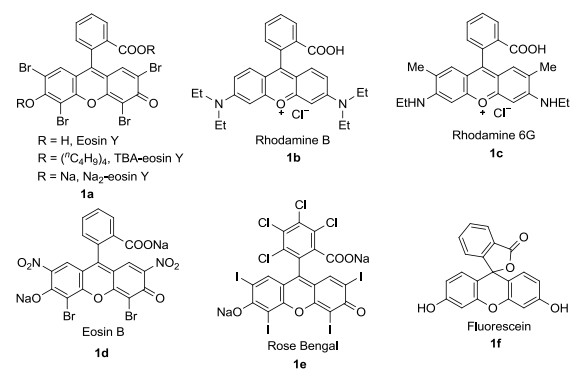
图 1.
常见氧杂蒽类有机染料结构式
Figure 1.
Structures of common oxime organic dyes

可见光是一种廉价易得、储存丰富、绿色的可再生资源[1].可见光诱导的反应因反应条件温和、操作简单等优点被广泛关注[2].典型的光照源主要为LED灯或家用日光灯, 相比于经典光化学反应中的紫外线反应器更廉价、便捷[3].光诱导反应的引发离不开光催化剂, 因此, 光催化剂的种类被不断地拓展.目前常见的光催化剂主要有钌、钯、铷、铱等过渡金属[3], TiO2、ZnO、V2O5等无机金属氧化物[4], 金属修饰的半导体[5], 以及各种有机染料[6].与过渡金属催化剂相比, 有机染料光催化剂具有廉价易得、低毒、易处理等特点[7].与无机金属光催化剂相比, 有机染料光催化剂对可见光吸收好, 并且可以通过合理的设计和合成对它们的光氧化还原性质进行精细调整[8].与金属修饰的半导体光催化剂相比, 有机染料光催化剂溶解性好、价格便宜、对环境污染小[9].
氧杂蒽类有机染料分子中含有大π共轭体系(Figure 1), 可以在能量较低的可见光范围进行π-π*跃迁[10]. 2013年, Albini课题组[11]报道了常用的光催化剂, 例如芳香族酮、醌类、杂环类、染料在光催化有机合成中的应用趋势.随后, Fukuzumi课题组[12]和Nicewicz课题组[13]对多种有机染料光催化剂在有机合成中的应用和反应机理进行了总结, 后者又对此类光催化剂的光物理和电化学特性进行了补充.其中, König课题组[14]和Singh课题组[15]分别针对Eosin Y作光催化剂在有机合成中应用进行了报道. 2017年, 胡新根等[16]总结了可见光促进的氧合反应中氧气的作用机理.上述报道均体现了有机染料在光促进有机合成领域的广泛应用价值.本文对Eosin Y在有机合成中的应用进行了补充, 并对Rose Bengal、Rhodamine 6G、Eosin B、Rose Bengal、Fluorescein这五种常见氧杂蒽类有机染料在可见光催化有机合成中的反应实例和具有代表性的反应机理进行了整理和归纳.
由于不同的有机染料结构具有不同的光物理性质, 这导致其在不同光介导电子转化(PET)过程中的催化效果具有一定差别[13].为了对相关反应中染料光敏剂类型及照射波长选择提供参考, 现将四种代表性染料的多种光物理性质(最大吸收波长、荧光寿命、基态能量、基态氧化还原电位、单线态激发态氧化还原电位、三线态激发态氧化还原电位)罗列于表 1.
 下载:
导出CSV
下载:
导出CSV
| Dye type | λmaxabs/nm | τf/ns | Excited state Energies/eV | Ground state redox potentials (V vs SCE) | Excited state redox potentials (V vs SCE): S1 | Excited state redox potentials (V vs SCE): T1 | |||||||
| E0, 0S1 | E0, 0T1a | E1/2red | E1/2ox | EredS1 | EoxS1 | EredT1 | EoxT1 | ||||||
| Eosin Y | 520a 533b |
2.1a 2.66a |
2.31a | 1.91 | -1.08a, c -1.13a, c |
+0.76a, c +0.72a, c |
+1.23a, c | -1.58a, c | +0.83a, c | -1.15a, c | |||
| Rose Bengal | 549 | 0.50 | 2.17a | 1.8 | -0.99a, c -0.78 |
+0.84a, c | +1.18a, c | -1.33a, c | +0.81a, c | -0.96a, c | |||
| Rhodamine B | 550d | 2.45a | 2.22a | 1.80 | -0.96a, c | +0.91a, c | +1.26a, c | -1.31a, c | +0.84a, c | -0.89a, c | |||
| Rhodamine 6G | 530b | 4.13a | 2.23 | 2.09 | -1.14c | +1.23 | +1.18c | -1.09a | +0.95a | -0.86a | |||
| a In MeOH. b In EtOH. c Potential originally reported relative to the Ag/AgCl reference electrode; referenced to SCE by subtracting 0.039 V from the reported valued. d In H2O. | |||||||||||||
2011年, Zeitler课题组[17]报道了醛的光催化非对称α-烷基化反应(Eq. 1).该方法以Eosin Y为光催化剂, 选择性好, 分离产率高.根据文献数据并通过实验验证, 以Hantzsch酯为氢源可以避免潜在的副反应.
叔胺中氮原子邻位的C(sp3)—H键与亲核试剂反应是一种简便、有效地形成C—C键的方法. 2011年, Tan等[18]以Rose Bengal为光催化剂, 在绿光照射下将N-芳基-四氢异喹啉分别与硝基烷基(Eq. 2)和酮(Eq. 3)反应, 完成了三级胺的α-官能团化.后者与经典Mannich反应相比, 不需要另外制备亚胺中间体, 操作更简单.同年, 该课题组[19]将Rose Bengal负载在石墨氧化物上作为光敏剂, 在绿光照射下将N-芳基-四氢异喹啉与氰化物(Eq. 4)和三氟甲基(Eq. 5)反应, 对比单纯的光敏剂, 产率得到了显著的提升.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
次年, Wu课题组[20]和Xu课题组[21]继续将N-芳基-四氢异喹啉与不同缺电子试剂反应.前者以TBA-Eosin Y为光催化剂, 以氧气为氧化剂(Eq. 6);后者以KF、CuI为协同催化剂(Eqs. 7, 8).
|
|
(6) |
|
|
(7) |
|
|
(8) |
2013年, Rueping等[22]以四氢异喹啉为实验对象, 通过连续流反应装置, 多方面改进了光催化交叉脱氢偶联反应(Eqs. 9, 10).该方法以Rose Bengal为光催化剂, 以绿光为光源, 在含水混合溶液中进行.与传统一锅煮的方法相比, 该方法光照面积大, 反应时间短, 副反应少, 反应效率高, 而且能对反应参数进行精确控制和预测.
|
|
(9) |
|
|
(10) |
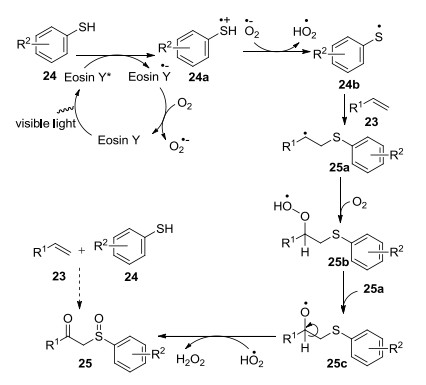
2014年, Yadav课题组[23]以烯烃和硫醇为底物, 以Eosin Y为光敏剂, 氧气为氧化剂, 在光催化下合成了β-羰基亚砜(Eq. 11).其中硫醇作供电子试剂, 氧气作吸电子试剂, 反应中的副产物过氧化氢可以作氧化剂将硫化物转化成亚砜.
|
|
(11) |
推测机理如下: Eosin Y受光照转化成激发态Eosin Y*, 激发态Eosin Y*通过系统内部偶联迅速由单线态生成稳定的三线态, 染料三线态激发态3Eosin Y*通过还原猝灭转化成Eosin Y·-, 其再被O2氧化成Eosin Y完成一次光催化循环.其中, 伴随激发态3Eosin Y*被供电子试剂硫醇的猝灭过程, 同时生成自由基阳离子24a, 其去质子化生成稳定的自由基23b, 自由基24b与烯烃23反应生成产物25a, 25a与氧气反应生成过氧自由基25b, 继而得到烷氧基自由基25c, 后者提供一个氢自由基生成过氧化氢, 过氧化氢再将硫醇氧化成亚砜, 得到目标产物25 (Scheme 1).
2014年, Kappe和Rincon课题组[24]通过持续流微通道光催化法实现了酮的α-三氟甲基化(Eq. 12).反应分两步进行, 首先羰基化合物原位转化成硅醇酯, 再加入三氟甲基自由基前体与之混合, 在可见光照射下生成目标产物.以醛为底物进行三氟甲基化可以避免使用强碱, 而且, 该方法光敏剂用量少, 产率高, 官能团容忍性好, 所需时间极短, 因此是一种十分具有优势的合成方法.
|
|
(12) |
2014年, Li等[25]报道了吲哚3位的光催化酰基化反应(Eq. 13).以Rose Bengal为光催化剂, 氧气为氧化剂, 四甲基乙二胺(TMEDA)为碳源, 向反应体系中加入少量的水可以促进水解, 提高产率, 加入KI共催化使得产率进一步提高.此催化体系反应条件温和, 分离产率良好, 具有很好的官能团容忍性.
次年, Singh课题组[26]以溴氟乙酸乙酯、取代吲哚、取代苯胺为底物, 通过光催化活化偶联C(sp2)—C(sp3)键, 得到二吲哚基、二苯胺基乙酸酯衍生物(Eqs. 14, 15)以及非对称二芳基乙酸酯衍生物(Eq. 16).其中, 以溴氟乙酸乙酯和取代吲哚为底物反应时, 富电子芳环对反应有利, 具有一定的官能团容忍性.以溴氟乙酸乙酯和取代苯胺为底物时, 烯基和炔基取代的苯胺、氟取代苯胺、N-苄基-1-萘胺具有很好的产率.反应通过单电子转换机理进行, 是一条方便的构C(sp2)—C(sp3)键的反应路线.
|
|
(13) |
|
|
(14) |
|
|
(15) |
|
|
(16) |
2015年, Wang课题组[27]通过可见光催化反应得到了四氢呋喃的乙烯化产物(Eq. 17).反应以Eosin Y为光敏剂, 叔丁基过氧化氢(TBHP)作氧化剂, 四氢呋喃脱掉一个氢自由基生成一个寿命较长的α-碳自由基, 其再与炔烃反应完成C—H到C—C键的转化, 得到四氢呋喃的乙烯化产物.
|
|
(17) |
2016年, Dixon课题组[28]报道了(杂)芳胺、(杂)芳醛、缺电子烯烃的三组分光催化还原偶联反应, 生成了一系列γ-氨基酸衍生物(Eq. 18).该方法以Eosin Y为光敏剂, Hantzsch酯为终端还原剂.对照实验显示, 反应中形成的Hantzsch酯自由基阳离子(HE·+)的介入通过质子耦合电子转移可以显著降低亚胺的氧化还原电势, 促进亚胺的还原, 使反应进行地更顺利.
|
|
(18) |
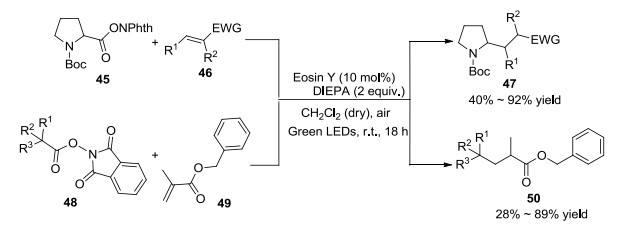
2016年, König课题组[29]报道了生物质来源化合物的脱羧烷基化(Scheme 2).该方法底物适用性广, 可以应用于N-Boc保护的脯氨酸与不同的缺电子烯烃反应, 也适用于不同N-(酰氧基)邻苯二甲酰胺衍生物与甲基丙烯酸苄酯反应.
2016年, Wang课题组[30]以芳基乙炔和氨基溴化β, β-二氰基芳香化合物为底物, 完成了芳基乙炔的双官能化反应(Eq. 19).实验结果表明, 对炔烃的直接双官能团化主要形成E式的溴化烯烃衍生物.
2016年, Wang课题组[31]通过可见光诱导芳基重氮盐与硝基烯烃的交叉偶联合成二苯乙烯衍生物(Eq. 20).反应过程中重氮盐先转化成芳基自由基, 芳基自由基被硝基烯烃捕获, 之后硝基作为离去基团离去, 完成消除反应生成目标产物.
2016年, Xie等[32]通过以硼为中心的碳硼烷基自由基进行B-C偶联, 合成B(3)芳基化的o-碳硼烷(Eq. 21).该方法产率高, 区域选择性高, 官能团容忍性好, 富电子芳烃比缺电子芳烃更利于发生反应.
2016年, König课题组[33]报道了在绿光照射下, 以Eosin Y为光催化剂, 三乙胺为缚酸剂, 实现了取代芳烃与多氟溴代芳烃的二芳基偶联反应(Eq. 22).反应条件温和, 分离产率高, 具有良好的区域选择性, 不需要进行官能团保护.
同年, 该课题组[34]又报道了以Rhodamine 6G为光催化剂, 通过两次持续光介导电子转移方式(con-PET), 完成了多卤代芳烃与吡咯衍生物的偶联反应(Scheme 3).该反应可以通过控制光的颜色实现选择性的C—C键偶联, 虽然产率一般, 但是对还原电位较低的卤代芳基具有很好的适用性.
|
|
(19) |
|
|
(20) |
|
|
(21) |
|
|
(22) |
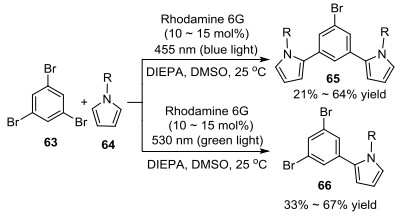
随后, König课题组[35]又以卤代芳杂环和吡咯为底物, 以Rhodamine 6G为光催化剂, 在可见光照射下实现了二芳基杂环多个位点的偶联(Eq. 23).该方法具有很好的官能团容忍性, 可用于合成具有良好药理学活性的苯并咪唑衍生物.
|
|
(23) |
N-亚磺酰亚胺分子中的C=N键被亲核试剂激活后可以进行非对应选择性反应, 多余部分很容易在温和的酸性条件下脱去, 因此是良好的手性助辅试剂. 2017年, Aleman等[36]报道了光催化N-亚磺酰亚胺的烷基化反应.在可见光条件下, 以Eosin Y为光催化剂, 将烷基试剂添加到N-亚磺酰亚胺中, 得到了具有手性结构的亚磺酰胺类衍生物(Eq. 24).该反应条件温和, 选择性良好, 具有一定的官能团容忍性.
|
|
(24) |
使用胺为碳源构建C—C键一直受到人们的关注.传统合成方法需要金属, 过氧化物, 无机氧化剂的参与.之前仅有的相关光催化反应得到的是亚甲基桥联的二-1, 3-二羰基化合物. 2017年, He等[37]以Rose Bengal为光敏剂, 通过可见光催化氧化叔胺和β-二羰基化合物, 合成了甲基或乙基桥联的二-1, 3-二羰基化合物(Eq. 25).反应通过氧化, 消除, Michael-加成机理进行, 具有很好的官能团容忍性.
|
|
(25) |
2017年, Yao等[38]报道了可见光光催化还原α-酮酯合成2, 3-二烷基化酒石酸酯(Eq. 26).反应以Eosin Y为光敏剂, 以Hantzsch酯1, 4-二氢吡啶为氢源, 通过光催化还原偶联和柱色谱光学拆分, 可以方便地得到目标化合物的对映体和非对映体.该方法具有很好的官能团容忍性, 已经扩大到克级.
|
|
(26) |
可见光光氧化还原催化的还原偶联机理包括质子耦合和电子转移.首先, Na2-Eosin Y经可见光照射生成激发态Eosin Y*, 1, 4-二吡啶(78, HEH)与激发态Eosin Y*之间发生单电子转化(SET)生成自由基阳离子HEH·+78a和自由基负离子Eosin Y·-. Eosin Y·-与α-酮酯76之间发生电子转移, HEH·+或HEH+再与α-酮酯76a之间发生质子转移, 生成中性羰基自由基76b, 中性羰基自由基76b马上经过二聚生成2, 3-二取代的酒石酸酯77.由于α-酮酯76具有高还原电位, 推测反应中短暂存在一个氢键复合物, 这同时促进了Eosin Y·-与羰基之间的电子转移(Scheme 4).
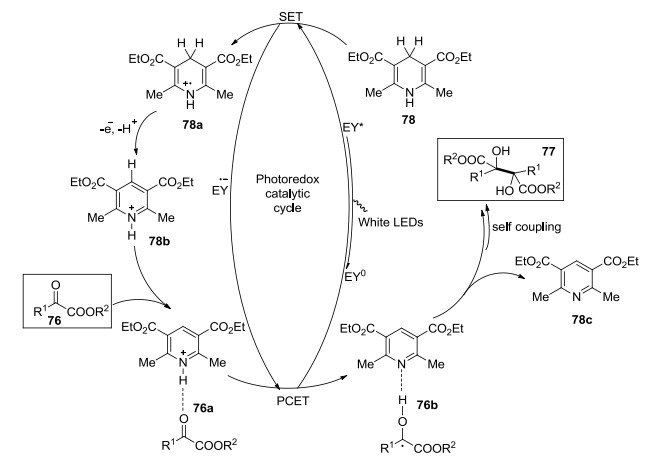
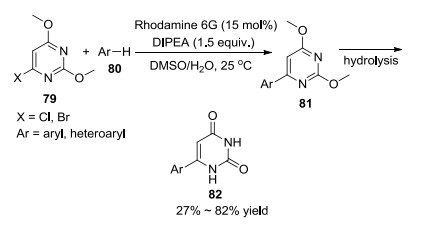
2017年, König课题组[39]以Rhodamine 6G为光催化剂, 先通过可见光催化合成芳基化的碱基, 再水解得到尿嘧啶(Scheme 5).此催化体系通过con-PET机理达到了较高的氧化还原电位, 溴代和氯代的碱基均可以在室温下完成转化.该方法操作简便, 可以在温和的条件下合成具有生物活性的芳基杂环衍生物.
2017年, Rastogi等[40]以叔胺和甲硅烷基重氮烯醇盐为底物通过曼尼希反应生成了α-重氮基-β-酮基酯(Eq. 27).反应以Rose Bengal为光催化剂, 在绿光照射下进行.实验结果显示, 底物不同位置连有吸电子基团对产率产生不同影响.
|
|
(27) |
2018年, Wu课题组[41]报道了以Eosin Y作为C—H活化氢原子转移(HAT)催化剂, 完成了系列缺电子烯烃的烷基化反应(Eq. 28).该方法底物适用性广, 适用于醚、硫醇、酰胺、醇、醛、卞基、烯丙基、环己烷、含氧天然产物以及各种富电子烯烃、缺电子烯烃、芳杂环或烷基取代的烯烃.其中, 当醇用硅胶处理后进行反应会发生环化, 得到烯胺-酮腈.部分反应具有区域选择性和立体选择性.为了进一步证明该方法的合成适用性, 使用连续流反应器可以将烷基化放大至克级, 得到了较好的产率.实验证明, 单负电子形式的Eosin Y是基于SET机理的氧化还原催化循环中的活性催化剂, 而此反应中, 中性Eosin Y是促进光HAT转化的活性催化剂.
|
|
(28) |
2010年, Tan等[42]报道了以羰基化合物和2, 2, 6, 6-四甲基哌啶氧化物(TEMPO)为底物, 以Rose Bengal为光催化剂, 在可见光诱导下实现了羰基α位的氧化胺化反应(Eq. 29).反应在乙腈或水中进行, 操作简单, 环境友好, 分离产率高, 可以获得传统方法难以获得的氟化物.
|
|
(29) |
合成亚砜的传统方法需要金属催化剂或氧化剂的参与, 而且通常得到大量过氧化副产物. 2013年, Yao等[43]报道了以分子氧为氧化剂, Rose Bengal为光催化剂, 进行硫醚的光催化磺酰化反应(Eq. 30).该方法原子经济性好, 原料转化率高, 反应程度得到了很好的控制.
|
|
(30) |
2013年, Tan课题组[44]进行了氨基醇与烯烃的光催化脱氢偶联反应(Eq. 31).反应以Rose Bengal为光催化剂, 以空气(O2)为氧化剂.氧气促进了反应的循环, 在体系中起到至关重要的作用.该方法操作简单, 是一种环境友好的烯丙基胺化方法, 可以避免金属催化剂及其他氧化剂引起的环境污染.
|
|
(31) |
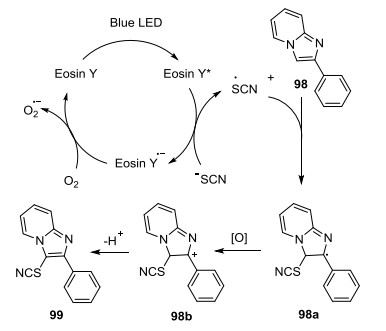
2015年, Hajra等[45]报道了可见光介导的咪唑杂环的硫氰酸化(Eq. 32).该方法对大量底物具有适用性, 也可用于咪唑并[1, 2-a]吡啶的硒氰化反应, 并且具有很好的官能团容忍性.推测反应机理如下: Eosin Y在蓝光下转化成激发态Eosin Y*, 硫氰酸根阴离子与Eosin Y*之间发生单电子转移, 同时得到光催化剂自由基负离子Eosin Y·-和硫氰酸自由基SCN·. Eosin Y·-被空气中的氧气氧化成基态, 完成光氧化还原循环.硫氰酸自由基与98反应生成自由基中间体98a, 98a再被氧化成中间体98b, 98b通过去质子化生成目标产物99 (Scheme 6).
|
|
(32) |
2015年, König课题组[46]报道以芳基亚磺酸盐和烯烃为底物, 在光催化体系下生成乙烯基砜(Eq. 33).反应以Eosin Y为光敏剂, 添加少量硝基苯作氧化剂, 该方法具有很好的官能团容忍性, 不同底物产率相差较大.
|
|
(33) |
反应过程中, 光敏剂Eosin Y先被光活化生成Eosin Y*, 活化的Eosin Y*被芳基亚磺酸盐100还原猝灭生成氧中心自由基, 氧中心自由基与相应的以硫中心自由基之间发生共振. 100a再进攻101的双键形成中间体101a.同时, 硝基苯被光催化剂自由基负离子还原形成硝基苯自由基负离子, 硝基苯负离子从101a吸收一个质子氧化成苯胺并得到产物乙烯基砜102 (Scheme 7).
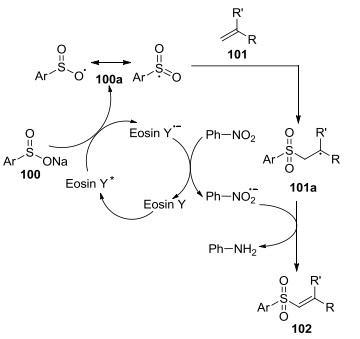
次年, Wang等[47]以烯烃与亚磺酸为底物, 经可见光引发, 直接氧化磺酰化生成β-酮砜(Eq. 34).实验结果显示, 硝基、氰基、卤素、甲基取代的烯烃以及具有较大位阻的萘磺酸也适用于此法, 但脂肪族烯烃不能发生反应.
|
|
(34) |
随后, Cai等[48]报道了以肉桂酸和磺酰肼为底物, 在可见光条件下反应合成乙烯砜(Eq. 35).该反应以染料Eosin Y为光敏剂, 氧气分子作为唯一的末端氧化剂, 需要添加KI和Cs2CO3作添加剂.平行实验结果显示, 苯乙烯或肉桂酸乙酯与磺酰肼不发生反应, 被保护的磺酰肼也不与肉桂酸发生反应, 这证实了端基羧基和肼是反应所必须的.此外, 荧光猝灭实验显示Eosin Y的荧光密度随着磺酰肼的增加显著下降, 这表明反应按单电子转化机理进行.
|
|
(35) |
2016年, Ananikov课题组[49]报道了光引发巯基-炔的点击反应合成C—S偶联产物(Eq. 36).该方法使用己烷作溶剂, 虽然Eosin Y在己烷中溶解性低, 但随着反应的进行溶解的光敏剂缓慢降解, 固相的染料随之逐渐溶解, 这种“饱和反馈”自动将光催化剂的浓度保持在最佳水平, 从而补偿了活性物质的损失.这使得产率和顺反异构的选择性大大提高.此外, 该课题组定制设计了3D打印化学光反应器, 使得反应温度控制更精确, 反应条件评估更快速, 体系的催化性能得到改善.
|
|
(36) |
2016年, Zhang等[50]通过光介导染料催化活化邻氨基苯甲酰胺5位C(sp3)—H制备α-烷基苯甲酰胺(Eq. 37).机理实验表示, 该反应通过芳基自由基的1, 5-氢原子转移, 形成N邻位的烷基自由基.该方法底物适用性广, 反应条件温和, 产率较好.
|
|
(37) |
2016年, Xie等[51]报道了o-(2, 4-二硝基苯基)肟光介导分子内环化合成菲啶的方法(Eq. 38).反应以Eosin Y为光敏剂, i-Pr2NEt为末端还原剂, 官能团容忍性好, 产率高.
|
|
(38) |
2016年, Guan等[52]以2-羧酸吲哚和偶氮二羧酸酯为底物, 经可见光介导, Rose Bengal催化得到了脱羧胺化产物(Eq. 39).该方法条件温和, 官能团适用性好, 但是产率一般.
2016年, Wu等[53]报道通过可见光驱动, Eosin B催化, 实现了噻唑衍生物和二苯基磷氧的氧化偶联(Eq. 40).此方法首次采用有机染料光催化的方法形成C(sp2)—P键, 反应条件温和, 绿色环保, 具有很好的官能团容忍性.
|
|
(39) |
|
|
(40) |
同年, König课题组[54]也报道了卤代芳烃与亚磷酸三烷基酯的氧化偶联反应(Eq. 41).该反应以Rhodamine 6G为光催化剂, N, N-二异丙基乙胺(DIPEA)为添加剂, 反应温和, 官能团容忍性好, 适用于向复杂、敏感的药理活性分子中导入磷酸酯基.
|
|
(41) |
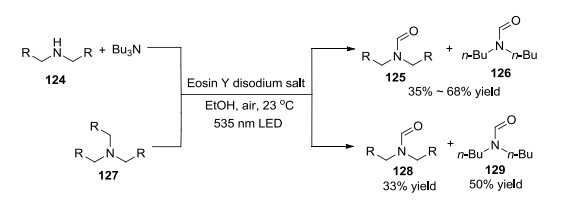
2017年, König课题组[55]通过可见光介导合成胺的N-甲酰化产物(Scheme 8).反应以Eosin Y为光敏剂, O2 (air)为氧化剂, 在室温下进行.该方法以副产物亚胺正离子为出发点, 其在空气中可直接被甲酰化而不需要添加其他甲酰化试剂, 体系简单, 但产率一般.
2017年, Aleman课题组[56]通过对硫醇-烯进行氧化串联合成烷基亚砜(Eq. 42).实验结果显示, 硫醇上连有吸电子基产率较高, 烯烃上连有较大基团时产率明显下降.为了在底物上引入氮原子, 将底物拓展至Boc保护的含氮烯烃, 依然得到了较高的产率.
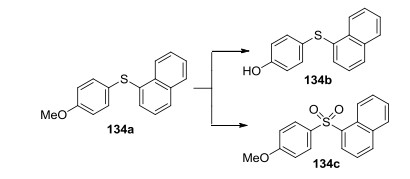
2017年, Lee等[57]报道了可见光介导合成二硫化物(Eq. 43).该方法以Eosin Y为光敏剂, 以空气中的氧气为氧化剂, 产率高, 具有很好的官能团容忍性, 已经扩展到克级.此外, o-甲氧基苯萘硫醚(134a)可以进一步转化成4-(萘-1-基硫代)苯酚(134b)和砜(134c) (Scheme 9), 它们是淀粉样蛋白聚集抑制剂的类似物.
2017年, Jiang课题组[58]报道了可控的光促进的有机硫代硫酸盐的磺化氧化和亚硫酸化反应(Scheme 10).磺化氧化和亚磺酰化转化分别涉及串联电子/能量转移和单电子转移过程.该方法操作简单, 被应用于药物的全合成实验中, 而且已经扩大到了克级.此外, 该课题组构建了一个含有亚砜的药物库.

2017年, 陈加荣等[59]采用“脱质子光致电子转移策略”, 实现了邻烯基取代苯甲酰胺的分子内自由基氢胺化合成3, 4-二氢异喹啉酮(Eq. 44).该反应利用Eosin Y-Na为光敏剂, 添加少量NaOH, 反应条件温和、操作简单、原子经济性好.此方法已经放大到克级规模, 而且以太阳光为光源得到了很好的产率, 采用连续流反应器则可以大大缩短反应时间.
2017年, Zhang等[60]以三级脂肪胺与芳基磺酰氯为底物, 经可见光照射, 曙红Eosin Y催化, 控制反应条件分别得到了乙烯基砜和磺酰胺(Eqs. 45, 46).
首先, Eosin Y被光照激发生成激发态Eosin Y*, Eosin Y*被三乙胺144捕获, 经还原猝灭形成Eosin Y·-和胺自由基阳离子144a. Eosin Y·-将一个电子转移给O2或芳基磺酰氯141, 生成芳基磺酰基自由基141a和氧气自由基负离子O2·-, Eosin Y·-自身转化成基态Eosin Y. O2·-从三乙胺自由基正离子144a攫取一个氢原子生成HO2·-和亚胺正离子144b, 亚胺正离子144b脱质子转化成烯胺144c.由于中间体亚胺正离子144b不稳定, 在氧气和水存在时容易水解生成二级胺144d.磺酰基自由基141a与烯胺144c反应转化成中间体自由基143a, 自由基143a再被氧化、脱质子生成目标产物乙烯基砜143, 磺酰基自由基141a进攻二级胺144d生成目标产物磺酰胺145 (Scheme 11).
|
|
(42) |
|
|
(43) |
|
|
(44) |
|
|
(45) |
|
|
(46) |

2017年, Zhang等[61]以三级胺与乙炔二羧酸二烷基酯为底物, 通过可见光介导进行脱烷基偶联(Eq. 47).反应以Eosin Y为光敏剂, 分子氧为氧化剂, 适用于一系列三烷基胺, 产率较好.
|
|
(47) |
2017年, Emanuelsson等[62]报道了苯甲胺的光催化氧化偶联反应(Eq. 48).作者通过特制实验装置, 实现了自然光环境的模拟.实验发现, 光照强度、反应温度、光催化剂用量、底物浓度、比表面积对反应速率影响较大, 最高速率常数达1.59×10-3 /s(由1H NMR谱监测).此外, 环上连有供电子基时转化率增加.
|
|
(48) |
2017年, Wang等[63]报道了以空气中的氧气为氧化剂, Rose Bengal为光催化剂, 实现了烯烃和硫醇的光催化磺酰化偶联(Eq. 49).该反应受电子效应影响小, 官能团容忍性好.
|
|
(49) |
2017年, Noel等[64]报道了以Eosin Y为光催化剂, 在可见光介导下实现了半胱氨酸的芳基化反应(Eq. 50).该方法在微波反应器中进行, 通过形成重氮盐提升反应的速率.此外, 在生物相容性反应条件(室温, 磷酸盐缓冲液)下, 该方法可以应用在标准肽的区域选择性芳基化反应中.
|
|
(50) |
2018年, Wang课题组[65]报道了炔烃的氢化硅烷化反应合成系列二取代和三取代的链烯基硅烷(Eq. 51).反应以Eosin Y为光敏剂, 硫醇为自由基猝灭剂, 含氢硅烷和H2O作氢源, 实验表明, 仅当有硫醇存在时, 才能实现水中氢原子的转移.该法适用于各种末端和内部炔烃, 产率高, 具有高度的区域选择性和立体选择性.
|
|
(51) |
2015年, Xiao等[66]报道了可见光催化的重氮盐的烷氧羰基化反应.反应以取代苯基重氮盐、脂肪醇以及手性醇、CO为底物, 生成相应的芳基羧酸酯(Eq. 52).烯醇和炔醇的双键和三键部分不发生反应, 说明了CO的高选择性.此外, 该方法光敏剂用量少, 不需要金属参与, 在室温下进行, 产率高, 官能团容忍性好, 可以拓展到其他芳基重氮盐.
|
|
(52) |
推测反应机理如下:首先在底物与染料激发态Dye*之间发生单电子转移生成苯基自由基157a, 同时释放氧化的染料自由基阳离子(DyeC·+), 活泼的苯基自由基157a捕捉一个CO分子, 生成苯酰基自由基158a. 158a再被DyeC·+氧化生成苄基偶氮氧鎓158b, 此时完成了染料的催化循环, 最后, 158b被甲醇捕捉生成目标产物158同时生成氟硼酸(Scheme 12).其中, 芳基自由基会攫取氢原子生成副产物芳烃.实验发现可以通过提高CO的压力显著降低副反应的发生.

2015年, Liu等[67]以芳基重氮盐、CO、杂环芳烃为底物, 以Eosin Y为光催化剂, 在可见光催化条件下合成了二芳基酮(Eq. 53).当存在一定数量的CO时, 芳基自由基可以被有效地转化成酰基自由基, 再经过系列自由基反应得到羰基化产物.
|
|
(53) |
同年, Wangelin课题组[68]以芳基重氮盐、CO、甲醇为底物, 在可见光催化下反应合成了芳基酯(Eq. 54).此法不需要氧化还原剂, 也适用于烷基酯的制备, 已经应用到工业化生产中.
|
|
(54) |
2016年, Wacharasindhu课题组[69]报道了光催化介导硫醇形成二硫化物的方法(Eq. 55).反应以Rose Bengal为光催化剂, 水为溶剂, 不需要金属催化剂或氧化剂, 反应条件温和, 分离产率高.
|
|
(55) |
2016年, Zhang等[70]报道了以Rose Bengal为光催化剂, 空气(O2)作氧化剂, 在可见光介导下实现了硫醇和P(O)化合物的氧化偶联(Eq. 56).该方法避免了使用强氧化剂和过渡金属催化剂对环境的污染, 也不需要高温条件, 是一条绿色环保、操作简便的合成路径.
|
|
(56) |
迈耶斯双环内酰胺构架在天然药物中广泛存在, 由γ-酮酸与氨基醇进行环合是常用的方法之一. 2012年, Vassilikogiannakis等[71]报道了可见光介导呋喃衍生物和1, 2-氨基醇的环化反应(Eq. 57).反应以Rose Bengal为光催化剂, 二甲硫醚为添加剂, 空气为氧化剂, 通过降低三氟乙酸的用量或在反应过程中加入三乙胺可避免目标化合物开环而进行分子间聚合.该方法官能团容忍性好, 不需要对基团进行保护.
|
|
(57) |
2012年, Zhou等[72]在羰基二氧化物和环氧化物的环加成实验中发现, 在90 ℃, 常压条件下, Rhodamine 6G的活性几乎是Rhodamine B的29倍, 加入三乙胺可以显著提高催化效果(Eq. 58), 推测是因为三乙胺可以消除催化剂中的氢离子与醇盐阴离子中间体的相互作用, 从而提高环状碳酸盐的产率.
|
|
(58) |
2013年, Tan等[73]以荧光素为光催化剂, 将N-苯基甘氨酸缓慢滴加到N-苯基马来酰亚胺中, 经光催化得到了脱羧环合产物(Eq. 59).该反应官能团容忍性好, 副反应少, 可作为合成天然产物的有效方法.
|
|
(59) |
2014年, Rueping课题组[74]以马来酰亚胺或活性炔烃和四氢异喹啉为底物, 通过可见光氧化还原催化合成吡咯并[2, 1-α]异喹啉(Eqs. 60, 61).反应以Rose Bengal为光敏剂, 分三步进行:催化氧化, [3+2]环加成, 氧化芳构化.为了提升芳构化过程的选择性, 待起始底物完全被消耗后加入N-溴代丁二酰亚胺(NBS), 但是, 以活性炔烃为底物时, 加入NBS对反应没有影响.该方法产率较好, 具有一定的官能团容忍性.
|
|
(60) |
|
|
(61) |
2014年, Liu等[75]报道通过可见光催化氧化酚亚胺得到2-芳基-苯并噁唑(Eq. 62).反应以TBA-Eosin Y为光催化剂, 以二氮杂二环(DBU)为碱, 在三氟甲苯中进行.电化学实验表明向反应体系中加入碱, 反应的氧化电位由1.10 V (vs. NHE)下降到0.58 V (vs. NHE), 推测是由于碱先将二氢嘧啶转化成负离子所致.负离子作为一个更强的电子供体, 与激发态TBA-eosin Y*之间的电子转化变得更强烈, 从而加快了反应速率.此外, TBA-eosin Y可以与分子氧作用同时生成过氧负离子和单线态激发态染料分子, 而碱可以抑制二氢嘧啶和单线态激发态染料分子作用, 从而提高了反应的选择性.
|
|
(62) |
2014年, Zhou等[76]报道用苯肼代替以往的芳基重氮盐与2-腈基联苯反应, 通过[4+2]环加成生成了一系列菲啶衍生物(Eq. 63).该方法用Eosin B为光催化剂, 需要添加碳酸钾.随后, Fan等[77]报道了芳基磺酰氯与2-腈基联苯反应合成菲啶衍生物的方法(Eq. 64).该反应以Eosin Y为光催化剂, 需要添加少量磷酸钾.
|
|
(63) |
|
|
(64) |
2014年, Miyabe课题组[78]报道含氮二烯化合物与卤代烷烃(i-C3F7I)反应得到了加成环合产物(Eq. 65).反应需要先将Rhodamine B和含氮二烯化合物溶解在乙醇中, 最后加入i-C3F7I进行荧光猝灭.此方法的优点在于可以在水存在下进行, 也是为数不多的以Rhodamine B为光催化剂进行的反应之一, 产率较高.
|
|
(65) |
2015年, Zhang等[79]通过光催化有氧氧化N, N-二甲基苯胺和马来酰亚胺, 环合得到四氢喹啉(Eq. 66).反应以Eosin Y为光敏剂, 以分子氧为氧化剂, 不需要金属参与, 对吸电子基和供电子基均有较好的容忍性.
|
|
(66) |
反应机理如下: Eosin Y被光照激发转化成单线态激发态1Eosin Y*, 单线态激发态1Eosin Y*马上经过分子内偶联转化成更稳定的三线态3Eosin Y*.底物188与3Eosin Y*之间发生单电子转化生成胺自由基阳离子188a, 同时, 3Eosin Y*被还原成EY·-.在氧气存在下, EY·-通过单电子转化氧化回到基态EY, 完成一次光催化循环, 同时生成过氧自由基负离子O2·-. 188a去质子化生成α-氨基烷基自由基188b, α-氨基烷基自由基188b再和189反应生成自由基189a, 189a经过环化形成中间体189b, 189b向O2·-转移一个氢自由基生成终产物190以及过氧根负离子, 后者再质子化形成过氧化氢(Scheme 13).
2015年, Tang等[80]通过可见光催化芳基脒C(sp3)—C(sp2)键偶联生成喹唑啉(Eq. 67).该方法与传统方法相比避免了使用复杂氧化剂或添加剂, 可生成大体积萘基取代的喹唑啉.
2015年, Leonori课题组[81]以低还原电位的o-芳基肟为N中心自由基前体, 以Eosin Y为光催化剂, 1, 4-环己二烯(CHD)为氢源, 得到了五元环化产物(Eq. 68).由于芳基肟具有高度结构模块化的特点, 这使得从产物出发逆推底物变得更容易.该反应不需要过渡金属催化剂, 具有很好的官能团容忍性, 分离产率良好.
|
|
(67) |
|
|
(68) |
2015年, Yadav课题组[82]报道了可见光介导的苯乙烯的二官能化, 合成5-芳基-2-亚胺基-1, 3-氧代硅烷(Eq. 69).相比芳环上连有吸电子基的苯乙烯, 芳环上连供电子基时反应更快, 产率略有增加.
|
|
(69) |
目前认为, 基态Eosin Y先吸收光转化成激发态EY*, 硫氰酸根负离子SCN-与染料自由基EY*之间发生单电子转化生成硫氰酸根自由基·SCN和染料自由基负离子EY·-, EY·-被氧气氧化成基态完成光氧化还原环.硫氰酸根自由基·SCN进攻苯乙烯195形成中间体195a, 195a联合O2形成过氧自由基195b, 195b再与195a反应形成烷氧基自由基195c. 195c被O2·-还原或从NH4+吸收质子形成β-羟基硫代酸酯195d. 195d最后经过快速环化生成目标产物196 (Scheme 14).
同年, Tripathi课题组[83]报道了半卡巴腙的光催化氧化环化合成1, 3, 4-噁二唑的方法(Eq. 70).反应以Eosin Y为光敏剂, 以CBr4为自由基引发剂, 具有产率高, 官能团容忍性好等特点.
|
|
(70) |
反应机理如下: Eosin Y被λ=535 nm的可见光照射转化成单线态激发态1Eosin Y*, 1Eosin Y*迅速通过分子内偶联转化成三线态激发态3Eosin Y*, 三线态激发态3Eosin Y*通过被缩氨基脲197a还原猝灭参与光氧化还原循环, 同时生成自由基阳离子197b, 197b经过去质子化生成共振稳定自由基197c.自由基197c再经过六元过渡态吸收一个质子生成自由基197d. 197d随后溴化形成衍生物197e, 197e经历anti-Baldwin’s规则进行环化生成目标产物198 (Scheme 15).

2015年, Wang课题组[84]以苯基丙炔酸酯与亚磺酸为底物, 经过可见光介导氧化环化生成香豆素衍生物(Eq. 71).反应以Eosin Y为光敏剂, 以叔丁基过氧化氢(TBHP)为氧化剂.实验发现, 亚磺酸结构确定时, 苯基丙炔酸酯苯环上连有吸电子基时具有较好的产率, 苯环上连有强供电子基(MeO)的底物几乎不反应, 而苯环3, 5位置上连有两个甲基时产率中等, 苯环的邻位或对位连有甲基时, 表现出一定的区域选择性或空间效应.另外, 确定苯基丙炔酸酯结构时, 亚磺酸的芳环上连有吸电子基或供电子基时产率维持在一定过的范围(58%~77%).
|
|
(71) |
2015年, Luo等[85]报道了可见光介导的多烯的有机光催化环化反应(Eqs. 72, 73).反应以Eosin Y为光催化剂, 在六氟-2-丙醇中进行, 需要添加催化量的LiBr以促进相应的烯醇的形成.基于带有1, 3-二羰基的多烯有转化成烯醇的趋势, 该课题组继续以其为底物进行了系列拓展.该方法具有产率高、立体选择性好等特点.
|
|
(72) |
|
|
(73) |
推测反应机理如下: Eosin Y先被绿色LEDs光照激活再迅速经过系统内部交换(ISC)转化成三线态激发态3Eosin Y*, 随后, 底物202与3Eosin Y*之间发生单电子转换生成自由基阳离子202a和染料自由基阴离子EY·-. 202a经过自由基级联反应伴随着氢转移生成自由基阳离子中间体202b.最后, 202b被还原生成目标产物203 (Scheme 16).其中, HFIP有双重作用:对自由基阳离子中间体有强烈的稳定作用, 因此能够促进立体选择性级联过程; HFIP参与并加速了上述的氢转换过程.
2016年, Yadav课题组[86]报道了光介导苯乙烯的高区域选择性双官能化合成1, 3-氧硫杂环己烷-2-硫酮(Eq. 74).反应以Eosin Y为光敏剂, 分子氧为氧化剂, 分两步进行.首先将CS2和CsCO3加入甲醇溶液中, 室温下搅拌3 h, 之后加入底物和光敏剂Eosin Y, 反应液与空气相通, 在Luxeon Rebel高功率绿色LED照射下继续反应8~10 h, 分离得到目标产物.与之前方法相比, 此法无需将苯乙烯预官能团化成环氧乙烷, 步骤更简单.
|
|
(74) |
2016年, Srivastava等[87]以邻苯二胺和醛为底物, 经光催化缩合得到了苯并咪唑衍生物(Eq. 75).该方法无需使用金属催化剂或酸碱调节剂, 官能团适用性好.
|
|
(75) |
反应机理概述如下, 伯胺208和醛209先形成亚胺209a, 激发的染料分子Rose Bengal*通过单电子转化方式从亚胺夺取一个电子生成自由基负离子Rose Bengal·-, Rose Bengal·-被氧气氧化回基态, 亚胺自由基阳离子将一个质子传递给O2·-生成亚胺自由基, 其再进行一次氢转移生成过氧化氢及目标产物210 (Scheme 17).
2016年, Lakhdar课题组[88]以芳基膦氧化物与炔烃为底物, 通过可见光介导合成苯并[b]磷杂环氧化物(Eq. 76).该方法以Eosin Y为光敏剂, 以N-乙氧基-2-甲基吡啶鎓(DEM)为氧化剂, 加入一定NaHCO3作碱, 条件温和, 具有很好的底物适用性, 已经扩展到克级.
机理实验发现光敏剂Eosin Y和氧化剂吡啶盐之间会形成电子供体-受体复合物(EDA).而EDA活性极高, 既可以氧化光催化剂, 也可以引发自由基的形成, 自由基再经过分子内反应从而避免了其在两种试剂间的扩散.
|
|
(76) |
2016年, Dhavale课题组[89]以芳基酮和苄基胺为底物, 在可见光的介导下生成2, 4, 6-三芳基吡啶(Eq. 77).反应联合Eosin Y-O2为光氧化还原催化剂, 加入BF3· Et2O, 产率高, 底物适用性好.
|
|
(77) |
反应机理如下:首先苄基胺被染料激发态Eosin Y*和O2氧化, 生成亚胺正离子215b, 亚胺正离子215b与苄胺偶联, 再脱去一分子氨, 生成亚胺中间体214b.同时, 在添加剂BF3·Et2O的促进下, 苯乙酮214转化成烯醇214a, 烯醇214a与亚胺中间体214b反应生成216a, 216a再被光催化氧化生成216b. 216b与烯醇214a反应生成216c. 216c与氨发生缩合生成216d, 216d再芳构化脱去一分子苄胺, 生成三芳基吡啶216 (Scheme 18).

2016年, König课题组[90]以N-芳基吡咯与芳基炔烃为底物, 通过光催化环化合成吡咯并[1, 2-a]喹啉和肼嘧啶.该反应在室温下进行, 不需要过渡金属或强碱的参与(Eq. 78).
2017年, Baruah等[91]以1-氨基烷基-2-萘酚为底物, 通过可见光促进的分子内交叉脱氢偶联合成1, 3-噁嗪(Eq. 79).反应通过亚胺离子中间体进行, 产率较高, 官能团容忍性好.
2017年, Wang等[92]报道了1, 7-烯炔的可见光介导的双环化, 合成功能化的含砜苯并[a]芴-5-酮(Eq. 80).该方法利用6-exo-dig/5-endo-trig成环机理, 通过自由基引发形成C—S和C—C键, 能够有效地构建多环连接的烷基芳基砜.
|
|
(78) |
|
|
(79) |
|
|
(80) |
首先, 亚磺酸225和碱发生酸碱中和合成亚磺酸负离子225a, 225a再与染料激发态Eosin Y*反应生成染料自由基负离子Eosin Y·-和磺酰基自由基225b. 225b通过自由基加成生成1, 7-烯炔的链烯基单元226a, 226a通过分子内6-exo-dig或5-endo-trig环化生成自由基中间体226b. 226b与染料自由基负离子Eosin Y·-发生单电子转化和质子转移生成最终产物226和Eosin Y-H, Eosin Y-H再与质子反应释放出H2重新生成Eosin Y (Scheme 19).

2017年, Xu等[93]报道了通过可见光催化构建C—P键生成3-磷酸化香豆素(Eq. 81).此法联合了磷酸化/环化反应, 以Eosin Y为光催化剂, 以叔丁基过氧化氢(t-BuOOH)为氧化剂, 在温和的条件下进行, 具有一定的官能团容忍性.
2017年, Jin等[94]报道了可见光促进的邻叠氮芳基炔烃与芳基重氮盐的环化反应, 合成2, 3-二芳基取代的吲哚(Eq. 82).该方法产率高, 底物适用范围广, 区域选择性高.
|
|
(81) |
|
|
(82) |
2017年, Wei课题组[95]分别将邻位羰基取代的N-炔丙基芳香胺与亚磺酸或硫醇反应, 以Na2-Eosin Y或Eosin Y为光敏剂, 对炔烃进行双官能团化反应, 合成3-磺酰基三烯酮和3-亚磺酰氮杂螺[4, 5]三烯酮(Eqs. 83, 84).
2017年, Maiti等[96]报道了光催化C-N, N-N偶联合成二氮丙啶衍生物(Scheme 20).反应以有机染料Rose Bengal为光催化剂, 不需要过渡金属试剂, 在温和的条件下进行.反应的官能团容忍性好, 受电子效应影响小.
|
|
(83) |
|
|
(84) |
2017年, He等[97]通过可见光介导分子间[2+2]环加成的方法合成了3-亚甲基吲哚酮衍生物(Eq. 85).反应以Rose Bengal为光催化剂, 光催化剂用量少, 产率高, 具有很好的非对映选择性和区域选择性.
|
|
(85) |
2018年, Zhang课题组[98]以N-炔丙基芳香胺, 二芳基碘鎓盐和二氧化硫底物, 通过三组分级联环加成合成3-芳基磺酰基喹啉(Eq. 86).该方法以Eosin Y为光催化剂, 需要添加少量NaHCO3, 通过一步构建C—S键和喹啉直接合成目标产物, 具有很好的底物适应性和官能团容忍性.为了证明此法的可扩展性和实用性, 该课题组又将实验拓展至克级, 产率达77%.
|
|
(86) |
2018年, Guo等[99]报道了酰肼和二酮的脱羧-环化合成2, 5-二取代的1, 3, 4-噁二唑(Eqs. 87, 88).采用优化后的条件, 该实验室又将不同酰肼与苯基乙醛酸反应, 依然得到了较高的产率.该方法适用于杂环酰肼和脂肪族酰肼, 其上基团的电子效应对反应产率影响小.
|
|
(87) |
|
|
(88) |
2009年, Orfanopoulos课题组[100]为了建立氧化反应的体系和机制, 尝试将N-取代吡咯与不同溶剂在多种光敏剂下反应.以Rose Bengal为光敏剂, 以分子氧为氧化剂, 得到了系列混合物(Eqs. 89, 90).在非质子溶剂中反应得到的主要产物为取代的醛, 在质子溶剂中反应得到的主要产物是选择性取代的醇, 控制反应时间可以得到单取代产物, 否则全部生成双取代产物.
|
|
(89) |
|
|
(90) |
2012年, Uhlig课题组[101]采用Rhodamine B为光催化剂, 将叠氮基引入不同烯烃中得到了系列产物(Eqs. 91, 92).以丙烯醇为底物, 得到是对映异构体; 以丙烯酯为底物, 得到的是混合物.该反应探索了自由基反应机理概括如下:叠氮基负离子N3-经电子转化形成叠氮基自由基N3·, N3·被烯烃加成生成碳自由基259a.碳自由基259a被氧气捕捉形成过氧自由基260c, 过氧自由基260c再经过电子转化形成过氧负离子260d并完成光催化剂的再生, 最后, 氧负离子260d获得氢质子生成过氧化物260a (Scheme 21).用此方法可以将叠氮基导入不饱和底物中, 具有一定的区域选择性和非对映立体选择性.
|
|
(91) |
|
|
(92) |
2014年, Sun等[102]报道了无金属光催化体系将溴甲(乙)基芳香化合物转化成相应的醇, 提高温度可进一步氧化成醛(Scheme 22).反应以Eosin Y为光敏剂, 与空气接触, 底物可以拓展为杂环取代的卤代烷烃.实验结果显示, 带有吸电子基的底物产率高于带有供电子基的底物.
2014年, Yadav课题组[103]在可见光介导下分别将醛肟和伯酰胺转化为腈(Eqs. 93, 94).反应以Eosin Y为光敏剂, 需要CBr4和催化量的N, N-二甲基甲酰胺(DMF)参与, 后两者先生成Vilsmeier-Haack中间体, 最后与底物反应得到目标产物.实验结果显示, 带有供电子基的底物产率较好.该方法适用于脂肪族、芳香族以及杂环化合物, 具有很好的官能团容忍性.
目前最受认可的反应机理如Scheme 23, 光敏剂Eosin Y吸收可见光转化成激发态Eosin Y*, 其最稳定的三线态3Eosin Y*通过与CBr4之间的单电子转移完成氧化猝灭, 并生成CBr3·. CBr3·与DMF反应生成自由基266a, 随后自由基266a与染料自由基阳离子Eosin Y·+之间发生单电子转移生成亚胺离子266b并完成了染料分子的氧化还原循环.亚胺离子266b转化成Vilsmeier-Haack试剂266c, 266c与底物266或268的羟基反应生成266d或266e, 最后生成腈267并释放出DMF.反应中会形成副产物COBr2, 其可以与DMF反应生成化合物266c.
|
|
(93) |
|
|
(94) |

2016年, Sun等[104]以Eosin Y为光催化剂, 4-氯苯硫醇为添加剂, 对芳基炔烃进行光催化氧化合成了1, 2-二酮(Eq. 95).该方法对光敏基团具有很好的容忍性, 分离产率高.
|
|
(95) |
2016年, Shah等[105]报道了以Eosin Y为光催化剂, 对苯乙醇进行光催化选择性氧化生成酮(Eq. 96).该方法催化体系简单、反应条件温和、产率较高、具有良好的选择性.
|
|
(96) |
反应历程如下: TBHP先被染料激发态Eosin Y*激发, 生成叔丁氧基自由基(t-BuO·)和羟基自由基(OH·), (t-BuO·)再从芳基醇苄位夺取氢自由基H·生成苄基自由基, 苄基自由基最后由羟基自由基(OH·)辅助生成目标羰基化合物(Scheme 24).

2016年, Lu等[106]报道可见光催化氧化脂肪胺生成酸或内酯(Eqs. 97, 98).该反应通过控制光照源和溶剂, 可以选择性氧化生成不同产物.以二噁烷为溶剂, 18 W白光为光源时得到芳基酮酸; 以DMF为溶剂, 8 W绿光为光源时得到芳基酮内酯.该方法对研究C—C键断裂或氧化具有重要的参考价值.
|
|
(97) |
|
|
(98) |
2017年, Fu等[107]报道了N-烷基吡啶鎓盐在光催化体系中的有氧氧化合成N-甲基吡啶酮、喹诺酮和异喹诺酮(Eq. 99).该方法官能团容忍性好, 已经扩大到克级, 产率和小试非常接近.此外, 为了证明此法的综合实用性, 联合此法通过三步反应合成4-脱氧酰亚胺, 总产率达74%;以1, 3-二甲基-1H-苯并[d]咪唑-3-鎓碘化物为底物合成1, 3-二甲基-1H-苯并[d]咪唑-2-(3H)-酮, 产率达93%.
|
|
(99) |
2018年, Nan等[108]报道了可见光促进的烯烃的双官能化合成α-硫氰酸酯酮(Eq. 100).该方法以烯烃、硫氰酸铵、氧气为底物, 通过构建C—S、C=O合成目标产物.实验结果显示, 位阻效应对反应影响小, 芳环o、m、p位置连有甲基时都有较好的产率.此外, 此方法也适用于芳香族烯烃, 如(E)-烯丙基苯, 而二苯乙烯和脂族烯烃无法合成目标产物.
|
|
(100) |
2014年, Liu等[75]报道了可见光催化氧化2-取代二氢嘧啶得到2-(甲硫基)嘧啶(Eq. 101).反应以TBA-Eosin Y为光催化剂, 在甲醇水溶液中进行, 以碳酸钠为碱.在反应体系中加碱可以提高底物与TBA-eosin Y之间的电子转化速度, 从而明显提高反应速率.
2017年, Balaraman课题组[109]以染料Rose Bengal为光催化剂, 以N, N-二甲基乙酰胺(DMA)为溶剂, 对多种N-杂环(喹啉、喹喔啉、喹唑啉吖、吲哚)实现了光催化氧化脱氢(Scheme 25).此方法催化剂用量少, 无需其他添加剂, 反应条件温和, 官能团容忍性好.
|
|
(101) |

2014年, Tan课题组[110]以CBr4为溴源, 中性的吗啉N-自由基作为吸氢体(不同底物使用量不同), 在可见光介导下对脂肪和芳香C(sp3)-H进行溴化(Scheme 26).以二氯化碳的水溶液为溶剂, 水层可以移除多余的溴单质, 这样保证有机相中的溴化物维持在较低的浓度, 促进反应正向进行.该反应产率一般, 但具有很好的区域选择性.
2014年, Tung等[111]报道了取代硝基苯的选择性光催化还原(Eq. 102).反应以Eosin Y为光敏剂, 原乙酸三乙酯(TEOA)为还原剂, 反应几乎完全转化, 定量得到目标产物.供电子基取代和吸电子基取代的底物都能很好地转化成目标产物.实验结果表明, 在苯环的取代基中, 仅硝基被还原, 羰基、炔基、腈基、卤素不会发生反应.这是因为硝基相比其他基团具有更强的吸电子能力, 这也是光催化反应与经典非均相催化还原不同的地方之一.
|
|
(102) |
2010年, Zeitler课题组[17]报道了光催化α-卤代苯乙酮的脱卤(Eq. 103).以Eosin Y为光催化剂, 选择性好, 分离产率高.实验结果比表明, 以Hantzsch酯为氢源可以避免潜在的副反应.
|
|
(103) |
2013年, Liu和Wu课题组[112]报道了β-芳基酮磺酸酯在可见光催化下进行的去磺酰化反应(Eq. 104).以TBA-Eosin Y为光敏剂, 二异丙基乙胺为还原剂, 部分反应添加乙酸后产率增加.该方法产率高, 受电子效应影响小, 官能团容忍性好, 但底物限于芳香族化合物.
|
|
(104) |
2015年, Cheng课题组[113]报道了在可见光催化体系中, 以Eosin Y为光敏剂, 过氧化氢为氧化剂, 添加一定的Brønsted acid NaHSO4, 可以除去对甲氧基苄基(PMB)醚中的保护基(Eq. 105).其中, H2O2分两次加入, 因为这样补偿了玻璃反应器表面上的H2O2的分解, 使得反应更具可重复性.该方法适用于不同的醇, 具有一定的官能团容忍性.在一级醇与三级醇的混合体系中, 三级醇优先发生反应.
|
|
(105) |
芳胺由于氨基的供电子效应容易发生亲电取代反应, 之后再通过重氮盐的形式脱去氨基是常用的反应路线. 2015年, Wangelin课题组[114]报道了芳基重氮盐经可见光催化实现了去官能团化(Eq. 106).该反应以Eosin B为光催化剂, 在非质子溶剂DMF中进行.此方法不需要金属试剂或还原试剂, 反应条件温和, 官能团容忍性好, 产率较高.
|
|
(106) |
缩醛反应是保护醛、酮常见的方法.以往此类可见光介导的反应需要结合光催化剂与胺催化剂或卡宾催化剂共同作用. 2016年, Lei等[115]仅依靠光催化剂Eosin Y就完成了缩醛(Eq. 107).此反应条件温和, 分离产率高, 官能团容忍性好, 当底物具有多个羰基时具有立体选择性, 也适用于对酸敏感的醛以及具有立体位阻的醛.
|
|
(107) |
2017年, Sumerlin课题组[116]通过光催光介导电子/能量转移-可逆加成-碎片链转移(PET-RAFT)聚合完成了蛋白质的嫁接(Eq. 108).实验以Eosin Y为光催化剂, 以叔胺(PMDEAT)为氧化剂, 不同单体(monomer)在转化率和分子质量方面各具优点.该方法拓展了温和条件下PET-RAFT聚合嫁接蛋白质的能力, 合成了多种蛋白质功能材料.
|
|
(108) |
有机染料是一种廉价、易得、反应条件温和的光催化剂.本文列举了氧杂蒽类有机染料光催化剂参与的C—C偶联、C—X (X=N、O、S、P)偶联、插羰偶联、S—P偶联、S—S偶联、环合偶联以及氧化、芳构化、去官能团化、官能团的保护、聚合等反应实例.目前应用最多的是Eosin Y和Rose Bengal, 主要应用于光介导单电子转化(PET)自由基反应中; 其次是Rhodamine 6G, 由于其具有较高的氧化还原电位, 有进一步应用于连续光介导单电子转化(con-PET)反应中的趋势.
氧杂蒽类有机染料参与的可见光催化有机反应虽然已经取得了重大的进展, 但仍然存在一些亟待改进和研究的问题, 比如: (1)反应一般限制在有孤对电子或共轭电子的化合物中, 因此光催化自由基反应的底物拓展是需要探索的领域; (2)目前反应大多发生在电离能较低的底物间, 未来研究可能需要对有机染料进行改进, 增大氧化还原电位区间; (3)氧杂蒽类有机染料多应用于还原猝灭反应中, 可以对相关染料进行结构改进, 研发适用于氧化猝灭机理的光催化剂; (4)目前反应主要合成消旋体, 寻找能够催化合成手性化合物的有机染料光催化剂是有待开发的领域; (5)有机染料催化的自由基反应受光照面积影响反应时间较长, 需要研发适合光催化有机合成的新型反应设备.
综上所述, 氧杂蒽类有机染料在光催化有机合成领域具有极大的探索空间和研究价值.
(a) Ciamician, G. Sci. New Ser. 1912, 36, 385.
(b) Grätzel, M. Nature 2001, 414, 338.
(c) Nocera, A. Chem. Rev. 2007, 107, 4022.
(d) Albini, A.; Fagnoni, M. ChemSusChem 2008, 1, 63.
(e) Yum, J.-H.; Chen, P.; Gratzel, M.; Nazeeruddin, M. ChemSusChem 2008, 1, 699.
(f) Sala, X.; Romero, I.; Rodriguez, M.; Escriche, L.; Llobet, A. Angew. Chem., Int. Ed. 2009, 48, 2842.
(a) Zen, J.-M.; Liu, S.-L.; Annamalai, M.-S. Angew. Chem., Int. Ed. 2003, 42, 577.
(b) Hirao, T.; Shiori, J.; Okahata, N. Bull. Chem. Soc. Jpn. 2004, 77, 1763.
(c) Herance, J.; Ferrer, B.; Bourdelande, J.; Marquet, J.; Garcia, H. Chemistry 2006, 12, 3890.
(d) Masahisa, O.; Han, N.; Munetaka, A. Royl. Soc. Chem. 2007, 8, 827.
(a) Li, L.-T.; Ma, S.-M. J. Org. Chem. 2001, 21, 75(in Chinese).
(李林涛, 麻生明, 有机化学, 2001, 21, 75.)
(b) Weng, J.-Q.; Liu, X.-H.; Zhang, G.-F.; Tan, C.-X.; Ding, C.-R.; Ou, X.-M. Chin. J. Org. Chem. 2011, 31, 374(in Chinese).
(翁建全, 刘幸海, 张国富, 谭成侠, 丁成荣, 欧晓明, 有机化学, 2011, 31, 374.)
(c) Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687.
(d) Hering, T.; Hari, D. P.; Konig, B. J. Org. Chem. 2012, 77, 10347.
(e) Schroll, P.; Hari, D. P.; Konig, B. Chem. Open 2012, 1, 130.
(f) Huang, L.; Zhao, J. Chem. Commun. 2013, 49, 3751.
(g) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. Chem. Rev. 2013, 113, 5322.
(h) Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387.
(i) Hari, D. P.; Hering, T.; Konig, B. Angew. Chem., Int. Ed. 2014, 53, 725.
(j) Liu, H.; Zhao, L.; Yuan, Y.; Xu, Z.; Chen, K.; Qiu, S.; Tan, H. ACS Catal. 2016, 6, 1732.
(h) Ren, X.; Guo, Q.; Chen, J.; Xie, H.; Xu, Q.; Lu, Z. Chemistry 2016, 22, 18695.
(a) Tang, S.-S.; Zhang, N.; Zhu, J. Ind. Catal. 2005, 13, 21(in Chinese).
(汤胜山, 张宁, 朱静, 工业催化, 2005, 13, 21.)
(b) Zhang, N.; Zhang, Y.-H.; Pan, X.-Y.; Fu, X.-Z.; Xu, Y.-J. Sci. China Chem. 2011, 41, 1097(in Chinese).
(张楠, 张燕辉, 潘晓阳, 付贤智, 徐艺军, 中国科学: 化学, 2011, 41, 1097.)
(b) Cui, E. T.; Lu, G. X. J. Phys. Chem. C 2013, 117, 26415.
(c) Meng, Q. Y.; Zhong, J. J.; Liu, Q.; Gao, X. W.; Zhang, H. H.; Lei, T.; Li, Z. J.; Feng, K.; Chen, B.; Tung, C. H.; Wu, L. Z. J. Am. Chem. Soc. 2013, 135, 19052.
(c) Kong, C.; Min, S. X.; Lu, G. X. ACS Catal. 2014, 4, 2763.
(d) Bhat, V. T.; Duspara, P. A.; Seo, S.; Abu Bakar, N. S. B.; Greaney, M. F. Chem. Commun. 2015, 51, 4383.
(e) Li, X. R.; Li, Y. Y.; Huang, Y. X.; Zhang, T.; Liu, Y. Z.; Yang, B.; He, C. X.; Zhou, X. C.; Zhang, J. M. Green Chem. 2017, 19, 2925.
(f) Liu, D.; Zhang, C.-H.; Han, N.; Du. M.-M.; Zhang, X.-L.; Zhao, P.-S.; Yang, P. Chin. J. Org. Chem. 2018, 38, 62(in Chinese).
(张成慧, 韩楠, 杜萌萌, 张效露, 赵朋杉, 杨萍, 刘迪, 有机化学, 2018, 38, 62.)
(a) Yuan, D.-Z.; Hhuang, B. Chin. J. Org. Chem. 2012, 32, 1368(in Chinese).
(袁定重, 黄斌, 有机化学, 2012, 32, 1368.)
(b) Yin, J.; Liao, G.-Z.; Zhu, D.-Y.; Lu, P.; Li, L.-S. China Environ. Sci. 2016, 36, 735(in Chinese).
(尹竞, 廖高祖, 朱冬韵, 卢平, 李来胜, 中国环境科学, 2016, 36, 735.)
(c) Dai, X.-W.; Wu, J.; Qi, X.-M.; Wu, Q.; Li, X. Res. Environ. Sci. 2014, 27, 827(in Chinese).
(代学伟, 吴江, 齐雪梅, 吴强, 何平, 李忺, 环境科学研究, 2014, 27, 827.)
唐甜, 魏鹏飞, 李琳, 杨丰科, 化学世界, 2018, 59, 144. http://www.cnki.com.cn/Article/CJFDTotal-HXSS201805002.htmTang, T.; Wei, P.-F.; Li, L.; Yang, F.-K. Chem. Word 2018, 59, 144(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-HXSS201805002.htm
Yadav, L.; Srivastava, V.; Yadav, A. Synlett 2013, 24, 465.
(a) Ravelli, D.; Fagnoni, M. ChemCatChem 2012, 4, 169.
(b) Xiao, T.; Dong, X.; Tang, Y.; Zhou, L. Adv. Synth. Catal. 2012, 354, 3195.
(c) Hari, D. P.; Konig, B. Angew. Chem., Int. Ed. 2013, 52, 4734.
(d) Huang, L.; Zhao, J. RSC Adv. 2013, 3, 23377.
(e) Arbeloa, E. M.; Previtali, C. M.; Bertolotti, S. G. J. Lumin. 2016, 180, 369.
楚增勇, 原博, 颜廷楠, 无机材料学报, 2014, 29, 785. http://www.cnki.com.cn/Article/CJFDTotal-WGCL201408001.htmChu, Z.-Y.; Yuan, B.; Yan, Y.-N. J. Inorg. Mater. 2014, 29, 785(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-WGCL201408001.htm
Sandrine, F.; Daniel, L.; Carlo, A. J. Phys. Chem. A 2008, 112, 7264. doi: 10.1021/jp8011624
Ravelli, D.; Fagnoni, M.; Albini, A. Chem. Soc. Rev. 2013, 42, 97. doi: 10.1039/C2CS35250H
Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem. 2014, 12, 6059. doi: 10.1039/C4OB00843J
(a) Nicewicz, D. A.; Nguyen, T. M. ACS Catal. 2013, 4, 355.
(b) Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075.
Hari, D. P.; Konig, B. Chem. Commun. 2014, 50, 6688. doi: 10.1039/C4CC00751D
Srivastava, V.; Singh, P. P. RSC Adv. 2017, 7, 31377. doi: 10.1039/C7RA05444K
程骁恺, 胡新根, 陆展, 有机化学, 2017, 37, 251. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345790.shtmlCheng, X.-K.; Hu, X.-G.; Lu, Z. Chin. J. Org. Chem. 2017, 37, 251(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345790.shtml
Neumann, M.; Fuldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951. doi: 10.1002/anie.201002992
Pan, Y.; Kee, C. W.; Chen, L.; Tan, C. H. Green Chem. 2011, 13, 2682. doi: 10.1039/c1gc15489c
Pan, Y.; Wang, S.; Kee, C. W.; Dubuisson, E.; Yang, Y.; Loh, K. P.; Tan, C. H. Green Chem. 2011, 13, 3341. doi: 10.1039/c1gc15865a
Liu, Q.; Li, Y. N.; Zhang, H. H.; Chen, B.; Tung, C. H.; Wu, L. Z. Chemistry 2012, 18, 620. doi: 10.1002/chem.v18.2
Fu, W.; Guo, W.; Zou, G.; Xu, C. J. Fluorine Chem. 2012, 140, 88. doi: 10.1016/j.jfluchem.2012.05.009
Vila, C.; Bootwicha, T.; Rueping, M. ACS Catal. 2013, 3, 1676. doi: 10.1021/cs400350j
Keshari, T.; Yadav, V. K.; Srivastava, V. P.; Yadav, L. D. S. Green Chem. 2014, 16, 3986. doi: 10.1039/C4GC00857J
Cantillo, D.; Frutos, O.; Rincon, J. A.; Mateos, C.; Kappe, C. O. Org. Lett. 2014, 16, 896. doi: 10.1021/ol403650y
Li, X.; Gu, X.; Li, Y.; Li, P. ACS Catal. 2014, 4, 1897. doi: 10.1021/cs5005129
Jadhav, S. D.; Bakshi, D.; Singh, A. J. Org. Chem. 2015, 80, 10187. doi: 10.1021/acs.joc.5b01736
Li, J.; Zhang, J.; Tan, H. B.; Wang, D. Z. Org. Lett. 2015, 17, 2522. doi: 10.1021/acs.orglett.5b01053
Fuentes de Arriba, A. L.; Urbitsch, F.; Dixon, D. J. Chem. Commun. (Camb) 2016, 52, 14434. doi: 10.1039/C6CC09172E
Schwarz, J.; König, B. Green Chem. 2016, 18, 4743. doi: 10.1039/C6GC01101B
Wang, K.; Meng, L. G.; Wang, L. J. Org. Chem. 2016, 81, 7080. doi: 10.1021/acs.joc.6b00973
Zhang, N.; Quan, Z. J.; Zhang, Z.; Da, Y. X.; Wang, X. C. Chem. Commun (Camb). 2016, 52, 14234. doi: 10.1039/C6CC08182G
Zhao, D.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 3166. doi: 10.1002/anie.201511251
Meyer, A. U.; Slanina, T.; Yao, C. J.; König, B. ACS Catal. 2016, 6, 369. doi: 10.1021/acscatal.5b02410
Ghosh, I.; BurkhardKonig Angew. Chem., Int. Ed. 2016, 55, 7676. https://www.ncbi.nlm.nih.gov/pubmed/27198967
Ghosh, I.; Marzo, L.; Das, A.; Shaikh, R.; König, B. Acc. Chem. Res. 2016, 49, 1566. doi: 10.1021/acs.accounts.6b00229
Garrido, A. F.; Choubane, H.; Daaou, M.; Maestro, M. C.; Aleman, J. Chem. Commun. 2017, 53, 7764. doi: 10.1039/C7CC03724D
Wu, L. L.; Tang, L.; Zhou, S. G.; Peng, Y. J.; He, X. D.; Guan, Z.; He, Y. H. Tetrahedron 2017, 73, 6471. doi: 10.1016/j.tet.2017.09.050
Zhu, J.; Yuan, Y.; Wang, S.; Yao, Z.-J. ACS Omega 2017, 2, 4665. doi: 10.1021/acsomega.7b00749
Graml, A.; Ghosh, I.; König, B. J. Org. Chem. 2017, 82, 3552. doi: 10.1021/acs.joc.7b00088
Pramanik, M. M. D.; Nagode, S. B.; Kant, R.; Rastogi, N. Org. Biomol. Chem. 2017, 15, 7369. doi: 10.1039/C7OB01756A
Fan, X. Z.; Rong, J. W.; Wu, H. L.; Zhou, Q.; Deng, H. P.; Tan, J. D.; Xue, C. W.; Wu, L. Z.; Tao, H. R.; Wu, J. Angew. Chem., Int. Ed. 2018, 57, 1. doi: 10.1002/anie.201712460
Liu, H.; Feng, W.; Kee, C. W.; Zhao, Y.; Leow, D.; Pan, Y.; Tan, C. H. Green. Chem. 2010, 12, 953. doi: 10.1039/b924609f
Gu, X.; Li, X.; Chai, Y.; Yang, Q.; Li, P.; Yao, Y. Green Chem. 2013, 15, 357. doi: 10.1039/c2gc36683e
Teo, Y. C.; Pan, Y.; Tan, C. H. ChemCatChem. 2013, 5, 235. doi: 10.1002/cctc.201200435
Mitra, S.; Ghosh, M.; Mishra, S.; Hajra, A. J. Org. Chem. 2015, 80, 8275. doi: 10.1021/acs.joc.5b01369
Meyer, A. U.; Jäger, S.; Prasad, Hari. D.; König, B. Adv. Synth. Catal. 2015, 357, 2050. doi: 10.1002/adsc.v357.9
Yang, D.; Huang, B.; Wei, W.; Li, J.; Lin, G.; Liu, Y.; Ding, J.; Sun, P.; Wang, H. Green Chem. 2016, 18, 5630. doi: 10.1039/C6GC01403H
Cai, S.; Xu, Y.; Chen, D.; Li, L.; Chen, Q.; Huang, M.; Weng, W. Org. Lett. 2016, 18, 2990. doi: 10.1021/acs.orglett.6b01353
Zalesskiy, S. S.; Shlapakov, N. S.; Ananikov, V. P. Chem. Sci. 2016, 7, 6740. doi: 10.1039/C6SC02132H
Huang, F.-Q.; Dong, X.; Qi, L.-W.; Zhang, B. Tetrahedron Lett. 2016, 57, 1600. doi: 10.1016/j.tetlet.2016.02.108
Liu, X.; Qing, Z.; Cheng, P.; Zheng, X.; Zeng, J.; Xie, H. Molecules 2016, 21, 1690. doi: 10.3390/molecules21121690
Zhang, M. J.; Schroeder, G. M.; He, Y. H.; Guan, Z. RSC Adv. 2016, 6, 96693. doi: 10.1039/C6RA17524D
Luo, K.; Chen, Y. Z.; Yang, W. C.; Zhu, J.; Wu, L. Org. Lett. 2016, 18, 452. doi: 10.1021/acs.orglett.5b03497
Shaikh, R. S.; Düsel, S. J. S.; König, B. ACS Catal. 2016, 6, 8410. doi: 10.1021/acscatal.6b02591
Ghosh, T.; Das, A.; König, B. Org. Biomol. Chem. 2017, 15, 2536. doi: 10.1039/C7OB00250E
Guerrero-Corella, A.; Maria Martinez-Gualda, A.; Ahmadi, F.; Ming, E.; Fraile, A.; Aleman, J. Chem. Commun. (Camb). 2017, 53, 10463. doi: 10.1039/C7CC05672A
Hong, B.; Lee, J.; Lee, A. Tetrahedron Lett. 2017, 58, 2809. doi: 10.1016/j.tetlet.2017.06.006
Li, Y.; Wang, M.; Jiang, X. ACS Catal. 2017, 7, 7587. doi: 10.1021/acscatal.7b02735
喻晓叶, 周帆, 陈加荣, 肖文精, 化学学报, 2017, 75, 86. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb201701012Yu, X.; Zhou, F.; Chen, J.; Xiao, W. Acta Chim. Sinica 2017, 75, 86(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb201701012
Zhang, R.; Cai, Y.; Sun, D.; Xu, S.; Zhou, Q. Synlett 2017, 28, 1630. doi: 10.1055/s-0036-1588828
Zhang, Z.; Liu, F.; Bao, Z.; Su, B.; Xing, H.; Yang, Q.; Yang, Y.; Ren, Q. Synlett 2017, 28, 1116. doi: 10.1055/s-0036-1588704
Tibbetts, J. D.; Carbery, D. R.; Emanuelsson, E. A. C. ACS Sustainable Chem. Eng. 2017, 5, 9826. doi: 10.1021/acssuschemeng.7b01754
Cui, H.; Wei, W.; Yang, D.; Zhang, Y.; Zhao, H.; Wang, L.; Wang, H. Green Chem. 2017, 19, 3520. doi: 10.1039/C7GC01416C
Bottecchia, C.; Rubens, M.; Gunnoo, S. B.; Hessel, V.; Madder, A.; Noel, T. Angew. Chem., Int. Ed. 2017, 56, 12702. doi: 10.1002/anie.201706700
Zhu, J.; Cui, W. C.; Wang, S.; Yao, Z. J. Org. Lett. 2018, 20, 3174. doi: 10.1021/acs.orglett.8b00909
Guo, W.; Lu, L. Q.; Wang, Y.; Wang, Y. N.; Chen, J. R.; Xiao, W. J. Angew. Chem., Int. Ed. 2015, 54, 2265. doi: 10.1002/anie.201408837
Gu, L.; Jin, C.; Liu, J. Green Chem. 2015, 17, 3733. doi: 10.1039/C5GC00644A
Majek, M.; Wangelin, A. J. Angew. Chem., Int. Ed. 2015, 54, 2270. doi: 10.1002/anie.201408516
Tankam, T.; Poochampa, K.; Vilaivan, T.; Sukwattanasinitt M.; Wacharasindhu, S. Tetrahedron 2016, 72, 788. doi: 10.1016/j.tet.2015.12.036
Sun, J. G.; Yang, H.; Li, P.; Zhang, B. Org. Lett. 2016, 18, 5114. doi: 10.1021/acs.orglett.6b02563
Kalaitzakis, D.; Montagnon, T.; Alexopoulou, I.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2012, 51, 8868. doi: 10.1002/anie.v51.35
Gong, Q.; Luo, H.; Cao, D.; Zhang, H.; Wang, W.; Zhou, X. B. Korean. Chem. Soc. 2012, 33, 1945. doi: 10.5012/bkcs.2012.33.6.1945
Chen, L.; Chao, C. S.; Pan, Y.; Dong, S.; Teo, Y. C.; Wang, J.; Tan. C. H. Org. Biomol. Chem. 2013, 11, 5922. doi: 10.1039/c3ob41091a
Vila, C.; Lau, J.; Rueping, M. Beilstein J. Org. Chem. 2014, 10, 1233. doi: 10.3762/bjoc.10.122
Wang, L.; Ma, Z. G.; Wei, X. J.; Meng, Q. Y.; Yang, D. T.; Du, S. F.; Chen, Z. F.; Wu, L. Z.; Liu, Q. Green Chem. 2014, 16, 3752. doi: 10.1039/C4GC00337C
Xiao, T.; Li, L.; Lin, G.; Wang, Q.; Zhang, P.; Mao, Z. W.; Zhou, L. Green Chem. 2014, 16, 2418. doi: 10.1039/C3GC42517G
Gu, L.; Jin, C.; Liu, J.; Ding, H.; Fan, B. Chem. Commun. 2014, 50, 4643. doi: 10.1039/C4CC01487A
Yoshioka, E.; Jichu, T.; Fukazawa, T.; Nagai, T.; Takemoto, Y.; Kohtani, S.; Miyabe, H. Synlett 2014, 26, 265.
Liang, Z.; Xu, S.; Tian, W.; Zhang, R. J. Org. Chem. 2015, 11, 425. https://www.ncbi.nlm.nih.gov/pubmed/25977716
Shen, Z. C.; Yang, P.; Tang, Y. J. Org. Chem. 2015, 81, 309.
Davies, J.; Booth, S. G.; Essafi, S.; Dryfe, R. A. W.; Leonori, D. Angew. Chem., Int. Ed. 2015, 54, 14017. doi: 10.1002/anie.201507641
Yadav, A. K.; Yadav, L. D. S. Green Chem. 2015, 17, 3515. doi: 10.1039/C5GC00642B
Yadav, L.; Kapoorr, R.; Singh, S.; Tripathi, S. Synlett 2015, 26, 1201. doi: 10.1055/s-00000083
Yang, W.; Yang, S.; Li, P.; Wang, L. Chem. Commun. (Camb.) 2015, 51, 7520. doi: 10.1039/C5CC00878F
Yang, Z.; Li, H.; Zhang, L.; Zhang, M. T.; Cheng, J. P.; Luo, S. Chemistry 2015, 21, 14723. doi: 10.1002/chem.201503118
Yadav, A. K.; Yadav, L. D. S. Green Chem. 2016, 18, 4240. doi: 10.1039/C6GC00924G
Kovvuri, J.; Nagaraju, B.; Kamal, A.; Srivastava, A. K. ACS Comb. Sci. 2016, 18, 644. doi: 10.1021/acscombsci.6b00107
Quint, V.; Morlet-Savary, F.; Lohier, J. F.; Lalevee, J.; Gaumont, A. C.; Lakhdar, S. J. Am. Chem. Soc. 2016, 138, 7436. doi: 10.1021/jacs.6b04069
Rohokale, R. S.; Koenig, B.; Dhavale, D. D. J. Org. Chem. 2016, 81, 7121. doi: 10.1021/acs.joc.6b00979
Das, A.; Ghosh, I.; König, B. Chem. Commun. 2016, 52, 8695. doi: 10.1039/C6CC04366F
Borpatra, P. J.; Deb, M. L.; Baruah, P. K. Tetrahedron Lett 2017, 58, 4006. doi: 10.1016/j.tetlet.2017.09.018
Huang, M.-H.; Zhu, Y.-L.; Hao, W.-J.; Wang, A.-F.; Wang, D.-C.; Liu, F.; Wei, P.; Tu, S.-J.; Jiang, B. Adv. Synth. Catal. 2017, 359, 2229. doi: 10.1002/adsc.v359.13
Liu, D.; Chen, J. Q.; Wang, X. Z.; Xu, P. F. Adv. Synth. Catal. 2017, 359, 2773. doi: 10.1002/adsc.v359.16
Jin, C.; Su, L.; Ma, D.; Cheng, M. New J. Chem. 2017, 41, 14053. doi: 10.1039/C7NJ03144K
Wei, W.; Cui, H.; Yang, D.; Yue, H.; He, C.; Zhang, Y.; Wang, H. Green Chem. 2017, 19, 5608. doi: 10.1039/C7GC02330H
Mondal, R. R.; Khamarui, S.; Maiti, D. K. Org. Lett. 2017, 19, 5964. doi: 10.1021/acs.orglett.7b02844
Wu, L. L.; Yang, G. H.; Guan, Z.; He, Y. H. Tetrahedron 2017, 73, 1854. doi: 10.1016/j.tet.2017.02.035
Sun, D.; Yin, K.; Zhang, R. Chem. Commun. (Camb.) 2018, 54, 1335. doi: 10.1039/C7CC09410H
Diao, P.; Ge, Y.; zhang, W.; Xu, C.; Zhang, N.; Guo, C. Tetrahedron Lett. 2018, 59, 767. doi: 10.1016/j.tetlet.2018.01.037
Alberti, M. N.; Vougioukalakis, G. C.; Orfanopoulos, M. J. Org. Chem. 2009, 74, 7274. doi: 10.1021/jo9012942
Griesbeck, A. G.; Steinwascher, J.; Reckenthäler, M.; Uhlig, J. Res. Chem. Intermed. 2012, 39, 33. doi: 10.1007%2Fs11164-012-0629-3
Li, J.; Wang, H.; Liu, L.; Sun, J. RSC Adv. 2014, 4, 49974. doi: 10.1039/C4RA09190F
Yadav, A. K.; Srivastava, V. P.; Yadav, L. D. S. RSC Adv. 2014, 4, 4181. doi: 10.1039/C3RA46553E
Liu, X.; Cong, T.; Liu, P.; Sun, P. J. Org. Chem. 2016, 81, 7256. doi: 10.1021/acs.joc.6b00097
Devari, S.; Rizvi, M. A.; Shah, B. A. Tetrahedron Lett. 2016, 57, 3294. doi: 10.1016/j.tetlet.2016.06.046
Cheng, X.; Yang, B.; Hu, X.; Xu, Q.; Lu, Z. Chemistry 2016, 22, 17566. doi: 10.1002/chem.v22.49
Jin, Y.; Ou, L.; Yang, H.; Fu, H. J Am. Chem. Soc. 2017, 139, 14237. doi: 10.1021/jacs.7b07883
Yue, H.; Nan, G. Synlett 2018, 29, 1340. doi: 10.1055/s-0037-1609443
Sahoo, M. K.; Jaiswal, G.; Rana. J.; Balaraman, E. Chemistry 2017, 23, 14167. doi: 10.1002/chem.201703642
Kee, C. W.; Chan, K. M.; Wong, M. W.; Tan, C. H. Asian J. Org. Chem. 2014, 3, 536. doi: 10.1002/ajoc.v3.4
Yang, X. J.; Chen, B.; Zheng, L. Q.; Wu, L. Z.; Tung, C. H. Green Chem. 2014, 16, 1082. doi: 10.1039/C3GC42042F
Yang, D. T.; Meng, Q. Y.; Zhong, J. J.; Xiang, M.; Liu, Q.; Wu, L. Z. Eur. J.Org. Chem. 2013, 2013, 7528. doi: 10.1002/ejoc.201301105
Liu, Z.; Zhang, Y.; Cai, Z.; Sun, H.; Cheng, X. Adv. Synth. Catal. 2015, 357, 589. doi: 10.1002/adsc.201400936
Majek, M.; Filace, F.; Wangelin, A. J. Chemistry 2015, 21, 4518. doi: 10.1002/chem.201406461
Yi, H.; Niu, L. B.; Wang, S. C.; Liu, T. Y.; Singh, A. K.; Lei, A. W. Org. Lett. 2016, 19, 122. https://www.ncbi.nlm.nih.gov/pubmed/28004943
Tucker, B. S.; Coughlin, M. L.; Figg, C. A.; Sumerlin, B. S. ACS Macro Lett. 2017, 6, 452. doi: 10.1021/acsmacrolett.7b00140

图式 1 光催化硫醇和烯烃合成β-酮亚砜的反应机理
Scheme 1 Reaction mechanism for synthesis of β-keto sulfoxides from mercaptan and olefin

图式 3 通过控制光的颜色选择性激发多卤代芳烃的C—X键
Scheme 3 Selective activation of aryl-halide bonds through different colors of light

图式 4 α-酮酯合成2, 3-二烷基化酒石酸酯的反应机理
Scheme 4 Mechanism of synthesis of 2, 3-dialkylated tartaric acid esters from α-ketoesters

图式 5 通过con-PET由卤化杂环合成芳基化的核碱基
Scheme 5 Synthesis of arylated nucleobases from halogenated heterocycles by con-PET

图式 6 咪唑杂环化合物的硫氰化反应机理
Scheme 6 Reaction mechanism of thiocyanation of imidazo-heterocycles

图式 7 由芳基亚磺酸盐合成乙烯基砜的反应机理
Scheme 7 Reaction mechanism of synthesis of vinyl sulfones from aryl sulfinates

图式 9 二芳基硫醚产物转化为4-(萘-1-基硫代)苯酚和砜
Scheme 9 Conversion from diaryl sulfide product to 4-(naph-thalen-1-ylthio)phenol and sulfone

图式 10 有机硫代硫酸盐的磺化氧化和亚硫酸化
Scheme 10 Sulfoxidation and sulfenylation of organic thiosulfate salts

图式 11 三级脂肪胺与芳基磺酰氯的反应机理
Scheme 11 Reaction mechanism of tertiary aliphatic amines with arenesulfonyl chlorides

图式 15 光催化合成香豆素衍生物的反应机理
Scheme 15 Mechanism of photocatalytic synthesis of coumarin derivatives

图式 18 合成2, 4, 6-三芳基吡啶的反应机理
Scheme 18 Mechanism of synthesis of 2, 4, 6-trisubstituted pyridines

图式 19 1, 7-烯炔的光催化双环化反应机理
Scheme 19 Mechanism of photocatalytic bicyclization of 1, 7-enynes

图式 23 由醛肟和伯酰胺合成腈的反应机理
Scheme 23 Mechanism of synthesis of nitriles from aldoximes and primary amides

图式 24 苄醇的区域选择性氧化机理
Scheme 24 Mechanism of chemo-selective oxidation of benzylic alcohols

图式 25 含N杂环的氧化脱羧反应机理
Scheme 25 Mechanism of oxidative dehydrogenation of N-heterocycles
表 1 代表性染料的光物理性质
Table 1. Photophysical properties of representative dyes
| Dye type | λmaxabs/nm | τf/ns | Excited state Energies/eV | Ground state redox potentials (V vs SCE) | Excited state redox potentials (V vs SCE): S1 | Excited state redox potentials (V vs SCE): T1 | |||||||
| E0, 0S1 | E0, 0T1a | E1/2red | E1/2ox | EredS1 | EoxS1 | EredT1 | EoxT1 | ||||||
| Eosin Y | 520a 533b |
2.1a 2.66a |
2.31a | 1.91 | -1.08a, c -1.13a, c |
+0.76a, c +0.72a, c |
+1.23a, c | -1.58a, c | +0.83a, c | -1.15a, c | |||
| Rose Bengal | 549 | 0.50 | 2.17a | 1.8 | -0.99a, c -0.78 |
+0.84a, c | +1.18a, c | -1.33a, c | +0.81a, c | -0.96a, c | |||
| Rhodamine B | 550d | 2.45a | 2.22a | 1.80 | -0.96a, c | +0.91a, c | +1.26a, c | -1.31a, c | +0.84a, c | -0.89a, c | |||
| Rhodamine 6G | 530b | 4.13a | 2.23 | 2.09 | -1.14c | +1.23 | +1.18c | -1.09a | +0.95a | -0.86a | |||
| a In MeOH. b In EtOH. c Potential originally reported relative to the Ag/AgCl reference electrode; referenced to SCE by subtracting 0.039 V from the reported valued. d In H2O. | |||||||||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们