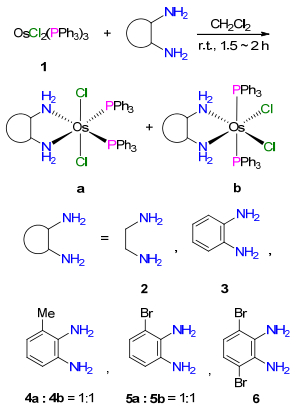
图式 1.
化合物2~6的合成
Scheme 1.
Syntheses of compounds 2~6

随着化石能源的逐渐枯竭, 环境问题日益加剧, 氢能源以其质量能量密度高, 燃烧后生成水对环境无污染等优点成为备受关注的新型替代能源[1].然而, 氢气在常温常压下密度小, 安全高效地储氢成为氢能利用的关键.为此, 化学储氢很好地解决了常态下储氢体积能量密度的问题[2].在众多潜在的化学储氢材料中, 近年来尤以氨硼烷(NH3•BH3, AB)受到了广泛关注, 关于氨硼烷释氢性质的研究成为热点研究领域[3].
氨硼烷在常温常压下是稳定的白色晶状固体, 其分子内氢含量高达19.6%, 受热可逐步脱氢生成硼氮化合物.但是, 直接热解释氢还存在放氢温度高, 释氢过程伴有杂质气体产生、体积膨胀, 再生困难等缺点, 严重制约其实际应用.为此, 一系列改善氨硼烷释氢性能的研究工作在世界范围内迅速展开, 如将氨硼烷负载到多孔材料上[4], 金属替代化学激活[5], 使用添加剂或催化剂[6]等.其中, 利用金属催化脱氢是改善释氢量、释氢反应选择性和动力学的最有效方法之一.近些年, 国际上已有关于氨/胺硼烷脱氢催化剂的大量研究报道, 涉及前、中、后过渡系的钛、锆、铬、钼、钨、铼、铁、钌、锇、铑、铱、镍、钯、铂等多种金属[7].其中, 第三过渡系金属锇催化氨硼烷脱氢的研究目前仅有Esteruelas小组关于锇氢配合物作为催化剂的报道[8].因此, 对锇基催化剂的研发, 以及锇催化氨硼烷脱氢反应的特点和规律值得进一步深入发掘和系统研究.
较之其它铂系金属元素, 锇在催化领域的应用非常有限, 最著名的莫过于Sharpless双羟化反应及其类似反应, 但是由于需要用到剧毒且易挥发的OsO4作为催化剂, 应用并不广泛[9].最近, 锇配合物在催化H2转化反应中展现出优越的性能[8, 10].值得一提的是, Esteruelas小组最近利用PNN型Pincer配体锇配合物作为催化剂实现了醇脱氢偶联[11], 以及五N配体(BPIs)Os配合物在没有受体分子和碱参与下催化醇或胺脱氢[12]. 2011年, Baratta小组[13]利用二胺-二膦配体Os配合物实现催化醇脱氢到酮.此外, 二胺-二膦配体Fe配合物已被Baker小组证实具有催化氨硼烷脱氢活性.以上研究表明含N配体锇配合物可催化断裂O—H, N—H等极性共价键.氨硼烷分子中同时存在B—H和N—H共价键, 可在催化剂作用下发生断裂[14].因此, 我们认为选用合适的N配体形成锇配合物同样也可用于催化氨硼烷释氢.
为了探索氨硼烷脱氢的新型锇催化剂, 本文选取乙二胺、(取代/非取代)邻苯二胺和(取代/非取代)1, 10-菲啰啉等双齿N基配体合成系列锇配合物2~11, 并将它们应用于催化氨硼烷脱氢反应中.结果表明, 这些锇配合物均具有良好的催化氨硼烷脱氢活性.其中, 3-甲基-邻苯二胺取代的锇配合物4的催化活性高于已报道的锇氢配合物催化剂, 是目前最高效的氨硼烷脱氢锇催化剂.
如Scheme 1所示, 将OsCl2(PPh3)3 (1)与乙二胺或取代/非取代邻苯二胺于二氯甲烷中室温下以物质的量比1:1.2反应1.5~2 h, 经二胺配体取代一个三苯基膦, 可生成二胺双齿配合物OsCl2(PPh3)2(diamine)(2~6)[15], 将反应液浓缩后, 经过滤、洗涤分离可高产率得到OsCl2(PPh3)2(diamine) (2~6)的黄色固体粉末.
化合物2~6经过核磁、元素分析等系统表征.具体数据详见实验方法部分.由结果可知, 当取代/非取代邻苯二胺作为配体时, 化合物3~6存在a和b两种异构体.与之对应的是, 当3-甲基邻苯二胺和3-溴-邻苯二胺作为配体时, 化合物4和5的常温1H NMR, 13C NMR和31P NMR对应的每种H、C和P原子均显示两组信号峰, 积分面积大约1:1, 表明a和b两种异构体同时生成.而当邻苯二胺和3, 6-二溴邻苯二胺作为配体时, 常温1H NMR, 13C NMR和31P NMR对应的每种H、C和P原子只显示一组信号峰, 低温核磁尝试使3和6的两种异构体谱峰裂分开没有成功, 表明在化合物3和6中, PPh3和Cl配体在金属中心发生快速位置交换.我们推测可能是由于3-甲基邻苯二胺和3-溴邻苯二胺分子中取代基位置的不对称性, 使生成的配合物分子中PPh3和Cl在金属中心的交换受阻, 从而使两种异构体并存.
进一步, 化合物2, 3, 4a和4b的结构经X射线单晶衍射得以确认, 其中2和4a的结构如图 1~2所示, 晶体学数据见表 1.从晶体结构可以看出, 化合物2仅有a构型, 表明a构型可能是热力学更稳定的产物[16].虽然配合物3和4b的晶体质量不够好, 但是粗结构显示3也有a和b两种空间结构, 进一步证明了该化合物固体有两种构型异构体, 但是在室温溶液中PPh3和Cl配体会发生快速位置交换, 从而使两种异构体的信号峰合并. 4a和4b的单晶结构证实, 由于邻苯二胺分子中的不对称取代, 可以使PPh3和Cl配体交换受阻, a和b两种异构体并存.这种配体之间的位置交换异构在配合物中是一种常见现象[17].
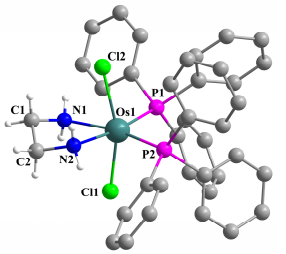
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.420(2), Os(1)—Cl(2) 2.439(2), Os(1)—P(1) 2.315(2), Os(1)—P(2) 2.309(2), Os(1)—N(1) 2.199(8), Os(1)—N(2) 2.196(8), N(1)—C(1) 1.482(13), C(1)—C(2) 1.488(17), C(2)—N(2) 1.467(13); P(1)—Os(1)—P(2) 97.43(8), N(1)—Os(1)—N(2) 76.8(3).
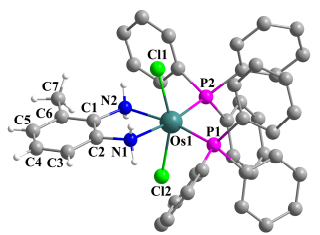
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.4410(10), Os(1)—Cl(2) 2.4151(10), Os(1)—P(1) 2.2827(11), Os(1)—P(2) 2.2962(11), Os(1)—N(1) 2.175(4), Os(1)—N(2) 2.161(3), N(1)—C(2) 1.451(6), C(1)—C(2) 1.358(7), C(2)—C(3) 1.414(7), C(3)—C(4) 1.375(8), C(4)—C(5) 1.349(8), C(5)—C(6) 1.414(8), C(6)—C(1) 1.394(7), C(6)—C(7) 1.404(8), C(1)—N(2) 1.459(6); P(1)—Os(1)—P(2) 97.31(4), N(1)—Os(1)—N(2) 77.65(15).
 下载:
导出CSV
下载:
导出CSV
| Complex | 2 | 4a | 10•1.5CH2Cl2 | 11•2CHCl3 |
| Empirical formula | C38H38Cl2N2OsP2 | C43H40Cl2N2OsP2 | C48H36Cl4N2OsP2•1.5CH2Cl2 | C52H46Cl2N2OsP2•2CHCl3 |
| Formula weight | 845.74 | 907.81 | 1162.11 | 1260.68 |
| Temperature/K | 294.00(10) | 99.99(11) | 100.00(10) | 100.01(10) |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/c | P-1 | P-1 | P-1 |
| a/Å | 10.93990(10) | 11.2009(4) | 10.7120(2) | 12.4079(4) |
| b/Å | 30.8282(2) | 12.1855(3) | 10.8899(2) | 13.2774(4) |
| c/Å | 20.30830(10) | 13.9418(3) | 22.1971(3) | 15.9101(5) |
| α/(°) | 90 | 93.727(2) | 96.6240(10) | 94.114(3) |
| β/(°) | 93.8270(10) | 99.049(2) | 94.0840(10) | 99.552(3) |
| γ/(°) | 90 | 103.329(2) | 115.657(2) | 90.277(3) |
| V/Å3 | 6833.85(8) | 1818.41(9) | 2297.14(8) | 2577.73(14) |
| Z | 4 | 2 | 2 | 2 |
| ρcalcd/(g•cm-3) | 1.644 | 1.658 | 1.680 | 1.624 |
| μ/mm-1 | 9.602 | 3.776 | 9.948 | 2.990 |
| F(000) | 3360.0 | 904.0 | 1150.0 | 1256.0 |
| Crystal size/mm3 | 0.38×0.32×0.21 | 0.15×0.14×0.03 | 0.42×0.35×0.16 | 0.12×0.10×0.02 |
| θ range/(°) | 3.60 to 72.92 | 3.46 to 25.00 | 4.05 to 72.49 | 3.31 to 25.00 |
| Reflns collected | 38010 | 13377 | 28165 | 24893 |
| Indep reflns | 13413 | 6405 | 8980 | 9065 |
| Data/restraints/params | 13413/6/811 | 6405/18/452 | 8980/81/596 | 9065/0/608 |
| GOF on F2 | 1.067 | 1.095 | 1.086 | 1.020 |
| R1/wR2 [I>2σ(I)] | 0.0665/0.2039 | 0.0305/0.0673 | 0.0358/0.0947 | 0.0258/0.0529 |
| R1/wR2 [all data] | 0.0824/0.2111 | 0.0334/0.0691 | 0.0362/0.0952 | 0.0286/0.0540 |
由以上结果不难推测, 虽然邻苯二胺类配体与乙二胺配体的电子效应接近, 但是邻苯二胺类配体的空间位阻比乙二胺大, 从而提高了配体在金属中心发生位置交换的能垒, 邻苯二胺环上取代基位置的不对称性, 会使该能垒进一步加大, 最终导致该类配体配合物产生异构.由化合物2仅有a构型可知, 乙二胺配体配合物是热力学主导的产物.
此外, 2和4a单晶结构的共同特点是, 锇中心在空间上呈现扭曲八面体的配位构型.在化合物2中, 两个Cl配体占据轴向位置相互处于对位, 而两个PPh3以及乙二胺的两个N原子位于赤道面上. Os—N键长与已报道的其它锇或钌二胺配合物的键长类似[16, 18]. 3a和3b, 4a和4b的晶体结构清楚地显示了a、b两种构型的差别主要是PPh3和Cl配体位置的不同, 3a和4a结构类似于2, 两个Cl配体占据轴向位置相互处于对位, 两个PPh3以及两个N原子位于赤道面上, 而3b和4b中刚好相反, 两个PPh3配体占据轴向位置相互处于对位, 两个Cl配体以及两个N原子位于赤道面上.
1, 10-菲啰啉是一类重要的芳香二齿N配体[19], 其共轭芳香环上1号和10号位的N原子可与金属配位形成稳定的配合物, 在分子发光[20]、催化[21]等领域具有广泛应用.基于以上脂肪族乙二胺和共轭邻苯二胺的研究结果, 我们进一步尝试了取代/非取代菲啰啉(简写作Phen*)与OsCl2(PPh3)3 (1)的反应, 如Eq. 1所示.反应在室温下二氯甲烷中以OsCl2(PPh3)3与菲啰啉物质的量比为1:1.2进行4~5 h, 将反应液浓缩, 经过滤、洗涤分离可得到固体产物OsCl2(PPh3)2(Phen*) (7~11).
|
|
化合物7~11均经过了1H NMR, 31P NMR, 13C NMR和元素分析的表征.其中, 化合物10和11的结构进一步得到单晶结构的确认(图 3和图 4), 晶体学数据见表 1.基于这几个化合物结构的相似性, 我们仅以化合物10为例进行讨论, 其它化合物的表征详见实验方法部分.从图 3可以看出, 菲啰啉取代金属中心一个PPh3配体与Os配位, 形成锇中心在空间上略微扭曲的八面体配位构型.与二胺类配体不同的是, 赤道面被菲啰啉环上两个N原子, 一个PPh3配体的P原子和一个Cl配体占据, 轴向上PPh3配体和Cl配体处于相对位置.该晶体构型的形成与1, 10-菲啰啉及其衍生物的大位阻效应有关, 使得类似于上述邻苯二胺类配体形成的a、b两种构型在空间上都过于拥挤[22]. Os—N键长[Os(1)—N(1) 2.104(3) Å, Os(1)—N(2) 2.050(3) Å]落在了典型的1, 10-菲啰啉取代的锇配合物的键长范围内[23].与晶体结构一致, 1H NMR中显示菲啰啉环上的H有三组信号, 分别位于δ 8.8(双峰), 8.3(单峰)和7.1(双峰), 分别对应C(1)—H和C(10)—H, C(5)—H和C(6)—H, C(2)—H和C(9)—H. 31P NMR在δ -17.8显示一个宽峰, 表明两个PPh3配体在溶液中会发生快速交换, 使两种P信号合并. 13C NMR在δ 156.9~124.2范围内显示有芳环碳原子的多组峰.
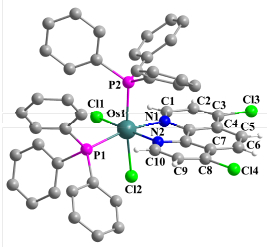
Selected bond length (Å) and bond angles (°): Os(1)—C(l1) 2.4207(9), Os(1)—Cl(2) 2.4561(8), Os(1)—P(1) 2.3405(9), Os(1)—P(2) 2.3323(9), Os(1)—N(1) 2.104(3), Os(1)—N(2) 2.050(3), N(1)—C(1) 1.330(5), C(1)—C(2) 1.401(5), C(2)—C(3) 1.364(6), C(3)—C(4) 1.416(6), C(4)—C(5) 1.426(6), C(5)—C(6) 1.350(6), C(6)—C(7) 1.433(5), C(7)—C(8) 1.408(6), C(8)—C(9) 1.370(6), C(9)—C(10) 1.397(5), C(10)—N(2) 1.344(5); P(1)—Os(1)—P(2) 104.40(3), N(1)—Os(1)—N(2) 78.50(12), Cl(1)—Os(1)—Cl(2) 84.43(3).
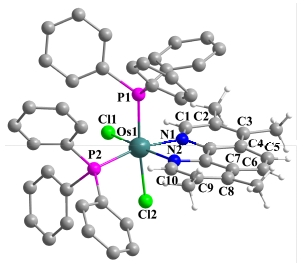
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.4269(7), Os(1)—Cl(2) 2.4624(7), Os(1)—P(1) 2.3037(8), Os(1)—P(2) 2.3318(8), Os(1)—N(1) 2.109(2), Os(1)—N(2) 2.060(2), N(1)—C(1) 1.332(4), C(1)—C(2) 1.401(4), C(2)—C(3) 1.385(5), C(3)—C(4) 1.422(4), C(4)—C(5) 1.438(4), C(5)—C(6) 1.353(4), C(6)—C(7) 1.436(4), C(7)—C(8) 1.419(4), C(8)—C(9) 1.375(5), C(9)—C(10) 1.397(4), C(10)—N(2) 1.336(4); P(1)—Os(1)—P(2) 102.74(3), N(1)—Os(1)—N(2) 78.41(10), Cl(1)—Os(1)—Cl(2) 87.46(2).
进一步, 我们尝试了化合物2~11在催化氨硼烷释氢反应方面的性能.起始的结果表明氨硼烷在无催化剂存在下脱氢难以进行, 60 ℃加热18 h仅有0.35 equiv. H2释放出来, 室温条件下催化脱氢反应也进行地很慢(表 2).进而, 依据氨硼烷和催化剂的溶解性并参考文献报道, 我们直接选取四氢呋喃(THF)与乙二醇二甲醚(DME)混合溶液(二者体积比1:1.6)为溶剂[24], 以配合物3和7为催化剂, 负载量设为5 mol%对催化反应温度进行了筛选, 发现在60 ℃时催化效果最好(表 2).此外, 通过汞中毒实验, 我们确认了催化反应为均相反应.
下载:
导出CSV
| 催化剂 | 反应温度/℃ | 反应时间/℃ | H2总释放量/equiv. |
| None | 60 | 18 | 0.35 |
| 3 | 40 | 9.2 | 1.70 |
| 3 | 50 | 8.4 | 1.84 |
| 3 | 60 | 7.6 | 2.00 |
| 7 | 40 | 16.5 | 1.85 |
| 7 | 50 | 12 | 1.93 |
| 7 | 60 | 7.3 | 2.14 |
随后, 我们使用优化条件即60 ℃ THF/DME混合溶液(1.0:1.6)为溶剂, 催化剂负载量为5 mol%测试了锇配合物2~11的催化性能, 结果如Eq. 2和表 3所示.
下载:
导出CSV
| 催化剂 | 反应时间/h | H2总释放量/ equiv. | 释放1 equiv. H2时间/min | TOFa/h-1 |
| 2 | 37.5 | 1.97 | 155 | 7.7 |
| 3 | 7.6 | 2.00 | 23.5 | 51.1 |
| 4 | 1.75 | 2.29 | 6 | 200 |
| 5 | 2.6 | 1.88 | 8 | 150 |
| 6 | 2.4 | 2.00 | 6 | 200 |
| 7 | 7.3 | 2.14 | 52 | 23.1 |
| 8 | 4.4 | 2.29 | 67 | 17.9 |
| 9 | 10.7 | 2.12 | 97 | 12.4 |
| 10 | 6.0 | 2.28 | 63 | 19.0 |
| 11 | 17.5 | 2.10 | 185 | 6.5 |
| a Calculation based on the time when 1 equiv. H2 was released. | ||||
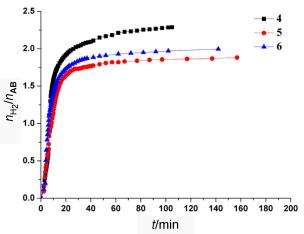
从表 3可以看出, 锇配合物2~11作为催化剂可以使氨硼烷释放1.88~2.29 equiv. H2.整体来讲, 无论是取代还是非取代邻苯二胺或菲啰啉锇配合物(3~10, 除了11), 其催化效率要优于乙二胺配体配合物2.其中, 取代型邻苯二胺锇配合物(4~6)的催化效率要明显高于其它催化剂, 尤其是3-甲基邻苯二胺取代的锇配合物4作为催化剂时, 其转化频率(TOF)值高达200 h-1, 催化性能不仅优于具有类似结构的其它锇配合物3, 5和6, 且它的释氢速度和释氢量均是锇催化剂2~11中最高的, 同时也高于已报道的多数其它金属催化剂, 是目前最高效的锇催化剂[8, 25].从图 5可以更清楚地看出, 4~6作为催化剂在刚开始几分钟释氢迅速, 6~8 min即可释放1 equiv. H2, 0.5 h后释氢速度明显变慢, 2.5 h后几乎不再释放H2, 反应结束.以上结果表明, 双齿N基配体本身的电子效应使得共轭或芳香环供电子配体较之脂肪供电子配体能显著提高锇配合物的催化反应活性.
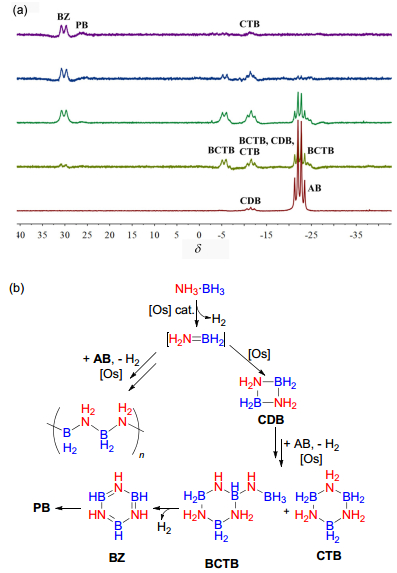
随着催化反应进行, 反应溶液中或多或少会有灰白色沉淀析出, 经分离表征可知为BN链状聚合物[NH2BH2]n, 其红外(IR)光谱中N—H的特征吸收峰在3200~3400 cm-1, B—H的特征吸收峰在2400 cm-1, B—N的特征吸收峰在1421 cm-1, 与文献报道一致[26].对反应溶液的原位11B NMR监控(图 6a)显示, 反应过程中经历了几个重要中间体.根据文献报道并结合实验事实, 推测锇催化氨硼烷转化的整个历程为(图 6b):首先, 氨硼烷在催化剂作用下迅速放出氢气, 得到瞬态产物硼氮烯BH2=NH2.随后硼氮烯有两条转化路径, 一部分多聚成BN链状聚合物[NH2BH2]n, 另一部分先二聚生成硼氮四元环化合物CDB, 与之对应的核磁硼信号在δ -11.4处出现一个三重峰(BH2); 接着, CDB再与AB作用生成BCTB和CTB, 并放出氢气, 这个过程放出氢气的量较多, 持续时间也较长, 生成的BCTB有三组硼信号, 分别是δ -23.8的四重峰(BH3), δ -11.3的三重峰(BH2)和δ -5.6的两重峰(BH), CTB和CDB的信号包埋在δ -11.3处三重峰(BH2)中; 最后, BCTB和CTB再脱氢变成δ 30.9处的硼吖嗪(BZ), 同时部分硼吖嗪会继续聚合成聚硼吖嗪(PB), 在δ 27.2处出现一个宽峰[27].
此外, 锇催化剂2~11在催化反应过程中均会发生分解, 通过原位核磁监测可以观察到反应结束时多数催化剂转化为同一个化合物, 它在11B NMR中δ -38.0处显示一组多重峰, 相对应的31P NMR在δ 21.4处显示一个宽峰, 依据文献可知为BH3•PPh3的信号[24, 28].因此, 我们推测催化反应有可能是先经历一分子PPh3配体的解离, 金属中心产生空位, 进而诱发后续的B—H和N—H键断裂脱氢, 即化合物2~11有可能是催化前体化合物, 真正的催化活性物种及其催化反应机理有待进一步研究.
通过OsCl2(PPh3)3与双齿N配体乙二胺、(取代/非取代)邻苯二胺和(取代/非取代)1, 10-菲啰啉发生反应, 合成了系列双齿N配体锇配合物2~11, 并对它们进行了系统的结构表征, 表明PPh3配体和Cl配体可以在金属中心的轴向和赤道面发生位置交换产生异构.在60 ℃, 体积比为1:1.6的四氢呋喃(THF)与乙二醇二甲醚(DME)混合溶液中, 催化剂负载量为5 mol%的条件下, 配合物2~11均具有高效的催化氨硼烷脱氢反应活性.总的来讲, 双齿N配体的电子效应使得共轭或芳香环供电子配体(3~10)较之脂肪供电子配体(2)能显著提高锇配合物的催化活性.其中, 3-甲基邻苯二胺取代的锇配合物4催化氨硼烷脱氢活性最高, 它也是目前最高效的锇催化剂.
除非特别说明, 所有实验均在手套箱中或采用标准的Schlenk操作技术, 在高纯氮气保护的无水无氧条件下进行.
所用溶剂均为分析纯, 四氢呋喃、正己烷使用前在氮气氛中经钠-二苯甲酮回流干燥处理.二氯甲烷使用前经氢化钙回流干燥处理. OsCl2(PPh3)3[17]根据文献方法合成, 氨硼烷从郑州联硼能源材料科技有限公司采购, 其它试剂从Sigma-Aldrich, Alfa-Aesar和aladdin购买.
核磁共振谱1H NMR, 31P NMR, 13C NMR在Bruker AV400 (400 MHz)或Bruker AV600 (600 MHz)核磁共振仪上测定, 1H NMR, 13C NMR采用TMS定标, 31P NMR采用85% H3PO4定标.元素分析使用Elementer Analysensystem GmbH仪器公司生产的Vario EL型元素分析仪.红外光谱使用美国伯乐公司生产的FTS-40傅里叶变换红外光谱仪测定.如无特别说明, 操作温度为298 K.熔点使用上海仪电物理光学仪器有限公司制造的SGW-X4B显微熔点仪测试.
向乙二胺(12 mg, 0.20 mmol)和OsCl2(PPh3)3 (178 mg, 0.17 mmol)的混合物中加入二氯甲烷溶剂(15 mL), 室温下搅拌反应2 h, 溶液由墨绿色变成棕黄色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成黄色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次后, 真空下干燥, 收集得到黄色固体粉末132 mg, 产率92%. m.p. 277.6~278.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.3 (t, J=8.0 Hz, 12H, PPh3), 7.1 (t, J=7.2 Hz, 6H, PPh3), 7.0 (t, J=7.2 Hz, 12H, PPh3), 3.3 (br s, 4H, NH2), 2.6 (br s, 4H, CH2); 13C NMR (101 MHz, CDCl3) δ: 138.0 (s, PPh3), 137.6 (s, PPh3), 134.6 (t, JCP=7.0 Hz, PPh3), 128.7 (s, PPh3), 127.2 (t, JCP=5.0 Hz, PPh3), 43.8 (s, CH2); 31P NMR (162 MHz, CDCl3) δ: -12.9 (s, PPh3). Anal. calcd for OsC38H38N2P2Cl2: C 53.96, H 4.53, N 3.31; found C 54.25, H 4.39, N 3.52.单晶培养:将正己烷溶剂缓慢注入到该化合物的二氯甲烷溶液上层, 通过缓慢扩散得到黄色块状晶体.
向邻苯二胺(22 mg, 0.20 mmol)和OsCl2(PPh3)3 (178 mg, 0.17 mmol)的混合物中加入二氯甲烷溶剂(15 mL), 室温下搅拌反应2 h, 溶液由墨绿色变成棕红色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成黄色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次后, 真空下干燥, 收集得到鹅黄色固体粉末136 mg, 产率90%. m.p. 268.7~269.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.4 (br s, 12H, PPh3), 7.2 (t, J=7.2 Hz, 6H, PPh3), 7.1 (t, J=7.2 Hz, 12H, PPh3), 7.0 (br s, 2H, Ar), 6.8 (br s, 2H, Ar), 5.0 (br s, 4H, NH2); 13C NMR (151 MHz, CDCl3) δ: 140.3 (s, Ar), 138.0 (d, J=7.4 Hz, Ar), 137.5 (d, J=7.7 Hz, Ar), 134.6 (t, J=7.0 Hz, PPh3), 128.9 (s, PPh3), 127.6 (s, PPh3), 127.4 (t, J=7.8 Hz, PPh3); 31P NMR (162 MHz, CDCl3)δ: -12.7 (s, PPh3). Anal. calcd for OsC42H38N2P2Cl2: C 56.44, H 4.29, N 3.13; found C 56.75, H 4.31, N 3.04.单晶培养:将正己烷溶剂缓慢注入到该化合物的二氯甲烷溶液上层, 通过缓慢扩散得到黄色块状3a和3b的混合晶体.
向3-甲基-邻苯二胺(24 mg, 0.20 mmol)和OsCl2(PPh3)3 (178 mg, 0.17 mmol)的混合物中加入二氯甲烷溶剂(15 mL), 室温下搅拌反应2 h, 溶液由墨绿色变成棕红色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成黄色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次后, 真空下干燥, 收集得到4a与4b混合黄色固体粉末133 mg, 产率为86%.由于同分异构体4a与4b无法分离, 因此将两种化合物的核磁位移一并罗列. m.p. 312.1~313.2 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.5 (q, J=8.4 Hz, 12H, PPh3), 7.2 (t, J=7.2 Hz, 6H, PPh3), 7.1 (t, J=7.2 Hz, 12H, PPh3), 6.9 (t, J=7.2 Hz, 1H, Ar), 6.8 (d, J=7.2 Hz, 1H, Ar), 6.6 (d, J=7.2 Hz, 1H, Ar), 5.0 (br s, 2H, NH2), 4.7 (br s, 2H, NH2), 1.8 (s, 3H, CH3); 13C NMR (151 MHz, CDCl3) δ: 139.3 (s, Ar), 138.2 (s, Ar), 137.0 (s, Ar), 136.8 (s, Ar), 136.7 (s, Ar), 136.5 (s, Ar), 134.6 (s, Ar), 133.7~133.5 (m, PPh3), 133.0 (s, Ar), 128.6 (s, Ar), 127.8 (s, PPh3), 126.6 (s, Ar), 126.5 (s, Ar), 126.4~126.3 (m, PPh3), 124.2 (s, Ar), 16.0 (s, CH3); 31P NMR (243 MHz, CDCl3) δ: -12.0 (s, PPh3), -12.7 (s, PPh3). Anal. calcd for OsC43H40N2P2Cl2: C 56.89, H 4.44, N 3.09; found C 57.01, H 4.34, N 3.10.单晶培养:将正己烷溶剂缓慢注入到该化合物的二氯甲烷溶液上层, 通过缓慢扩散得到黄色块状4a和4b的混合晶体.
向3-溴邻苯二胺(38 mg, 0.2 mmol)和OsCl2(PPh3)3 (178 mg, 0.17 mmol)的混合物中加入二氯甲烷溶剂(15 mL), 室温下搅拌反应2 h, 溶液由墨绿色变成棕红色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成黄色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次后, 真空下干燥, 收集得到5a与5b混合黄色固体粉末129 mg, 产率78%.由于同分异构体5a与5b无法分离, 因此将两种化合物的核磁位移一并罗列. m.p. 308.5~309.4 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.4 (br s, 12H, PPh3), 7.3 (br s, 1H, Ar), 7.2 (br s, PPh3), 7.1 (br s, 12H, PPh3), 6.9 (t, J=7.2 Hz, 1H, Ar), 6.8 (d, J=7.2 Hz, 1H, Ar), 5.1 (br s, 2H, NH2), 4.9 (br s, 2H, NH2); 13C NMR (151 MHz, CDCl3) δ: 141.2 (s, Ar), 139.6 (s, Ar), 136.8 (s, Ar), 136.6 (s, Ar), 136.5 (s, Ar), 136.2 (s, Ar), 133.5 (d, J=9.0 Hz, PPh3), 132.9 (s, Ar), 130.1 (s, Ar), 128.7 (s, Ar), 127.9 (d, J=3.0 Hz, PPh3), 126.8 (s, Ar), 126.5 (d, J=9.0 Hz, PPh3), 126.4 (d, J=9.0 Hz, PPh3), 126.0 (s, Ar), 122.6 (s, Ar); 31P NMR (243 MHz, CDCl3) δ: -12.7 (s, PPh3), -13.1 (s, PPh3). Anal. calcd for OsC42H37N2P2Cl2Br: C 51.86, H 3.83, N 2.88; found C 51.99, H 3.99, N 2.51.
向3, 6-二溴-1, 2-苯二胺(54 mg, 0.2 mmol)和OsCl2(PPh3)3 (178 mg, 0.17 mmol)的混合物中加入二氯甲烷溶剂(15 mL), 室温下搅拌反应1.5 h, 溶液由墨绿色变成棕红色, 伴随大量黄色固体析出, 经针桥过滤后将固体部分用正己烷(8 mL×3)洗涤三次, 真空干燥, 得到固体粉末115 mg.将滤液减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(15 mL), 生成黄色沉淀, 过滤, 保留固体.再经正己烷(8 mL×3)洗涤三次, 真空干燥, 得到固体粉末46 mg.两部分固体合并共161 mg, 产率90%. m.p. 281.4~282.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.5 (t, J=7.8 Hz, 12H, PPh3), 7.2 (t, J=7.2 Hz, 6H, PPh3), 7.1 (t, J=7.2 Hz, 12H, PPh3), 7.1 (s, 2H, Ar), 5.0 (s, 4H, NH2); 13C NMR (151 MHz, CDCl3) δ: 141.3 (s, Ar), 133.6 (s, PPh3), 131.1 (s, Ar), 128.2 (s, PPh3), 126.4 (br s, PPh3), 121.9 (s, Ar); 31P NMR (243 MHz, CDCl3) δ: -13.1 (s, PPh3). Anal. calcd for OsC42H36- N2P2Cl2Br2: C 47.97, H 3.45, N 2.66; found C 48.12, H 3.81, N 2.31.
向1, 10-菲啰啉(45 mg, 0.25 mmol)和OsCl2(PPh3)3 (220 mg, 0.21 mmol)混合物中加入二氯甲烷溶剂(20 mL), 室温下搅拌反应4 h, 溶液由墨绿色变成紫黑色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成紫红色沉淀, 过滤, 保留固体.再经正己烷(10 mL ×3)洗涤三次, 真空干燥, 收集得到紫红色固体粉末148 mg, 产率73%. m.p. 331.4~332.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.9 (s, 2H, Ar), 7.9 (d, J=8.0 Hz, 2H, Ar), 7.8 (s, 2H, Ar), 7.3 (t, J=7.2 Hz, 12H, PPh3), 7.1 (t, J=7.0 Hz, 6H, PPh3), 7.0 (d, J=8.4 Hz, 2H, Ar), 6.9 (t, J=7.2 Hz, 12H, PPh3); 13C NMR (101 MHz, DMSO) δ: 157.0 (s, Ar), 150.1 (s, Ar), 136.2 (s, Ar), 135.7 (s, PPh3), 130.3 (s, Ar), 129.4 (s, PPh3), 127.8 (s, Ar), 127.3 (br s, PPh3), 124.6 (s, Ar); 31P NMR (162 MHz, CDCl3) δ: -16.2 (s, PPh3). Anal. calcd for OsC48H38- N2P2Cl2: C 59.69, H 3.97, N 2.90; found C 59.48, H 4.11, N 2.88.
向4-甲基-1, 10-菲啰啉(49 mg, 0.25 mmol)和OsCl2(PPh3)3 (220 mg, 0.21 mmol)混合物中加入二氯甲烷溶剂(20 mL), 室温下搅拌反应4 h, 溶液由墨绿色变成紫黑色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成紫红色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次, 真空干燥, 收集得到紫红色固体粉末158 mg, 产率77%. m.p.>350 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.9 (d, J=7.8 Hz, 1H, Ar), 8.8 (d, J=7.8 Hz, 1H, Ar, ), 8.0 (d, J=13.2 Hz, 1H, Ar), 7.9 (d, J=12.0 Hz, 1H, Ar), 7.8 (d, J=13.8 Hz, 1H, Ar), 7.3 (d, J=10.2 Hz, 12H, PPh3), 7.1 (t, J=10.8 Hz, 6H, PPh3), 7.0 (t, J=10.5 Hz, 13H, PPh3 and Ar), 6.9 (d, J=7.8 Hz, 1H, Ar), 2.8 (s, 3H, CH3); 13C NMR (151 MHz, CDCl3) δ: 156.2 (s, Ar), 155.3 (s, Ar), 150.0 (s, Ar), 149.4 (s, Ar), 143.1 (s, Ar), 134.9 (br s, PPh3), 133.1 (s, Ar), 131.1 (s, Ar), 128.7 (s, Ar), 127.6 (s, PPh3), 125.9 (d, J=2.2 Hz, , PPh3), 125.6 (s, Ar), 123.7 (s, Ar), 122.6 (s, Ar), 122.4 (s, Ar), 17.3 (s, CH3); 31P NMR (243 MHz, CDCl3) δ: -14.5 (s, PPh3). Anal. calcd for OsC49H40N2P2Cl2: C 60.06, H 4.11, N 2.86; found C 60.11, H 4.08, N 2.78.
向4, 7-二甲基-1, 10-菲啰啉(52 mg, 0.25 mmol)和OsCl2(PPh3)3 (220 mg, 0.21 mmol)混合物中加入二氯甲烷溶剂(20 mL), 室温下搅拌反应4 h, 溶液由墨绿色变成紫黑色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成紫红色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次, 真空干燥, 收集得到紫红色固体粉末148 mg, 产率71%. m.p.>350 ℃; 1H NMR (600 MHz, CD2Cl2) δ: 8.8 (s, 2H, Ar), 8.0 (s, 2H, Ar), 7.3 (d, J=6.0 Hz, 12H, PPh3), 7.1 (t, J=6.6 Hz, 6H, PPh3), 7.0 (t, J=6.6 Hz, 12H, PPh3), 6.9 (br s, 2H, Ar), 2.8 (s, 6H, CH3); 13C NMR (151 MHz, CD2Cl2) δ: 156.4 (s, Ar), 150.2 (s, Ar), 144.5 (s, Ar), 136.0 (s, PPh3), 129.7 (s, Ar), 128.6 (s, PPh3), 126.7 (br s, PPh3), 124.6 (s, Ar), 123.3 (s, Ar), 18.4 (s, CH3); 31P NMR (243 MHz, CD2Cl2) δ: -14.5 (s, PPh3). Anal. calcd for OsC50H42N2P2Cl2: C 60.42, H 4.26, N 2.82; found C 60.55, H 4.16, N 2.66.
向4, 7-二氯-1, 10-菲啰啉(63 mg, 0.25 mmol)和OsCl2(PPh3)3 (220 mg, 0.21 mmol)混合物中加入二氯甲烷溶剂(20 mL), 室温下搅拌反应4 h, 溶液由墨绿色变成蓝紫色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成蓝紫色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次, 真空干燥, 收集得到蓝紫色固体粉末150 mg, 产率69%. m.p. 317.2~318.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.9 (d, J=6.1 Hz, 2H, Ar), 8.3 (s, 2H, Ar), 7.3 (br s, 12H, PPh3), 7.2 (t, J=6.8 Hz, 6H, PPh3), 7.1 (d, J=6.1 Hz, 2H, Ar), 7.0 (t, J=6 Hz, 12H, PPh3); 13C NMR (151 MHz, CDCl3) δ: 156.8 (s, Ar), 151.7 (s, Ar), 141.0 (s, Ar), 135.9 (s, PPh3), 129.0 (s, PPh3), 128.7 (s, Ar), 127.1 (br s, PPh3), 124.5 (s, Ar), 124.2 (s, Ar); 31P NMR (243 MHz, CDCl3) δ: -17.8 (s, PPh3). Anal. calcd for OsC48H36N2P2Cl4: C 55.71, H 3.51, N 2.71; found C 55.95, H 3.63, N 2.59.单晶培养:将正己烷溶剂缓慢注入到该化合物的二氯甲烷溶液上层, 通过缓慢扩散得到灰蓝色片状晶体
向3, 4, 7, 8-四甲基-1, 10-菲啰啉(59 mg, 0.25 mmol)和OsCl2(PPh3)3 (220 mg, 0.21 mmol)混合物中加入二氯甲烷溶剂(20 mL), 室温下搅拌反应5 h, 溶液由墨绿色变成紫红色, 减压浓缩溶剂至1 mL, 持续搅拌下加入正己烷(20 mL), 生成紫红色沉淀, 过滤, 保留固体.再经正己烷(10 mL×3)洗涤三次, 真空干燥, 收集得到紫红色固体粉末150 mg, 产率为70%. m.p.>350 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.9 (s, 2H, Ar), 7.3 (br s, 12H, PPh3), 7.3 (s, 2H, Ar), 7.2 (br s, 6H, PPh3), 7.0 (br s, 12H, PPh3), 2.9 (s, 6H, CH3), 1.8 (s, 6H, CH3).13C NMR (151 MHz, CDCl3) δ: 158.8 (s, Ar), 149.6 (s, Ar), 135.8 (s, PPh3), 133.9 (s, Ar), 130.3 (s, Ar), 128.3 (s, PPh3), 127.5 (s, Ar), 126.9 (br s, PPh3), 122.7 (s, Ar), 16.9 (s, CH3), 13.5 (br s, CH3); 31P NMR (243 MHz, CDCl3) δ: -14.9 (s, PPh3). Anal. calcd for OsC52H46N2P2Cl2: C 61.11, H 4.54, N 2.74; found C 61.38, H 4.75, N 2.66.单晶培养:将正己烷溶剂缓慢注入到该化合物的二氯甲烷溶液上层, 通过缓慢扩散得到黄色块状晶体.
无水无氧条件下, 在一个干燥的25 mL Schlenk瓶中先加入氨硼烷(1.77 mmol)的THF (5 mL)溶液, 再加入锇二齿N配合物催化剂(0.09 mmol)的DME (8 mL)溶液, 二者混合后将Schlenk瓶的侧支口关闭.接着将侧支口与测量气体的滴定管附带的塑料导管连接, 并将Schlenk瓶转移至60 ℃的油浴中, 控制磁子的转速为300 r/min, 打开支口和秒表, 收集氢气并记录时间.在不同的时间间隔内记录氢气的体积(空白实验表明, 单纯在60 ℃下加热5 mL THF和8 mL DME混合溶液, 由于溶剂挥发和体积膨胀所产生的气体体积小于0.05当量氢气的体积, 可以忽略不计).
挑选适当的晶体, 常温(294 K)或低温(100 K)下, 在日本理学SuperNova双微焦斑X射线单晶衍射仪上, 采用石墨单色化的Cu Kα (λ=1.54184 nm)或Mo Kα (λ=0.71073 nm)收集数据, 电压50 kV, 电流0.8 mA.全部数据均经过multi-scan吸收校正, 晶体结构采用olex2程序包解析, 对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正.数据存于英国剑桥数据中心, CCDC号分别为: 2, CCDC-1842871; 4a, CCDC- 1842872; 10, CCDC-1842873; 11, CCDC-1842874.
辅助材料(Supporting Information) 化合物3a, 3b和4b的晶体粗结构, [NH2BH2]n的红外光谱图以及化合物2~11的1H、31P和13C核磁共振谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Rand, D. A. J.; Dell, R. M. Hydrogen Energy: Challenges and Prospects, Royal Society of Chemistry, Cambridge, UK, 2008.
(a) Yadav, M.; Xu, Q. Energy Environ. Sci. 2012, 5, 9698.
(b) Dalebrook, A. F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Chem. Commun. 2013, 49, 8735. http://qe3gw7ge4p.search.serialssolutions.com/?ctx_ver=Z39.88-2004&ctx_enc=info%3Aofi%2Fenc%3AUTF-8&rfr_id=info:sid/xueshu.baidu.com&rft_val_fmt=info:ofi/fmt:kev:mtx:journal&rft.genre=article&rft.atitle=Ammonia-Borane%20Dehydrogenation%20Promoted%20by%20an%20Osmium%20Dihydride%20Complex%3A%20Kinetics%20and%20Mechanism&rft.jtitle=Acs%20Catalysis
(a) Staubitz, A.; Robertson, A. P. M.; Manners, I. Chem. Rev. 2010, 110, 4079.
(b) Zhang, X.; Kam, L.; Trerise, R.; Williams, T. J. Acc. Chem. Res. 2017, 50, 86. http://www.ncbi.nlm.nih.gov/pubmed/22335547
(a) Tang, Z.; Chen, X.; Chen, H.; Wu, L.; Yu, X. Angew. Chem., Int. Ed. 2013, 52, 5832.
(b) Tang, Z.; Chen, H.; Chen, X.; Wu, L.; Yu, X. J. Am. Chem. Soc. 2012, 134, 5464. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23646884
Wang, K.; Zhang, J.-G.; Man, T.-T.; Wu, M.; Chen, C.-C. Chem.-Asian. J. 2013, 8, 1076. doi: 10.1002/asia.201201241
(a) Appelt, C.; Chris Slootweg, J.; Lammertsma, K.; Uhl, W. Angew. Chem., Int. Ed. 2013, 52, 4256.
(b) Kalidindi, S. B.; Joseph, J.; Jagirdar, B. R. Energ. Environ. Sci. 2009, 2, 1274. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23471587
(a) Alcaraz, G.; Sabo-Etienne, S. Angew. Chem., Int. Ed. 2010, 49, 7170.
(b) Staubitz, A.; Robertson, A. P. M.; Sloan, M. E.; Manners, I. Chem. Rev. 2010, 110, 4023.
(c) Rossin, A.; Peruzzini, M. Chem. Rev. 2016, 116, 8848. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM20721992
(a) Esteruelas, M. A.; López, A. M.; Mora, M. ACS Catal. 2015, 5, 187; (b) Esteruelas, M. A.; Fernández, I.; López, A. M. Organometallics 2014, 33, 1104. http://openurl.ebscohost.com/linksvc/linking.aspx?stitle=Organometallics&volume=18&issue=9&spage=1606
(a) Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483.
(b) Döbler, C.; Mehltretter, G. M.; Sundermeier, U.; Beller, M. J. Am. Chem. Soc. 2000, 122, 10289.
(c) Döbler, C.; Mehltretter, G. M.; Sundermeier, U.; Beller, M. J. Organomet. Chem. 2001, 621, 70. (d) Heravi, M. M.; Zadsirjan, V.; Esfandyari, M.; Lashaki, T. B. Tetrahedron: Asymmetry 2017, 28, 987. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM25636054
(a) Esteruelas, M. A.; Honczek, N.; Oliván, M.; Onate, E.; Valencia, M. Organometallics 2011, 30, 2468.
(b) Bertoli, M.; Choualeb, A.; Lough, A. J.; Moore, B.; Spasyuk, D.; Gusev, D. G. Organometallics 2011, 30, 3479.
(c) Buil, M. L.; Esteruelas, M. A.; Herrero, J.; Izquierdo, S.; Pastor, I. M.; Yus, M. ACS Catal. 2013, 3, 2072.
(d) Chelucci, G.; Baldino, S.; Baratta, W. Acc. Chem. Res. 2015, 48, 363.
(e) Bolaño, T.; Esteruelas, M. A.; Gay, M. P.; Oñate, E.; Pastor, I. M.; Yus, M. Organometallics 2015, 34, 3902.
(f) Barbato, C.; Baldino, S.; Ballico, M.; Figliolia, M.; Magnolia, S.; Siega, K.; Herdtweck, E.; Strazzolini, P.; Chelucci, G.; Baratta, W. Organometallics 2018, 37, 65.. http://openurl.ebscohost.com/linksvc/linking.aspx?stitle=Inorganic%20Chemistry&volume=30&issue=5&spage=1159
Spasyuk, D.; Vicent, C.; Gusev, D. G. J. Am. Chem. Soc. 2015, 137, 3743. doi: 10.1021/ja512389y
Buil, M. L.; Esteruelas, M. A.; Gay, M. P. Organometallics 2018, 37, 603. doi: 10.1021/acs.organomet.7b00906
(a) Baratta, W.; Bossi, G.; Putignano, E.; Rigo, P. Chem.-Eur. J. 2011, 17, 3474.
(b) Chelucci, G.; Baldino, S.; Baratta, W. Coord. Chem. Rev. 2015, 300, 29. http://www.ncbi.nlm.nih.gov/pubmed/21341330
Baker, R. T.; Gordon, J. C.; Hamilton, C. W. J. Am. Chem. Soc. 2012, 134, 5598. doi: 10.1021/ja210542r
When the article was prepared, a similar synthetic method for complex 2 was reported by Baratta. Please see Ref.[10f] for details.
Nascimento, R. D.; Silva, A. K.; Lião, L. M. J. Mol. Struct. 2018, 1151, 277. doi: 10.1016/j.molstruc.2017.09.044
Hoffman, P. R.; Caulton, K. G. J. Am. Chem. Soc. 1975, 97, 4221. doi: 10.1021/ja00848a012
(a) Lay, P. A.; Sargeson, A. M.; Skelton, B, W. J. Am. Chem. Soc. 1982, 104, 6161.
(b) Clapham, S. E.; Morris, R. H. Organometallics 2005, 24, 479.
(c) McQueen, J. S.; Nagao, N.; Eberspacher, T. Inorg. Chem. 2003, 42, 3815.
(d) Ettner, N.; Hillen, W.; Ellestad, G. A. J. Am. Chem. Soc. 1993, 115, 2546.
(e) Peacock, A. F. A.; Habtemariam, A.; Moggach, S. A. Inorg. Chem. 2007, 46, 4049.
(f) Gong, L.; Lin, Y.; Wen, T. B. Organometallics 2009, 28, 1101.
(g) Martínez-Peña, F.; Pizarro, A. M. Chem.-Eur. J. 2017, 23, 16231. doi: 10.1002/chin.198305307/abstract
Luman, C. R.; Castellano, F. N. In Comprehensive Coordination Chemistry Ⅱ, 2nd ed., Vol. 1, Eds.: Meyer, T. J.; McCleverty, J. A., Elsevier Ltd., Pergamon, 2003, p. 25. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM17330969
(a) Akerboom, S.; van den Elshout, J. J. M. H.; Mutikainen, I. Eur. J. Inorg. Chem. 2013, 2013, 6137.
(b) Nakagawa, A.; Ito, A.; Sakuda, E. Eur. J. Inorg. Chem. 2017, 3794.
(c) Glazer, E. C.; Magde, D.; Tor, Y. J. Am. Chem. Soc. 2007, 129, 8544. doi: 10.1002/ejic.201301000/full
Sjögren, M. P. T.; Frisell, H. B. Organometallics 1997, 16, 942. doi: 10.1021/om960260i
(a) Zheng, A.-X.; Si, J.; Tang, X.-Y.; Miao, L.-L.; Yu, M.; Hou, K.-P.; Wang, F.; Li, H.-X.; Lang, J.-P. Inorg. Chem. 2012, 51, 10262.
(b) Zheng, A.-X.; Wang, H.-F.; Lü, C.-N.; Ren, Z.-G.; Li, H.-X.; Lang, J.-P. Dalton Trans. 2012, 41, 558.
(c) Li, F.-L.; Yang, S.-P.; Zhang, W.-H.; Liu, Q.; Yu, H.; Chen, J.-X.; Lang, J.-P. ChemistrySelect 2016, 1, 2979. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM22985442
(a) Liu, B.; Zhao, Q.; Wang, H. Chin. J. Chem. 2012, 30, 2158.
(b) Nakamura, A.; Sato, T.; Kuroda, R. Chem. Commun. 2004, 2858.
(c) Carlson, B.; Phelan, G. D.; Kaminsky, W. J. Am. Chem. Soc. 2002, 124, 14162.
(d) Cheng, Y. K.; Cheung, J.; Che, K.-K.; Chi, M. Chem. Commun. 1997, 623.
(e) Carlson, B.; Phelan, G. D.; Benedict, J. B. Inorg. Chim. Acta 2006, 359, 1093. http://iopscience.iop.org/0256-307X/30/10/104205
Bhattacharya, P.; Krause, J. A.; Guan, H. J. Am. Chem. Soc. 2014, 136, 11153. doi: 10.1021/ja5058423
Duman, S.; Özkar, S. Int. J. Hydrogen Energy 2013, 38, 180. doi: 10.1016/j.ijhydene.2012.10.041
(a) Rossin, A.; Rossi, A.; Peruzzini, M. ChemPlusChem 2014, 79, 1316.
(b) Metters, O. J.; Chapman, A. M.; Robertson, A. P. M.; Woodall, C. H.; Gates, P. J.; Wass, D. F.; Manners, I. Chem. Commun. 2014, 50, 12146.
(c) Robertson, A. P. M.; Leitao, E. M.; Jurca, T.; Haddow, M. F.; Helten, H.; Lloyd-Jones, G. C.; Manners, I. J. Am. Chem. Soc. 2013, 135, 12670.
(d) Pons, V.; Baker, R. T. Angew. Chem., Int. Ed. 2008, 47, 9600.
(e) Staubitz, A.; Presa Soto, A.; Manners, I. Angew. Chem., Int. Ed. 2008, 47, 6212. http://openurl.ebscohost.com/linksvc/linking.aspx?stitle=Chempluschem&volume=79&issue=9&spage=1316
(a) Kalviri, H. A.; Gärtner, F.; Ye, G. Chem. Sci. 2015, 6, 618. (b) Shaw, W. J.; Linehan, J. C.; Szymczak, N. K. Angew. Chem., Int. Ed. 2008, 47, 7493. http://www.ncbi.nlm.nih.gov/pubmed/28706630
Sayalero, S.; Pericas, M. A. Synlett 2006, 2585. doi: 10.1002/chin.200707153/full

图 1 化合物2的晶体结构图
Figure 1 Molecular structure of 2
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.420(2), Os(1)—Cl(2) 2.439(2), Os(1)—P(1) 2.315(2), Os(1)—P(2) 2.309(2), Os(1)—N(1) 2.199(8), Os(1)—N(2) 2.196(8), N(1)—C(1) 1.482(13), C(1)—C(2) 1.488(17), C(2)—N(2) 1.467(13); P(1)—Os(1)—P(2) 97.43(8), N(1)—Os(1)—N(2) 76.8(3).

图 2 化合物4a的晶体结构图
Figure 2 Molecular structure of 4a
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.4410(10), Os(1)—Cl(2) 2.4151(10), Os(1)—P(1) 2.2827(11), Os(1)—P(2) 2.2962(11), Os(1)—N(1) 2.175(4), Os(1)—N(2) 2.161(3), N(1)—C(2) 1.451(6), C(1)—C(2) 1.358(7), C(2)—C(3) 1.414(7), C(3)—C(4) 1.375(8), C(4)—C(5) 1.349(8), C(5)—C(6) 1.414(8), C(6)—C(1) 1.394(7), C(6)—C(7) 1.404(8), C(1)—N(2) 1.459(6); P(1)—Os(1)—P(2) 97.31(4), N(1)—Os(1)—N(2) 77.65(15).

图 3 化合物10的晶体结构图
Figure 3 Molecular structure of 10
Selected bond length (Å) and bond angles (°): Os(1)—C(l1) 2.4207(9), Os(1)—Cl(2) 2.4561(8), Os(1)—P(1) 2.3405(9), Os(1)—P(2) 2.3323(9), Os(1)—N(1) 2.104(3), Os(1)—N(2) 2.050(3), N(1)—C(1) 1.330(5), C(1)—C(2) 1.401(5), C(2)—C(3) 1.364(6), C(3)—C(4) 1.416(6), C(4)—C(5) 1.426(6), C(5)—C(6) 1.350(6), C(6)—C(7) 1.433(5), C(7)—C(8) 1.408(6), C(8)—C(9) 1.370(6), C(9)—C(10) 1.397(5), C(10)—N(2) 1.344(5); P(1)—Os(1)—P(2) 104.40(3), N(1)—Os(1)—N(2) 78.50(12), Cl(1)—Os(1)—Cl(2) 84.43(3).

图 4 化合物11的晶体结构图
Figure 4 Molecular structure of 11
Selected bond length (Å) and bond angles (°): Os(1)—Cl(1) 2.4269(7), Os(1)—Cl(2) 2.4624(7), Os(1)—P(1) 2.3037(8), Os(1)—P(2) 2.3318(8), Os(1)—N(1) 2.109(2), Os(1)—N(2) 2.060(2), N(1)—C(1) 1.332(4), C(1)—C(2) 1.401(4), C(2)—C(3) 1.385(5), C(3)—C(4) 1.422(4), C(4)—C(5) 1.438(4), C(5)—C(6) 1.353(4), C(6)—C(7) 1.436(4), C(7)—C(8) 1.419(4), C(8)—C(9) 1.375(5), C(9)—C(10) 1.397(4), C(10)—N(2) 1.336(4); P(1)—Os(1)—P(2) 102.74(3), N(1)—Os(1)—N(2) 78.41(10), Cl(1)—Os(1)—Cl(2) 87.46(2).

图 5 锇配合物4~6(负载量5 mol%)在60 ℃, 1:1.6的THF/DME混合液中催化氨硼烷释氢过程
Figure 5 H2 release from ammonia borane (in 1:1.6 THF/ DME) at 60 ℃ catalyzed by complexes 4~6 (5 mol%)

图 6 锇催化氨硼烷脱氢机理
Figure 6 Proposed mechanisms for the Os catalyzed dehydrogenation of ammonia-borane
表 1 化合物2, 4a, 10•1.5CH2Cl2和11•2CHCl3的晶体学数据
Table 1. Crystal data and refinement details for 2, 4a, 10•1.5CH2Cl2 and 11•2CHCl3
| Complex | 2 | 4a | 10•1.5CH2Cl2 | 11•2CHCl3 |
| Empirical formula | C38H38Cl2N2OsP2 | C43H40Cl2N2OsP2 | C48H36Cl4N2OsP2•1.5CH2Cl2 | C52H46Cl2N2OsP2•2CHCl3 |
| Formula weight | 845.74 | 907.81 | 1162.11 | 1260.68 |
| Temperature/K | 294.00(10) | 99.99(11) | 100.00(10) | 100.01(10) |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/c | P-1 | P-1 | P-1 |
| a/Å | 10.93990(10) | 11.2009(4) | 10.7120(2) | 12.4079(4) |
| b/Å | 30.8282(2) | 12.1855(3) | 10.8899(2) | 13.2774(4) |
| c/Å | 20.30830(10) | 13.9418(3) | 22.1971(3) | 15.9101(5) |
| α/(°) | 90 | 93.727(2) | 96.6240(10) | 94.114(3) |
| β/(°) | 93.8270(10) | 99.049(2) | 94.0840(10) | 99.552(3) |
| γ/(°) | 90 | 103.329(2) | 115.657(2) | 90.277(3) |
| V/Å3 | 6833.85(8) | 1818.41(9) | 2297.14(8) | 2577.73(14) |
| Z | 4 | 2 | 2 | 2 |
| ρcalcd/(g•cm-3) | 1.644 | 1.658 | 1.680 | 1.624 |
| μ/mm-1 | 9.602 | 3.776 | 9.948 | 2.990 |
| F(000) | 3360.0 | 904.0 | 1150.0 | 1256.0 |
| Crystal size/mm3 | 0.38×0.32×0.21 | 0.15×0.14×0.03 | 0.42×0.35×0.16 | 0.12×0.10×0.02 |
| θ range/(°) | 3.60 to 72.92 | 3.46 to 25.00 | 4.05 to 72.49 | 3.31 to 25.00 |
| Reflns collected | 38010 | 13377 | 28165 | 24893 |
| Indep reflns | 13413 | 6405 | 8980 | 9065 |
| Data/restraints/params | 13413/6/811 | 6405/18/452 | 8980/81/596 | 9065/0/608 |
| GOF on F2 | 1.067 | 1.095 | 1.086 | 1.020 |
| R1/wR2 [I>2σ(I)] | 0.0665/0.2039 | 0.0305/0.0673 | 0.0358/0.0947 | 0.0258/0.0529 |
| R1/wR2 [all data] | 0.0824/0.2111 | 0.0334/0.0691 | 0.0362/0.0952 | 0.0286/0.0540 |
 下载: 导出CSV
下载: 导出CSV
表 2 THF/DME混合溶液中不同温度和催化剂条件下氨硼烷释氢结果
Table 2. Dehydrogenation of ammonia borane in THF/DME under different temperatures and catalysts
| 催化剂 | 反应温度/℃ | 反应时间/℃ | H2总释放量/equiv. |
| None | 60 | 18 | 0.35 |
| 3 | 40 | 9.2 | 1.70 |
| 3 | 50 | 8.4 | 1.84 |
| 3 | 60 | 7.6 | 2.00 |
| 7 | 40 | 16.5 | 1.85 |
| 7 | 50 | 12 | 1.93 |
| 7 | 60 | 7.3 | 2.14 |
下载: 导出CSV
表 3 化合物2~11在60 ℃ THF/DME混合溶液中催化氨硼烷释氢
Table 3. Dehydrogenation of ammonia borane with catalysts 2~11 (5 mol%) in THF/DME at 60 ℃
| 催化剂 | 反应时间/h | H2总释放量/ equiv. | 释放1 equiv. H2时间/min | TOFa/h-1 |
| 2 | 37.5 | 1.97 | 155 | 7.7 |
| 3 | 7.6 | 2.00 | 23.5 | 51.1 |
| 4 | 1.75 | 2.29 | 6 | 200 |
| 5 | 2.6 | 1.88 | 8 | 150 |
| 6 | 2.4 | 2.00 | 6 | 200 |
| 7 | 7.3 | 2.14 | 52 | 23.1 |
| 8 | 4.4 | 2.29 | 67 | 17.9 |
| 9 | 10.7 | 2.12 | 97 | 12.4 |
| 10 | 6.0 | 2.28 | 63 | 19.0 |
| 11 | 17.5 | 2.10 | 185 | 6.5 |
| a Calculation based on the time when 1 equiv. H2 was released. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

