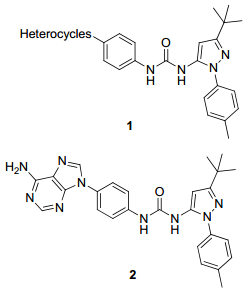
图 1.
已进入临床研究的FAK抑制剂
Figure 1.
Structures of the FAK inhibitors in clinic studies

黏着斑激酶(Focal adhesion kinase, FAK)是一种分子量约125 kDa的细胞内非受体酪氨酸激酶, 在多条信号通路, 尤其是整合素介导的信号传导过程中起中枢作用, 对细胞生长、增殖、迁移和粘附等有重要影响[1~3].研究表明, FAK在多种肿瘤中过度表达, 如卵巢癌、头颈鳞状细胞癌、肝癌等[4], 因此, 寻求高活性高选择性的FAK抑制剂成为肿瘤治疗的研究热点之一.
FAK抑制剂的研发主要集中在与腺嘌呤核苷三磷酸(ATP)结合口袋相互作用的小分子. FAK催化裂口有一个保守的活化环, 具有DFG (Asp-Phe-Gly)基序, 在调节激酶活性中具有重要作用, DFG中苯丙氨酸的苯环位置决定着激酶是否处于活性构象[5].当DFG处于ATP结合口袋外时(DFG-in), FAK处于激活状态; 当DFG的构象发生重排并移动进入到ATP结合口袋, 阻碍了该口袋与ATP的结合而形成非活性激酶构象(DFG-out), 同时使与ATP结合位点直接相邻的疏水结合位点暴露了出来[6, 7].
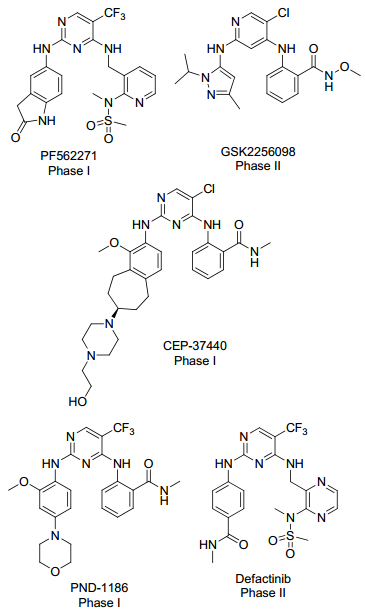
目前, 已有不少FAK抑制剂进入临床研究, 多为作用于DFG-in构象的小分子, 称为Ⅰ型抑制剂, 现进入临床研究的有PF562271[8], GSK2256098[9], CEP- 37440[10], PND-1186[11]以及Defactinib[12] (图 1)这5个小分子抑制剂.由于激酶ATP结合口袋高度保守, Ⅰ型抑制剂普遍存在着选择性不高等问题[13, 14].
而对于作用于激酶DFG-out构象的Ⅱ型抑制剂, 由于该类抑制剂除了与ATP结合口袋的铰链区相作用外, 部分分子片段可以伸入到DFG基序移动后留下的疏水口袋, 与变构疏水腔的残基相作用; 而不同激酶的变构疏水腔的差异性较大, 因此Ⅱ型抑制剂选择性多高于Ⅰ型抑制剂[13].
目前FAKII型抑制剂文献报道较少, 仅有Merck公司通过高通量筛选, 获得的一类如图 2所示结构为N-苯基吡唑脲类的Ⅱ型抑制剂, 其中, 活性最好的化合物是杂环结构为6-氨基-9H-嘌呤-9-基的化合物2, 其报道的酶抑制活性半数抑制率(IC50)为266 nmol/L[15].
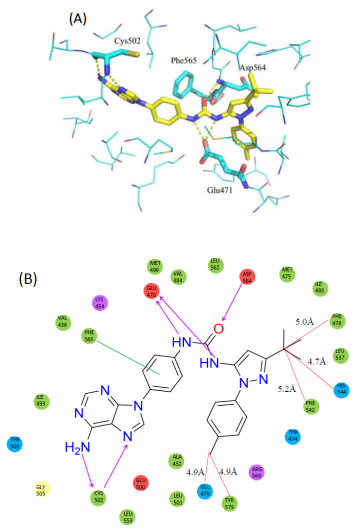
从化合物2与FAK激酶域的复合晶体结构中可以看出(图 3):化合物2的腺嘌呤结构, 朝向铰链区, 和铰链上的Cys502形成氢键作用; 连接链的苯环和DFG基序中苯丙氨酸形成π-π堆积作用, 脲基与Asp564和Glu471形成氢键作用; N-苯基吡唑结构伸入到变构疏水口袋中, 3-位的叔丁基被His544, Phe542及Phe478三个芳香性残基包围, 1-位苯基对位的甲基指向关键氨基酸Gln470及Tyr570.
鉴于激酶DFG-out构象中变构疏水区域的结构特异性, 本工作以化合物2为先导化合物, 基于FAK变构疏水腔的结构特征(图 3), 对作用于该区域相应的分子片段, 即尾部N-苯基吡唑结构, 进行了系统的结构修饰和构效关系研究.
化合物的设计主要从以下几个方面进行: (1)苯基与吡唑环之间二面角对活性的影响, 设计在苯环的2-或者6-位引入卤素或甲基, 增加两个芳环之间的二面角, 探索最合适的二面角; (2)苯基对位取代基对活性的影响, 苯环上的甲基与Tyr579与Gln470距离较近, 因此将甲基修饰成酰胺结构, 在保持基团疏水性的同时, 考察与周围氨基酸残基可否形成氢键作用; (3)吡唑环上基团对活性的影响, 3-位叔丁基周围的His544, Phe542及Phe478均为芳香性氨基酸残基, 将叔丁基修饰为苯基或具有部分芳香性的小环, 考察其与周围氨基酸残基可否形成π-π堆积作用.
9H-嘌呤-6-胺(3)和4-氯硝基苯在高温碱性条件下经亲核取代得9-N-对硝基苯基-9H-嘌呤-6-胺(4)[16], 化合物4经过铁粉还原得到关键中间体9-N-对氨基苯基- 9H-嘌呤-6-胺(5).不同取代的苯肼盐酸盐6和相应的酰基乙腈环合得到1-N-苯基吡唑-5-胺7a~7i, 7a~7g与氯甲酸苯酯缩合得到引入苯氧酰基的关键中间体8a~8g[15]. 9-N-对氨基苯基-9H-嘌呤-6-胺(5)和化合物8a~8g在碱性条件下以47%~81%的收率缩合得到目标化合物9a~9g (Scheme 1).
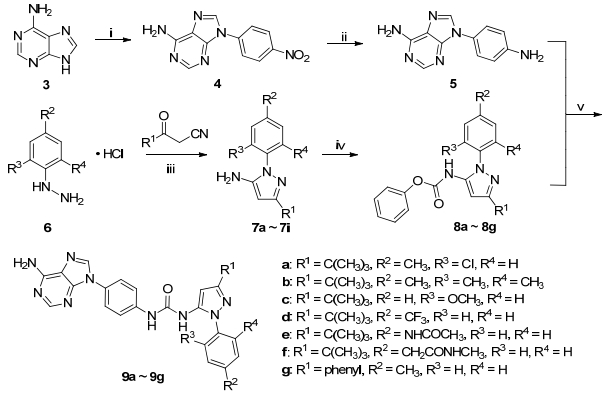
Reagents and conditions: (ⅰ) 4-chloronitrobenzene, K2CO3, DMF, 150 ℃, 3 d, 29%; (ⅱ) Fe, HCl, EtOH, 95 ℃, overnight, 68%; (ⅲ) HCl, EtOH, 95 ℃, 8 h, 27%~82%; (ⅳ) phenyl chloroformate, pyridine, EA, r.t. 1 h, quantitatively; (ⅴ) TEA, THF, r.t., 7 h, 47%~81%
值得注意的是, 当吡唑环上的取代基为环丙基(7h)和环丁基(7i)时, 这两个化合物和氯甲酸苯酯的反应未获得预期的氨基酰化产物, 板层析显示反应无明显主产物, 因此我们调整了合成方案, 如Scheme 2所示, 将另一个关键中间体化合物4的氨基酰化, 即合成9-N-对苯氧甲酰氨基苯基-9H-嘌呤-6-胺(10), 化合物10分别与7h和7i缩合以39%和62%的收率获得化合物9h和9i.
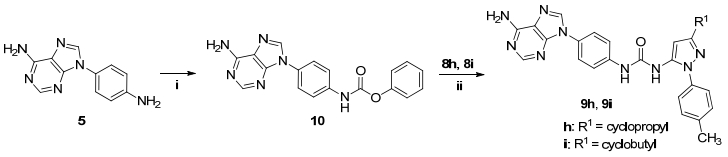
Reagents and conditions: (ⅰ) phenyl chloroformate, pyridine, THF, r.t. 3 h, 56%; (ⅱ) TEA, THF, r.t., 7 h, 39%~62%
利用ATPase-LUM assay, 对合成目标化合物9a~9i进行FAK抑制活性测试, 活性结果(表 1)显示, 化合物2尾部N-苯基吡唑环上基团的改变, 对活性影响非常显著, 说明该变构疏水腔对基团改变比较敏感. (1)当苯环的2-和/或6-位引入基团后, 如化合物9a~9c, 酶抑制活性均显著降低, 推测苯环与吡唑环相连位置的邻位有取代基时, 使得苯环和吡唑之间单键不能自由旋转, 较大二面角的存在造成尾部片段呈现一种刚性结构, 从而不能更好地契合FAK的变构疏水腔. (2)苯基上对位的甲基修饰成三氟甲基(9d)、乙酰氨基(9e)和N-甲基氨甲酰甲基(9f)后, 酶活性与先导化合物在同一水平, 其中9e活性相比于先导化合物提高了两倍, 说明化合物的酶活性对苯环对位基团没有严格的要求. (3)当吡唑环上的叔丁基修饰成苯基(9g)和环丙基(9h)时, 酶活性降低, 而修饰成环丁基(9i)时, 酶活性保持, 说明吡唑3-位的基团大小对活性的影响非常大, 基团过大和过小都不能很好地契合疏水腔.
 下载:
导出CSV
下载:
导出CSV
| Compd. | Inhibition rate/% | FAK IC50/(μmol•L-1) |
| 9a | 48 | NT |
| 9b | 11.5 | NT |
| 9c | 35 | NT |
| 9d | 86 | 0.094 |
| 9e | 94.5 | 0.041 |
| 9f | 98.5 | 0.144 |
| 9g | 37 | NT |
| 9h | 64 | NT |
| 9i | 91 | 0.076 |
| 2 | 97.5 | 0.077 |
| a Test concentration: 10 μmol•L-1; NT: not tested. | ||
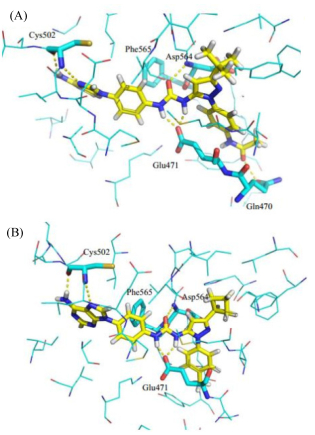
我们对活性显著的目标化合物9e和9i与FAK激酶域进行了分子对接(图 4), 从图中可以看出9e和9i头部保持和Cys502的相互作用, 脲基和Glu471及Asp564形成氢键作用以稳定FAK的非激活构象.除此之外, 9e的N-苯基吡唑结构中苯环对位的酰胺键和疏水腔的关键氨基酸Gln470可以形成氢键作用, 认为该作用力存在使得化合物活性提高.从图 4B可以看出9i尾部N-苯基吡唑环上的环丁基嵌入His544、Phe542及Phe478形成的疏水腔中.而取代基为苯环的化合物9g的活性降低, 说明吡唑3-位苯基并未和周围氨基酸残基形成π-π堆积作用, 反而由于体积较大而和周围氨基酸残基有空间位阻.
以FAKII型抑制剂化合物2为先导化合物, 基于FAK变构疏水腔的结构特征, 对尾部结构N-苯基吡唑进行了设计和结构优化, 共合成9个结构修饰的目标化合物.生物活性评价显示FAK的变构疏水腔对基团改变非常敏感, 这与Ⅱ型抑制剂选择性较高相一致.其中, 化合物9e具有优于先导化合物的FAK抑制活性, IC50值为41 nmol/L, 表明N-苯基吡唑结构中苯环对位有更多结构优化空间, 可通过与周边残基形成氢键从而提高化合物活性, 这一初步构效关系研究结论为后续结构优化奠定了研究基础并提供了研究方向.
1H NMR和13C NMR谱分别以Bruker-DPX 400 MHz和600 MHz核磁共振仪测定; 低分辨质谱以Agilent G1946D质谱仪测定; 高分辨质谱以Bruker Daltonics APEXIII 7.0 TESLA FTMS质谱仪测定; 熔点用SGW X-4型显微熔点仪测定; 薄层色谱(TLC)用板为青岛海洋的0.2 mm GF254高效薄层层析硅胶板; 柱层析所用的硅胶规格为300~400目, 生产商为黄海化学公司; 多功能酶标仪型号为SpectraMax M5.实验所用试剂均购自商业试剂公司, 纯度在97%以上; 所用溶剂为分析纯; 所用氘代试剂购自美国CIL公司, 以TMS为内标, 纯度在99.8%以上.生物活性测试所用FAK激酶购自默克密理博, Kinase-Glo Plus Luminescence kinase assay kit购自Promega, Poly(Glu, Tyr)购自Sigma- Aldrich, ATP购自Invivogen.
9H-嘌呤-6-胺100 mg (0.74 mmol)、对硝基氯苯140 mg (0.89 mmol)和碳酸钾205 mg (1.48 mmol)溶于4 mL二甲亚砜(DMSO)中, 外浴150 ℃搅拌过夜.反应结束后, 反应液加入到20 mL水中, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 分得黄色固体54 mg, 收率29%. m.p.>250 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 7.56 (s, 2H), 8.28 (s, 1H), 8.37 (d, J=9.00 Hz, 2H), 8.47 (d, J=9.00 Hz, 2H), 8.48 (s, 1H); ESI-MS m/z: 257.1 [M+H]+.
化合物4 172 mg (0.67 mmol)及铁粉151 mg (2.69 mmol)投于6 mL乙醇中, 加入盐酸使反应液pH为1, 外浴95 ℃下搅拌. 3 h后停止反应, 向反应液加入饱和碳酸钠溶液调节溶液pH大于7, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 分得棕色固体105 mg, 收率68%. m.p.>250 ℃ (Lit.[16]>260 ℃); 1H NMR (DMSO-d6, 400 MHz) δ: 7.56 (s, 2H), 8.28 (s, 1H), 8.37 (d, J=9.00 Hz, 2H), 8.47 (d, J=9.00 Hz, 2H), 8.48 (s, 1H); ESI-MS m/z: 227.1 [M+H]+; HRMS (ESI) calcd for C11H9N6O2 [M+H]+ 257.0782, found 257.0779.
2, 4, 6-三取代苯肼盐酸盐(1 mmol)和取代酰基乙腈(1.2 mmol)投于0.4 mol/L的盐酸乙醇溶液中, 外浴95 ℃下搅拌.反应完成(通过TLC监测)后, 将反应液浓缩, 加入氢氧化钠溶液调节pH到11, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 得到化合物7a~7i.
1-(2-氯-4-甲基苯基)-3-叔丁基-1H-吡唑-5-胺(7a):白色固体, 收率73%. m.p. 134~135 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.30 (s, 9H), 2.37 (s, 3H), 3.53 (s, 2H), 5.51 (s, 1H), 7.16 (d, J=8.00 Hz, 1H), 7.31 (s, 1H), 7.35 (d, J=8.00 Hz, 1H); ESI-MS m/z: 264.2 [M+H]+; HRMS (ESI) calcd for C14H19ClN3 [M+H]+ 264.1262, found 264.1264.
1-(2, 4, 6-三甲基苯基)-3-叔丁基-1H-吡唑-5-胺(7b):白色固体, 收率59%. m.p. 115~116 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.29 (s, 9H), 2.00 (s, 6H), 2.29 (s, 3H), 3.36 (s, 2H), 5.47 (s, 1H), 6.92 (s, 2H); ESI-MS m/z: 258.3 [M+H]+; HRMS (ESI) calcd for C16H24N3 [M+ H]+ 258.1965, found 258.1967.
1-(2-甲氧基苯基)-3-叔丁基-1H-吡唑-5-胺(7c):白色固体, 收率46%. m.p. 171~173 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.31 (s, 9H), 3.80 (s, 2H), 3.86 (s, 3H), 5.51 (s, 1H), 7.70~7.08 (m, 2H), 7.30~7.34 (m, 1H), 7.45 (d, J=8.64 Hz, 1H); ESI-MS m/z: 246.1 [M+H]+; HRMS (ESI) calcd for C14H20N3O [M+H]+ 246.1601, found 246.1602.
1-(4-三氟甲基苯基)-3-叔丁基-1H-吡唑-5-胺(7d):白色固体, 收率52%. m.p. 124~125 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.31 (s, 9H), 3.76 (s, 2H), 5.57 (s, 1H), 7.70 (d, J=8.42 Hz, 2H), 7.77 (d, J=8.42 Hz, 2H); ESI-MS m/z: 284.2 [M+H]+; HRMS (ESI) calcd for C14H17F3N3 [M+H]+ 284.1369, found 284.1368.
1-(4-乙酰氨基苯基)-3-叔丁基-1H-吡唑-5-胺(7e):黄色固体, 收率54%. m.p. 97~99 ℃; 1H NMR (DMSO- d6, 400 MHz) δ: 1.20 (s, 9H), 2.05 (s, 3H), 5.14 (s, 2H), 5.34 (s, 1H), 7.44 (d, J =8.44 Hz, 2H), 7.64 (d, J =8.44 Hz, 2H), 10.05 (s, 1H); ESI-MS m/z: 273.2 [M+H]+; HRMS (ESI) calcd for C15H21N4O [M+H]+ 273.1710, found 273.1711.
1-(4-(N-甲基氨甲酰甲基)苯基)-3-叔丁基-1H-吡唑- 5-胺(7f):黄色固体, 收率27%. m.p. 143~145 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.20 (s, 9H), 2.58 (s, 3H), 3.40 (s, 2H), 5.18 (s, 2H), 5.36 (s, 1H), 7.31 (d, J=7.06 Hz, 2H), 7.47 (d, J=7.06 Hz, 2H), 8.00 (s, 1H); ESI-MS m/z: 287.2 [M+H]+; HRMS (ESI) calcd for C16H22N4ONa [M+Na]+ 309.1686, found 309.1680.
1-(4-甲基苯基)-3-苯基-1H-吡唑-5-胺(7g):白色固体, 收率76%. m.p. 148~149 ℃ [Lit.[17] m.p. 145~148 ℃]; 1H NMR (CDCl3, 400 MHz) δ: 2.41 (s, 3H), 3.84 (s, 2H), 5.96 (s, 1H), 7.29~7.32 (m, 3H), 7.37~7.40 (m, 2H), 7.50 (d, J=8.16 Hz, 2H), 7.81 (d, J=7.60 Hz, 2H); ESI-MS m/z: 250.2 [M+H]+.
1-(4-甲基苯基)-3-环丙基-1H-吡唑-5-胺(7h):淡黄色固体, 收率77%. m.p. 127~128 ℃; 1H NMR (CDCl3, 400 MHz) δ: 0.64~0.65 (m, 2H), 0.82~0.84 (m, 2H), 1.80~1.86 (m, 1H), 2.31 (s, 3H), 3.66 (s, 2H), 5.19 (s, 1H), 7.18 (d, J=8.10 Hz, 2H), 7.33 (d, J=8.10 Hz, 2H); ESI-MS m/z: 214.2 [M+H]+; HRMS (ESI) calcd for C13H16N3 [M+H]+ 214.1339, found 241.1340.
1-(4-甲基苯基)-3-环丁基-1H-吡唑-5-胺(7i):淡黄色固体, 收率82%. m.p. 103~105 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.82~2.04 (m, 2H), 2.14~2.33 (m, 4H), 2.37 (s, 3H), 3.44~3.52 (m, 1H), 3.74 (s, 2H), 5.54 (s, 1H), 7.23 (d, J=8.22 Hz, 2H), 7.39 (d, J=8.22 Hz, 2H); ESI-MS m/z: 228.2 [M+H]+; HRMS (ESI) calcd for C14H18N3 [M+H]+ 228.1495, found 228.1496.
化合物7a~7i (1 mmol)和吡啶(2 mmol)投于乙酸乙酯中, 冰浴下滴加氯甲酸苯酯(1.1 mmol), 滴加完毕后撤去冰浴, 室温搅拌.反应完成后(通过TLC监测), 减压除去溶剂, 得到粗品8a~8i.向8a~8i中加入溶有化合物5 (0.8 mmol), 三乙胺(2 mmol)的四氢呋喃溶液, 室温下搅拌.反应完成后(通过TLC监测), 向反应液中加入15 mL水, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 得到化合物9a~9i.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(2-氯-4-甲基苯基)-3-叔丁基-1H-吡唑-5-基)脲(9a):淡棕色固体, 收率59%. m.p. 236~238 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.31 (s, 9H), 2.43 (s, 3H), 6.39 (s, 1H), 7.36 (d, J=8.20 Hz, 1H), 7.40 (s, 2H), 7.46 (d, J=8.20 Hz, 1H), 7.57 (s, 2H), 7.58 (d, J=8.58 Hz, 2H), 7.77 (d, J=8.58 Hz, 2H), 8.19 (s, 1H), 8.33 (s, 1H), 8.52 (s, 1H), 9.11 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 160.93, 156.13, 152.87, 150.57, 148.95, 141.13, 139.42, 138.61, 138.32, 132.66, 131.40, 130.29, 130.15, 129.11, 128.65, 123.53, 118.97, 118.36, 91.45, 31.93, 30.08, 20.33; ESI-MS m/z: 516.2 [M+H]+; HRMS (ESI) calcd for C26H27ClN9O [M+H]+ 516.2022, found 516.2019.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(2, 4, 6-三甲基苯基)-3-叔丁基-1H-吡唑-5-基)脲(9b):白色固体, 收率81%. m.p. 236~241 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.27 (s, 9H), 1.90 (s, 6H), 2.35 (s, 3H), 6.39 (s, 1H), 7.08 (s, 2H), 7.40 (s, 2H), 7.57 (d, J=8.28 Hz, 2H), 7.76 (d, J=8.28 Hz, 2H), 8.17 (s, 1H), 8.19 (s, 1H), 8.52 (s, 1H), 9.14 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 160.73, 156.13, 152.87, 150.51, 148.94, 139.42, 138.55, 138.36, 138.06, 136.55, 133.18, 129.06, 128.65, 123.50, 118.97, 118.30, 90.04, 31.90, 30.21, 20.57, 16.77; ESI-MS m/z: 510.3 [M+H]+; HRMS (ESI) calcd for C28H32N9O [M+H]+ 510.2724, found 510.2727.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(2-甲氧基苯基)-3-叔丁基-1H-吡唑-5-基)脲(9c):淡棕色固体, 收率65%. m.p. 194~196 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.27 (s, 9H), 3.83 (s, 3H), 6.39 (s, 1H), 7.10~7.13 (m, 1H), 7.30 (d, J=8.36 Hz, 2H), 7.36~7.40 (m, 3H), 7.50~7.54 (m, 1H), 7.59 (d, J=8.68 Hz, 2H), 7.77 (d, J=8.68 Hz, 2H), 8.09 (s, 1H), 8.19 (s, 1H), 8.52 (s, 1H), 9.33 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 160.52, 156.12, 153.74, 152.87, 150.61, 148.95, 139.43, 138.44, 130.08, 129.13, 126.44, 123.54, 120.65, 118.97, 118.35, 112.67, 91.54, 55.67, 31.88, 30.13; ESI-MS m/z: 498.3 [M+H]+; HRMS (ESI) calcd for C26H28N9O2 [M+H]+ 498.2360, found 498.2365.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-三氟甲基苯基)-3-叔丁基-1H-吡唑-5-基)脲(9d):棕色固体, 收率55%. m.p. 189~193 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.31 (s, 9H), 6.47 (s, 1H), 7.41 (s, 2H), 7.61 (d, J=8.40 Hz, 2H), 7.77 (d, J=8.40 Hz, 2H), 7.83 (d, J=8.04 Hz, 2H), 7.92 (d, J=8.04 Hz, 2H), 8.20 (s, 1H), 8.52 (s, 1H), 8.66 (s, 1H), 9.30 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 161.64, 156.13, 152.89, 151.64, 148.96, 141.78, 139.42, 138.45, 137.32, 129.16, 126.29 (dC-F, J=2.75 Hz), 123.76, 123.48, 118.98, 118.63, 97.24, 31.98, 29.89; ESI-MS m/z: 536.3 [M+H]+; HRMS (ESI) calcd for C26H25F3N9O [M+H]+ 536.2129, found 536.2127.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-乙酰氨基苯基)-3-叔丁基-1H-吡唑-5-基)脲(9e):淡棕色固体, 收率55%. m.p. 216~220 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.28 (s, 9H), 2.08 (s, 3H), 6.38 (s, 1H), 7.39 (s, 2H), 7.45 (d, J=7.40 Hz, 2H), 7.60 (d, J=8.72 Hz, 2H), 7.73~7.77 (m, 4H), 8.19 (s, 1H), 8.52 (s, 1H), 8.60 (s, 1H), 9.48 (s, 1H), 10.23 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 168.29, 160.30, 156.13, 152.88, 151.28, 148.95, 139.43, 138.55, 138.44, 136.96, 133.07, 129.03, 124.99, 123.51, 119.23, 118.96, 118.44, 94.49, 31.87, 30.08, 23.87; ESI-MS m/z: 525.2 [M+H]+; HRMS (ESI) calcd for C27H29N10O2 [M+H]+ 525.2469, found 525.2468.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-(N-甲基氨甲酰甲基)苯基)-3-叔丁基-1H-吡唑-5-基)脲(9f):棕色固体, 收率47%. m.p. 194~196 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.28 (s, 9H), 2.59 (d, J=4.52 Hz, 3H), 3.47 (s, 2H), 6.41 (s, 1H), 7.39~7.47 (m, 6H), 7.61 (d, J=8.76 Hz, 2H), 7.76 (d, J=8.76 Hz, 2H), 8.07 (d, J=3.96 Hz, 1H), 8.19 (s, 1H), 8.49 (s, 1H), 8.52 (s, 1H), 9.29 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 170.03, 160.52, 156.13, 152.88, 151.37, 148.96, 139.44, 138.54, 136.88, 136.71, 135.53, 129.66, 129.05, 124.07, 123.51, 118.97, 118.47, 94.98, 41.69, 31.87, 30.04, 25.49; ESI-MS m/z: 539.3 [M+H]+; HRMS (ESI) calcd for C28H31N10O2 [M+H]+ 539.2626, found 539.2627.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-甲基苯基)-3-苯基-1H-吡唑-5-基)脲(9g):淡棕色固体, 收率50%. m.p. 246~248 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 2.41 (s, 3H), 6.96 (s, 1H), 7.32~7.45 (m, 7H), 7.52 (d, J=7.76 Hz, 2H), 7.63 (d, J=8.54 Hz, 2H), 7.78 (d, J=8.54 Hz, 2H), 7.86 (d, J=7.76 Hz, 2H), 8.20 (s, 1H), 8.52 (s, 1H), 8.60 (s, 1H), 9.33 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 156.13, 152.89, 151.37, 149.75, 148.96, 139.44, 138.44, 138.24, 137.41, 135.59, 132.95, 129.71, 129.16, 128.52, 127.68, 124.92, 124.56, 123.53, 118.98, 118.60, 95.60, 20.52; ESI-MS m/z: 502.3 [M+H]+; HRMS (ESI) calcd for C28H24N9O [M+H]+ 502.2098, found 502.2098.
化合物5 50 mg (0.22 mmol)和吡啶18 μL (0.44 mmol)投于4 mL四氢呋喃中, 冰浴下滴加氯甲酸苯酯36 μL (0.29 mmol), 滴加完毕后撤去冰浴, 室温搅拌. 1 h后, 向反应液加入15 mL水, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 分得到43 mg白色固体, 收率56%. m.p.>250 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 7.26~7.30 (m, 3H), 7.42~7.47 (m, 4H), 7.70 (d, J=8.76 Hz, 2H), 7.84 (d, J=8.76 Hz, 2H), 8.20 (s, 1H), 8.54 (s, 1H), 10.50 (s, 1H); ESI-MS m/z: 347.1 [M+H]+; HRMS (ESI) calcd for C18H15N6O2 [M+H]+ 347.1251, found 347.1244
化合物10 (1 mmol), 化合物7h或7i (1.7 mmol)和三乙胺(2 mmol)投于4 mL四氢呋喃中, 加热回流.反应完成(通过TLC监测), 向反应液中加入15 mL水, 乙酸乙酯萃取, 合并有机相, 干燥并浓缩后经柱层析, 得到化合物9h和9i.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-甲基苯基)-3-环丙基-1H-吡唑-5-基)脲(9h):白色固体, 收率39%. m.p.>250 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 0.69~0.70 (m, 2H), 0.88~0.90 (m, 2H), 1.85~1.92 (m, 1H), 2.38 (s, 3H), 6.18 (s, 1H), 7.33~7.40 (m, 6H), 7.59 (d, J=8.58 Hz, 2H), 7.76 (d, J=8.58 Hz, 2H), 8.19 (s, 1H), 8.52 (s, 1H), 8.66 (s, 1H), 9.30 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 156.12, 153.89, 152.88, 151.29, 149.03, 139.43, 138.49, 137.10, 136.75, 135.73, 129.57, 129.06, 124.22, 123.51, 118.96, 118.49, 94.65, 20.46, 9.24, 7.52; ESI-MS m/z: 466.2 [M+H]+; HRMS (ESI) calcd for C25H24N9O [M+H]+ 466.2098, found 466.2100.
N1-(4-(6-氨基-9H-嘌呤-9-基)苯基)-N3-(1-(4-甲基苯基)-3-环丁基-1H-吡唑-5-基)脲(9i):白色固体, 收率62%. m.p. 190~192 ℃; 1H NMR (DMSO-d6, 400 MHz) δ: 1.81~2.03 (m, 2H), 2.13~3.51 (m, 4H), 2.38 (s, 3H), 3.43~3.51 (m, 1H), 6.41 (s, 1H), 7.34~7.42 (m, 6H), 7.60 (d, J=8.16 Hz, 2H), 7.77 (d, J=8.16 Hz, 2H), 8.19 (s, 1H), 8.46 (s, 1H), 8.52 (s, 1H), 9.27 (s, 1H); 13C NMR (DMSO-d6, 150 MHz) δ: 156.14, 155.65, 152.88, 151.31, 148.96, 139.43, 138.51, 137.19, 136.82, 135.75, 129.58, 129.07, 124.33, 123.51, 118.98, 118.49, 95.67, 33.67, 28.47, 20.46, 18.07; ESI-MS m/z: 480.3 [M+H]+; HRMS (ESI) calcd for C26H26N9O [M+H]+ 480.2255, found 480.2254.
在96孔板中, 依次加入腺嘌呤核苷三磷酸(ATP) (100 μmol/L, 5 μL), 底物Poly (Glu: Tyr) (100 μmol/L, 5 μL)及FAK (240 ng, 2.4 μL).用DMSO将化合物配成10 mmol/L母液, 再用缓冲液依次稀释得到一系列浓度的测试液, 向96孔板中依次添加各浓度的测试液, 每孔5 μL, 每个浓度3个复孔.同时设置不加药的阴性对照组.在摇床中孵化40 min, 加入50 μL的Kinase-Glo Plus检测液, 孵化10 min.酶标仪Luminecesce模式下检测发光, 收集数据.依据公式:酶活性(%)=[(Lu无酶-Lu加药)/(Lu无酶-Lu有酶无药)]×100%.计算得到各浓度下的活性, 再用Graphpad Prism 6软件处理, 得到IC50值.
辅助材料(Supporting Information) 化合物9a~9i的1H NMR, 13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Zhao, X.; Guan, J. Adv. Drug Delivery Rev. 2011, 63, 610. doi: 10.1016/j.addr.2010.11.001
Heinrich, T.; Seenisamy, J.; Emmanuvel, L.; Kulkarni, S. S.; Bomke, J.; Rohdich, F.; Greiner, H.; Esdar, C.; Krier, M.; Gradler, U.; Musilet, D. J. Med. Chem. 2013, 56, 1160. doi: 10.1021/jm3016014
Parsons, J. T. J. Cell Sci. 2003, 116, 1409. doi: 10.1242/jcs.00373
Sulzmaier, F. J.; Jean, C.; Schlaepfer, D. D. Nat. Rev. Cancer 2014, 14, 598. doi: 10.1038/nrc3792
Schneider, G.; Geppert, T.; Hartenfeller, M.; Reisen, F.; Klenner, A.; Reutlinger, M.; Hähnke, V.; Hiss, J. A.; Zettl, H.; Keppner, S.; Spänkuch, B.; Schneideret, P. Future Med. Chem. 2011, 3, 415. doi: 10.4155/fmc.11.8
彭文, 张小猛, 张仓, 王芳, 尤启冬, 中国新药杂志, 2012, 21, 890. http://www.cnki.com.cn/Article/CJFDTotal-ZXYZ201208017.htmPeng, W.; Zhang, X. W.; Zhang, C.; Wang, F.; You, Q. D. Chin. J. New Drug 2012, 21, 890(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-ZXYZ201208017.htm
Dietrich, J.; Hulme, C.; Hurley, L. H. Bioorg. Med. Chem. 2010, 18, 5738. doi: 10.1016/j.bmc.2010.05.063
Infante, J. R.; Camidge, D. R.; Mileshkin, L. R.; Chen, E. X.; Hicks, R. J.; Rischin, D.; Fingert, H.; Pierce, K. J.; Xu, H. P.; Roberts, W. G.; Shreeve, S. M.; Burris, H. A.; Siu, L. L. J. Clin. Oncol. 2012, 30, 1527. doi: 10.1200/JCO.2011.38.9346
Zhang, J.; He, D. H.; Zajac-Kaye, M.; Hochwald, S. N. Cell Cycle 2014, 13, 3143. doi: 10.4161/15384101.2014.949550
Ott, G. R.; Cheng, M.; Learn, K. S.; Wagner, J.; Gingrich, D. E.; Lisko, J. G.; Curry, M.; Mesaros, E. F.; Ghose, A. K.; Quail, M. R.; Wan, W. H.; Lu, L. H.; Dobrzanski, P.; Albom, M. S.; Angeles, T. S.; Wells-Knecht, K.; Huang, Z. Q.; Aimone, L. D.; Bruckheimer, E.; Anderson, N.; Friedman, J.; Fernandez, S. V.; Ator, M. A.; Ruggeri, B. A.; Dorsey, B. D. J. Med. Chem. 2016, 59, 7478. doi: 10.1021/acs.jmedchem.6b00487
Tanjoni, I.; Colin Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.; Mielgo, A.; Lim, S.; Liang, C. X.; Koenig, M.; Patel, N.; Kwok, C.; McMahon, G.; Stupack, D. G.; Schlaepfer, D. D. Cancer Biol. Ther. 2010, 9, 764. doi: 10.4161/cbt.9.10.11434
Kang, Y.; Hu, W.; Ivan, C.; Dalton, H. J.; Miyake, T.; Pecot, C. V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N. B.; Rupaimoole, R.; Taylor, M.; Pradeep, S.; Wu, S. Y.; Lu, C. H.; Wen, Y. F.; Huang, J. F.; Liu, J. S.; Sood A. K. J. Natl. Cancer Inst. 2013, 105, 1485. doi: 10.1093/jnci/djt210
Iwatani, M.; Iwata, H.; Okabe, A.; Skene, R. J.; Tomita, N.; Hayashi, Y.; Aramaki, Y.; Hosfield, D. J.; Hori, A.; Baba, A.; Miki, H. Eur. J. Med. Chem. 2013, 61, 49. doi: 10.1016/j.ejmech.2012.06.035
Johnson, L. N.; Noble, M.; Owen, D. J. Cell 1996, 85, 149. doi: 10.1016/S0092-8674(00)81092-2
Grädler, U.; Bomke, J.; Musil, D.; Dresing, V.; Lehmann, M.; Hölzemann, G.; Greiner, H.; Esdar, C.; Krier, M.; Heinrich, T. Bioorg. Med. Chem. Lett. 2013, 23, 5401. doi: 10.1016/j.bmcl.2013.07.050
Váňa, L.; Vrzal, L.; Dvořáková, H.; Himl, M.; Linhart, I. Synth. Commun. 2014, 44, 788. doi: 10.1080/00397911.2013.831902
Simay, A.; Takacs, K.; Horvath, K.; Dvortsak, P. Acta Chim. Acad. Sci. Hung. 1980, 105, 127.

图 3 化合物2与FAK激酶域的复合晶体3D和2D图(PDB ID: 4K9Y)
Figure 3 The 3D and 2D figures of crystal complexes of 2 and FAK kinase region (PDB ID: 4K9Y)

图式 1 目标化合物9a~9g的合成路线
Scheme 1 Synthetic route of compounds 9a~9g
Reagents and conditions: (ⅰ) 4-chloronitrobenzene, K2CO3, DMF, 150 ℃, 3 d, 29%; (ⅱ) Fe, HCl, EtOH, 95 ℃, overnight, 68%; (ⅲ) HCl, EtOH, 95 ℃, 8 h, 27%~82%; (ⅳ) phenyl chloroformate, pyridine, EA, r.t. 1 h, quantitatively; (ⅴ) TEA, THF, r.t., 7 h, 47%~81%

图式 2 化合物9h和9i的合成路线
Scheme 2 Synthetic route of compounds 9h and 9i
Reagents and conditions: (ⅰ) phenyl chloroformate, pyridine, THF, r.t. 3 h, 56%; (ⅱ) TEA, THF, r.t., 7 h, 39%~62%

图 4 化合物9e (A)及9i (B)和FAK激酶域的分子对接图
Figure 4 Docking structures of 9e (A) and 9i (B) with FAK kinase region
表 1 化合物9a~9i对FAK酶的抑制活性a
Table 1. Enzymatic activity assay of 9a~9i
| Compd. | Inhibition rate/% | FAK IC50/(μmol•L-1) |
| 9a | 48 | NT |
| 9b | 11.5 | NT |
| 9c | 35 | NT |
| 9d | 86 | 0.094 |
| 9e | 94.5 | 0.041 |
| 9f | 98.5 | 0.144 |
| 9g | 37 | NT |
| 9h | 64 | NT |
| 9i | 91 | 0.076 |
| 2 | 97.5 | 0.077 |
| a Test concentration: 10 μmol•L-1; NT: not tested. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们