; Ros Barcelo, A. FEBS Lett. 1998, 428, 299. doi: 10.1016/S0014-5793(98)00551-1
[68]
Shang, Y.; Ma, Y. S.; Zhou, Y.; Zhang, H. M.; Duan, L. X.; Chen, H. M.; Zeng, J. G.; Zhou, Q.; Wang, S. H.; Gu, W. J.; Liu, M.; Ren, J. W.; Gu, X. F.; Zhang, S. P.; Wang, Y.; Yasukawa, K.; Bouwmeester, H. J.; Qi, X. Q.; Zhang, Z. H.; Lucas, W. J.; Huang, S. W. Science 2014, 346, 1084. doi: 10.1126/science.1259215
Marques, J. V.; Kim, K. W.; Lee, C.; Costa, M. A.; May, G. D.; Crow, J. A.; Davin, L. B.; Lewis, N. G. J. Biol. Chem. 2013, 288, 466. doi: 10.1074/jbc.M112.400689
Moses, T.; Pollier, J.; Almagro, L.; Buyst, D.; Van Montagu, M.; Pedreno, M. A.; Martins, J. C.; Thevelein, J. M.; Goossens, A., Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 1634. doi: 10.1073/pnas.1323369111
[73]
(a) Naesby, M.; Nielsen, S. V. S.; Nielsen, C. A. F.; Green, T.; Tange, T. O.; Simon, E.; Knechtle, P.; Hansson, A.; Schwab, M. S.; Titiz, O.; Folly, C.; Archila, R. E.; Maver, M.; Fiet, S. V.; Boussemghoune, T.; Janes, M.; Kumar, A. S. S.; Sonkar, S. P.; Mitra, P. P.; Benjamin, V. A. K.; Korrapati, N.; Suman, I.; Hansen, E. H.; Thybo, T.; Goldsmith, N.; Sorensen, A. S. Microb. Cell Fact. 2009, 8, 45. (b) Klein, J.; Heal, J. R.; Hamilton, W. D. O.; Boussemghoune, T.; Tange, T. O.; Delegrange, F.; Jaeschke, G.; Hatsch, A.; Heim, J. ACS Synth. Biol. 2014, 3, 314.
[74]
Wu, J. J.; Zhou, T. T.; Du, G. C.; Zhou, J. W.; Chen, J. PLoS One 2014, 9, e101492. doi: 10.1371/journal.pone.0101492
[75]
Thodey, K.; Galanie, S.; Smolke, C. D. Nat. Chem. Biol. 2014, 10, 837. doi: 10.1038/nchembio.1613
[76]
Zhou, Y. J.; Buijs, N. A.; Zhu, Z. W.; Gomez, D. O.; Boonsombuti, A.; Siewers, V.; Nielsen, J. J. Am. Chem. Soc. 2016, 138, 15368. doi: 10.1021/jacs.6b07394
[77]
Wang, H. H.; Isaacs, F. J.; Carr, P. A.; Sun, Z. Z.; Xu, G.; Forest, C. R.; Church, G. M. Nature 2009, 460, 894. doi: 10.1038/nature08187
[78]
Guo, X.; Chavez, A.; Tung, A.; Chan, Y.; Kaas, C.; Yin, Y.; Cecchi, R.; Garnier, S. L.; Kelsic, E. D.; Schubert, M.; DiCarlo, J. E.; Collins, J. J.; Church, G. M. Nat. Biotechnol. 2018, 36, 540 doi: 10.1038/nbt.4147
[79]
Caspeta, L.; Chen, Y.; Ghiaci, P.; Feizi, A.; Buskov, S.; Hallstrom, B. M.; Petranovic, D.; Nielsen, J. Science 2014, 346, 75. doi: 10.1126/science.1258137
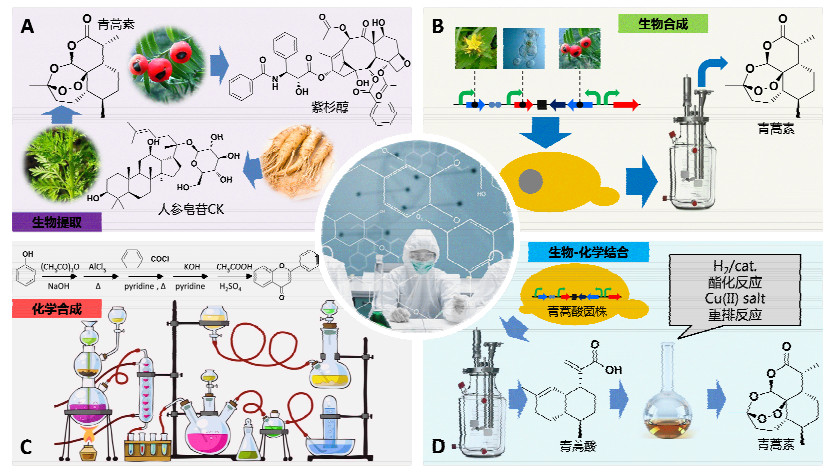
图 1
植物天然产物的制备方法
Figure 1
Methods to manufacture plant natural products
(A) Plant extraction, (B) synthetic biology approaches, (C) chemical synthesis, (D) combined synthetic biology and chemical synthesis
Figure 4
Biosyntheses of the bioactive polyphenol aromatic acid compounds salvianic acid A, salidroside and gastrodin from traditional Chinese medicines

 下载:
下载:

 下载:
下载:








