Scheme 1.
Synthesis of diaryl-o-carboranes

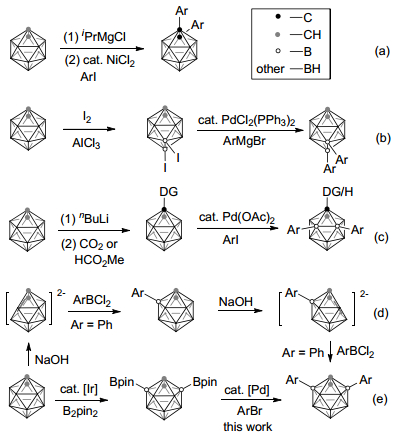
Carboranes are finding wide applications in medicine as boron neutron capture therapy (TNCT) agents, [1] coordination/organometallic chemistry as versatile ligands, [2] in materials as functional units, [3] and in supramolecular design as building blocks.[4] Especially, cage carbon-arylated o-carboranes are used as a class of AIE (aggregation induced emission) active and stimuli-responsive luminescent materials, where the carborane moiety plays a unique role.[5] In this regard, 1, 2-diaryl- o-carboranes were prepared by transition metal-catalyzed Kumada-type cross-coupling of 1, 2-di-magnesium chloride-o-carboranes with aryl iodides, [6] or via the condensation reaction of decaborane with diarylacetylenes.[7] For cage boron arylation, direct palladium-catalyzed B-H substitution only afforded a mixture of B(8)- or B(9)-mono-arylation compounds.[8] Pre-iodinaion is necessary for the most electron-rich B(9, 12)-position to achieve diarylation by a palladium-catalyzed coupling with aryl Grignard reagents.[9] More recently, 1-CO2H, 1-CHO and 1-CH2NH2 groups guided transition metal catalyzed cage B(4, 5)-diarylation[10] of o-carboranes has been achieved by direct B—H bond activation.[11] On the other hand, cage B(3, 6)-diarylaed-o-carboranes are generally achieved via multistep reaction of deboration-capping- deboration-capping, which is suffered from the narrow substrate scope and low yields (Scheme 1).[12]
Suzuki-Miyaura coupling is one of the most widely used selective C—C bond forming methods, in which organoboron reagents react with a series of electrophilic coupling partners, such as aryl or vinyl halide, triflate, diazonium/iodonium salt and carboxylic acid derivatives.[13] Very recently, we have developed an iridium-catalyzed regioselective B(3, 6)-diborylation of o-carborane via direct B—H activation.[14] These novel cage B-Bpin compounds can act as a new kind of boron nucleophiles. Herein we report our studies on the cross-coupling reactions of 3, 6-(Bpin)2-1, 2- C2B10H10 with aryl halides for an efficient synthesis of B(3, 6)-diarylated-o-carboranes.
The initial optimization of reaction conditions for the reaction of 3, 6-(Bpin)2-1, 2-C2B10H10 (1) with phenylbromide (2a) was summarized in Table 1. In the presence of 20 mol% Pd(PPh3)4 and 3 equiv. of Cs2CO3, a quantitative formation of coupling product 4a was observed by GC when cyclohexane was used as the solvent (Table 1, Entry 4). Further screening of other bases in cyclohexane solution did not afford better results (Table 1, Entries 5~8). Screening the Pd catalysts indicated that Pd2(dba)3/PPh3 offered a lower conversion, while Pd2(dba)3 showed no reactivity (Table 1, Entries 9 and 10). Among the Pd(Ⅱ) catalysts, Pd(OAc)2/PPh3 worked as well as Pd(PPh3)4 to achieve the quantitative conversion (Table 1, Entries 11~15). Lowering the catalyst loading or reaction temperature greatly affected the yield of 4a (Table 1, Entries 16 and 17), although the catalyst loading is relative higher than classic Suzuki-Miyaura cross-coupling with a C—C bond forming process. P(o-tol)3 offered poor result. It was noted that the combination of Pd(OAc)2 or PdCl2(cod) (cod=1, 5-cy-clooctadiene) with PCy3 resulted in the formation of 4a in the yields of 84% and 83% (Table 1, Entries 20 and 21).
 下载:
导出CSV
下载:
导出CSV
 |
|||||||
| Entry | Cat. | L | Base | Solvent | Yieldb/% | ||
| 1 | 3a | 4a | |||||
| 1 | Pd(PPh3)4 | — | Cs2CO3 | THF | 10 | — | 90 |
| 2 | Pd(PPh3)4 | — | Cs2CO3 | Dioxane | 9 | 16 | 75 |
| 3 | Pd(PPh3)4 | — | Cs2CO3 | DCE | 93 | 7 | — |
| 4 | Pd(PPh3)4 | — | Cs2CO3 | Cyclohexane | — | — | 100 |
| 5 | Pd(PPh3)4 | — | K2CO3 | Cyclohexane | 92 | 8 | — |
| 6 | Pd(PPh3)4 | — | AgOAc | Cyclohexane | — | — | — |
| 7 | Pd(PPh3)4 | — | K2HPO3 | Cyclohexane | 100 | — | — |
| 8 | Pd(PPh3)4 | — | KOAc | Cyclohexane | 100 | — | — |
| 9 | Pd2(dba)3 | — | Cs2CO3 | Cyclohexane | 100 | — | — |
| 10 | Pd2(dba)3 | PPh3 | Cs2CO3 | Cyclohexane | 7 | 7 | 86 |
| 11 | Pd(acac)2 | PPh3 | Cs2CO3 | Cyclohexane | 100 | — | — |
| 12 | Pd(MeCN)4(BF4)2 | PPh3 | Cs2CO3 | Cyclohexane | 72 | 20 | 8 |
| 13 | [PdCl(C3H5)]2 | PPh3 | Cs2CO3 | Cyclohexane | 28 | 21 | 51 |
| 14 | PdCl2(cod) | PPh3 | Cs2CO3 | Cyclohexane | 2 | 1 | 97 |
| 15 | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | — | — | 100 |
| 16c | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | 21 | 17 | 62 |
| 17d | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | 20 | 18 | 62 |
| 18 | Pd(OAc)2 | P(C6H4OMe-o)3 | Cs2CO3 | Cyclohexane | 39 | 22 | 39 |
| 19 | Pd(OAc)2 | PCy3 | Cs2CO3 | Cyclohexane | 8 | 8 | 84 |
| 20 | PdCl2(cod) | PCy3 | Cs2CO3 | Cyclohexane | 7 | 10 | 83 |
| a Reaction condition: 1 (0.1 mmol), PhBr (0.25 mmol), 20 mol% cat., 40 mol% L, base (0.3 mmol), 110 ℃, 8 h; DCE=dichloroethane; dba=dibenzylideneacetone, cod=1, 5-cyclooctadiene; Cy=cyclohexyl. b GC yield. c10 mol% Pd(OAc)2, 20 mol% PPh3. d 80 ℃. | |||||||
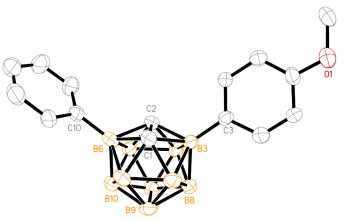
Under the optimal reaction conditions (Table 1, Entry 15), coupling reaction of 1 with aryl bromides was examined (Table 2). The reactions of p- and m-tolyl bromide proceeded as well as phenyl bromide to give, unfortunately, an inseperatable mixture of 4 and 5, resulting from the aryl-phenyl exchange between palladium center and the large amount of phosphine ligand (Table 2, Entries 2 and 3).[15] The exchange product 3-(p-MeOC6H4)-6-Ph-1, 2-C2B10H10 was isolated in 26% yield using p-MeOC6H4Br as the reagent under aforementioned reaction conditions (Table 2, Entry 4). Its structure was confirmed by single-crystal X-ray analyses (Figure 1). On the other hand, no aryl exchange product was formed using electron-withdrawing group substituted p-ClC6H4Br and p-CF3C6H4Br as coupling partners (Table 2, Entries 5 and 6), which agrees well with the trends observed by Novak on the substituent effects of the aryl-aryl interchange reaction.[15h] We next studied the reaction using PCy3 ligand to avoid this aryl exchange. However, another unexpected inseparable mixture of diarylated by-products 6 was obtained along with the desired compound 4 (Table 2, Entries 7~10). This structurally undetermined by-product may be ascribed to palladium- catalyzed direct B—H arylation of electron-rich boron vertexes. [8]
下载:
导出CSV
 |
|||
| Entry | Ar | L | Yieldb/% |
| 1 | C6H5 | PPh3 | 84 (4) |
| 2 | 4-MeC6H4 | PPh3 | 85 (4:5=64:21) |
| 3 | 3-MeC6H4 | PPh3 | 87 (4:5=73:14) |
| 4c | 4-MeOC6H4 | PPh3 | 48 (4), 26 (5) |
| 5 | 4-ClC6H4 | PPh3 | 69 (4) |
| 6 | 4-CF3C6H4 | PPh3 | 60 (4) |
| 7 | 4-MeC6H4 | PCy3 | 61 (4:6=49:12) |
| 8 | 3-MeC6H4 | PCy3 | 51 (4:6=48:3) |
| 9c | 4-MeOC6H4 | PCy3 | 25 (4:6=22:3) |
| 10 | 4-FC6H4 | PCy3 | 65 (4:6=62:3) |
| a Reaction condition: 1 (0.5 mmol), ArBr (1.25 mmol), Cs2CO3 (1.5 mmol), 20 mol% Pd(OAc)2, 40 mol% L, 110 ℃, 24 h. b Isolated yield, ratio determined by GC. c130 ℃. | |||
We next used p-tolyl halide as a model electrophilic coupling partner to further optimize the reaction conditions (Table 3). Alkyl phosphine ligand PnBu3 offered a very low conversion of 1 (Table 3, Entry 1). If PCy3 was employed as the ligand, PdCl2(cod) showed both high reactivity and selectivity for 4b (Table 3, Entries 2~4).
下载:
导出CSV
 |
||||||
| Entry | Cat. | L | Yieldb/% | |||
| 1 | 3b | 4b | 6b | |||
| 1 | Pd(OAc)2 | PnBu3 | 75 | 20 | 5 | — |
| 2 | Pd(OAc)2 | PCy3 | 8 | 9 | 65 | 16 |
| 3 | Pd2(dba)3 | PCy3 | 16 | 19 | 62 | 3 |
| 4 | PdCl2(cod) | PCy3 | 7 | 12 | 78 | 3 |
| 5 | PdCl2(PCy3)2 | — | 51 | 31 | 18 | 0 |
| a Reaction condition: 1 (0.1 mmol), p-tolyl halide (0.25 mmol), 20 mol% cat., 40 mol% L, Cs2CO3 (0.3 mmol), 110 ℃, 24 h. b GC yield. | ||||||
Under the further optimal reaction conditions (Table 3, Entry 4), the substrate scope was examined and the results were compiled in Table 4. Both electron-withdrawing and electron-donating groups on phenyl ring afforded the corresponding products in moderate isolated yields. 2-Na- phthyl bromide also worked well, giving 4x in 57% yield. Steric factors played an important role in these reactions. Only trace product was observed in the reaction with sterically hindered o-methyl/o-fluorophenyl or 1-naphthyl bromide (2d, 2p and 2y), while o-methoxyl and o-triflu- oromethyl groups on phenyl ring (2k and 2u) completely suppressed the reaction. For heteroatom containing substituents, the p- and m-OMe containing products 4i and 4j were isolated in relatively lower yields. And only a trace amount of product was observed from GC-MS for p-bromo-N, N-dimethylaniline 2l, probably owing to the interactions of the heteroatom with the metal center.
Compounds 4 and 5i were fully characterized by 1H NMR, 13C NMR and 11B NMR spectra, HRMS as well as elemental analyses. For 4, the NMR spectra exhibited symmetric structures. Their 11B{1H} NMR spectra showed the same pattern of 2:2:2:4. The substituted cage B was found at about δ -4 as a singlet in the 1H coupled 11B NMR spectra.
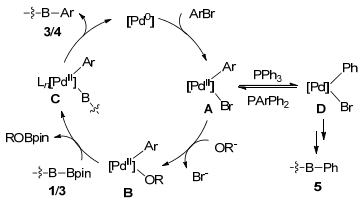
A proposed mechanism for the above 3, 6-diarylation of o-carborane is shown in Scheme 2. The catalysis is likely to be initiated by a Pd0 species that is generated from the reduction of PdⅡ with phosphine ligand.[16] Oxidative addition of Ar—Br on Pd(0), followed by the subsequent reaction with base, produces PdⅡ intermediate B. Transmetalation of B with B-carboranyl boronate 1 forms the carboranylpalladium intermediate C. Reductive elimination yields the arylation product 3 and releases the Pd0 species to complete the catalytic cycle. Compound 3 undergoes another catalytic cycle to afford cage B(3, 6)-diarylated product 4. On the other hand, in the presence of PPh3, aryl-phenyl exchange between the palladium center in A and phosphine ligands produces intermediate D, which undergoes a similar reaction cycle to give the unsymmetrical 3, 6-diarylated- o-carborane 5.
In summary, an efficient Pd-catalyzed coupling of borylated o-carborane with different aryl bromide has been achieved, giving a series of B(3, 6)-diaryl-o-carboranes.
This represents the first general method for the synthesis of B(3, 6)-diaryl-o-carboranes. This work may also shed some light on developing new methodologies for the B(3, 6)-func- tionalization using cage B-Bpin compounds as a new kind of boron nucleophile.
All reactions were carried out in oven-dried glassware under an atmosphere of dry N2 with the rigid exclusion of air and moisture using standard Schlenk techniques or in a glovebox. Organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. Compound 1a was prepared according to literature procedures.[14] All other chemicals were purchased from either Aldrich or J & K Chemical Co. and used as received unless otherwise specified. The melting points were measured on an Electrothermal Melt Temp instrument of Shanghai INESA Physico-Optical Instrument Co., Ltd and were uncorrected. 1H, 13C{1H}, 11B{1H}, 11B and 19F NMR spectra were recorded on a Bruker 400 spectrometer at 400, 101, 128 and 376 MHz, respectively. All signals were reported with references to the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts, to external BF3•OEt2 (0.00) for boron chemical shifts and to external CFCl3 (0.00) for fluorine chemical shifts. Mass spectra were obtained on a Waters Micromass GCT Premier CAB088 spectrometer. Elemental analyses were performed with an elementary VARIO EL Ⅲ elemental analyzer, Shanghai Institute of Organic Chemistry, CAS.
An oven-dried Schlenk flask equipped with a stir bar was charged with 1a (198 mg, 0.5 mmol), aryl bromide (1.5 mmol), PdCl2(cod) (29 mg, 0.1 mmol), PCy3 (56 mg, 0.2 mmol) and Cs2CO3 (490 mg, 1.5 mmol), followed by dry cyclohexane (5 mL). The flask was closed under an atmosphere of nitrogen and stirred at 110 ℃ (bath temperature) for 24 h. After cooled to room temperature, water (10 mL) and 30% H2O2 (5 mL) aqueous solution were successively added and the mixture was stirred at room temperature overnight to oxidize the phosphine ligand. After extraction with diethyl ether (10 mL×3), the ether solutions were combined and concentrated to dryness in vacuo. The residue was subjected to flash column chromatography on silica gel (230~400 mesh) using n-hexane and ethyl acetate (V/V=100/1) as eluent to give 4.
3, 6-(C6H5)2-1, 2-C2B10H10 (4a): White solid, yield 58%. m.p. 175~176 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.62 (m, 4H, ArH), 7.40 (m, 6H, ArH), 3.85 (s, 2H, cage CH). These data are identical with those reported in the literature.[14]
3, 6-(p-CH3C6H4)2-1, 2-C2B10H10 (4b): White solid, yield 67%. m.p. 192~194 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.51 (d, J=8.0 Hz, 4H, ArH), 7.19 (d, J=8.0 Hz, 4H, ArH), 3.81 (s, 2H, cage CH), 2.37 (s, 6H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δ: 140.0, 133.3, 129.2 (aromatic C), 59.3 (cage CH), 21.5 (CH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.3 (d, JBH=160 Hz, 2B, BcageH), -3.6 (s, 2B, ArBcage), -11.4 (d, JBH=164 Hz, 2B), -12.9 (d, JBH=174 Hz, 4B, BcageH). Anal. calcd for C16H24B10: C 59.23, H 7.46; found C 59.14, H 7.38.
3, 6-(m-(CH3)2C6H4)2-1, 2-C2B10H10 (4c): White solid, yield 56%. m.p. 207~209 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.42 (s, 2H, ArH), 7.41 (d, J=6.8 Hz, 2H, ArH), 7.25 (m, 4H, ArH), 3.83 (s, 2H, cage CH), 2.37 (s, 6H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δ: 138.0, 134.0, 130.7, 130.3, 128.4 (aromatic C), 59.2 (cage CH), 21.6 (CH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.3 (d, JBH=149 Hz, 2B, BcageH), -3.7 (s, 2B, ArBcage), -11.3 (d, JBH=182 Hz, 2B), -12.8 (d, JBH=177 Hz, 4B, BcageH). Anal. calcd for C16H24B10: C 59.23, H 7.46; found C 58.94, H 7.46.
3, 6-(3', 5'-(CH3)2C6H4)2-1, 2-C2B10H10 (4e): White solid, yield 65%. m.p. 228~229 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.23 (s, 4H, ArH), 7.06 (s, 2H, ArH), 3.82 (s, 2H, cage CH), 2.34 (s, 12H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δ: 137.9, 131.6, 131.1 (aromatic C), 59.3 (cage CH), 21.4 (CH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.4 (d, JBH=154 Hz, 2B, BcageH), -3.6 (s, 2B, ArBcage), -11.4 (d, JBH=178 Hz, 2B), -12.9 (d, JBH=178 Hz, 4B, BcageH). Anal. calcd for C18H28B10: C 61.33, H 8.01; found C 61.14, H 7.76.
3, 6-(p-C2H5C6H4)2-1, 2-C2B10H10 (4f): White solid, yield 58%. m.p. 95~96 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J=7.2 Hz, 4H, ArH), 7.25 (d, J=7.2 Hz, 4H, ArH), 3.84 (s, 2H, cage CH), 2.70 (q, J=7.2 Hz, 4H, CH2), 1.29 (t, J=7.2 Hz, 6H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δ: 146.3, 133.4, 128.0 (aromatic C), 59.3 (cage CH), 28.9 (CH2), 15.6 (CH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.9 (d, JBH=152 Hz, 2B, BcageH), -4.2 (s, 2B, ArBcage), -11.9 (d, JBH=175 Hz, 2B), -13.4 (d, JBH=177 Hz, 4B, BcageH). HRMS (ESI) calcd for C18H2810B211B8: 352.3194, found 352.3189.
3, 6-(p-(CH3)2CHC6H4)2-1, 2-C2B10H10 (4g): White solid, yield 66%. m.p. 135~137 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.56 (d, J=7.6 Hz, 4H, ArH), 7.25 (d, J=7.6 Hz, 4H, ArH), 3.83 (s, 2H, cage CH), 2.93 (m, 2H, CH(CH3)2), 1.27 (d, J=6.8 Hz, 12H, CH(CH3)2); 13C{1H} NMR (101 MHz, CDCl3) δ: 150.9, 133.4, 126.6 (aromatic C), 59.3 (cage CH), 34.2 (CH(CH3)2), 24.0 (CH(CH3)2), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.7 (d, JBH=148 Hz, 2B, BcageH), -3.9 (s, 2B, ArBcage), -11.8 (d, JBH=178 Hz, 2B), -13.3 (d, JBH=177 Hz, 4B, BcageH). Anal. calcd for C20H32B10: C 63.12, H 8.48; found C 63.07, H 8.52.
3, 6-(p-(CH3)3C-C6H4)2-1, 2-C2B10H10 (4h): White solid, yield 68%. m.p. 245~247 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.55 (d, J=8.0 Hz, 4H, ArH), 7.39 (d, J=8.0 Hz, 4H, ArH), 3.83 (s, 2H, cage CH), 1.32 (s, 18H, CH3); 13C{1H} NMR (101 MHz, CDCl3) δ: 153.1, 133.2, 125.4 (aromatic C), 59.3 (cage CH), 34.8 (C(CH3)3), 31.3 (C(CH3)3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.2 (d, JBH=152 Hz, 2B, BcageH), -3.8 (s, 2B, ArBcage), -11.2 (d, JBH=188 Hz, 2B), -12.8 (d, JBH=183 Hz, 4B, BcageH). Anal. calcd for C22H36B10: C 64.67, H 8.88; found C 64.68, H 8.79.
3, 6-(p-OCH3C6H4)2-1, 2-C2B10H10 (4i): White solid, yield 29%. m.p. 170~172 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.54 (d, J=8.0 Hz, 4H, ArH), 6.90 (d, J=8.0 Hz, 4H, ArH), 3.82 (s, 6H, OCH3), 3.79 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 161.1, 134.7, 114.0 (aromatic C), 59.4 (cage CH), 55.3 (OCH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.4 (d, JBH=142 Hz, 2B, BcageH), -3.0 (s, 2B, ArBcage), -11.5 (d, JBH=169 Hz, 2B), -13.0 (d, JBH=172 Hz, 4B, BcageH). Anal. calcd for C16H24B10O2: C 53.91, H 6.79; found C 54.02, H 6.86.
3, 6-(m-OCH3C6H4)2-1, 2-C2B10H10 (4j): White solid, yield 43%. m.p. 168~170 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.25 (m, 2H, ArH), 7.13 (m, 4H, ArH), 6.92 (m, 2H, ArH), 3.80 (s, 8H, OCH3 & cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 159.4, 129.7, 125.3, 119.3, 114.9 (aromatic C), 59.2 (cage CH), 55.4 (OCH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.4 (d, JBH=150 Hz, 2B, BcageH), -4.1 (s, 2B, ArBcage), -11.4 (d, JBH=165 Hz, 2B), -13.0 (d, JBH=170 Hz, 4B, BcageH). Anal. calcd for C16H24B10O2: C 53.91, H 6.79; found C 53.49, H 6.67.
3, 6-(p-ClC6H4)2-1, 2-C2B10H10 (4m): White solid, yield 61%. m.p. 187~188 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.54 (d, J=8.4 Hz, , 4H, ArH), 7.35 (d, J=8.4 Hz, 4H, ArH), 3.80 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 136.6, 134.6, 128.7 (aromatic C), 59.0 (cage CH), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -1.9 (d, JBH=148 Hz, 2B, BcageH), -4.0 (s, 2B, ArBcage), -11.0 (d, JBH=204 Hz, 2B), -12.8 (d, JBH=182 Hz, 4B, BcageH). Anal. calcd for C14H18B10Cl2: C 46.03, H 4.97; found C 46.21, H 5.03.
3, 6-(p-FC6H4)2-1, 2-C2B10H10 (4n): White solid, yield 69%. m.p. 164~166 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.60 (dd, J=8.0, 6.0 Hz, 4H, ArH), 7.07 (t, J=8.0 Hz, 4H, ArH), 3.81 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 164.3 (d, 1JCF=249 Hz), 135.2 (d, 3JCF=8 Hz), 115.6 (d, 2JCF=20 Hz, aromatic C), 59.3 (cage CH), the Bcage-C were not observed; 19F{1H} NMR (376 MHz, CDCl3) δ: -110.7; 11B NMR (128 MHz, CDCl3) δ: -2.1 (d, JBH=148 Hz, 2B, BcageH), -3.8 (s, 2B, ArBcage), -11.1 (d, JBH=189 Hz, 2B), -12.8 (d, JBH=178 Hz, 4B, BcageH). Anal. calcd for C14H18B10F2: C 50.59, H 5.46; found C 50.77, H 5.68.
3, 6-(m-FC6H4)2-1, 2-C2B10H10 (4o): White solid, yield 41%. m.p. 120~121 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.34 (m, 6H, ArH), 7.12 (m, 2H, ArH), 3.83 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 162.7 (d, 1JCF=247 Hz) (CF), 130.3 (d, 3JCF=7 Hz), 128.8 (d, 4JCF=3 Hz), 120.1 (d, 2JCF=20 Hz), 117.0 (d, 2JCF=21 Hz, aromatic C), 59.1 (cage CH), the Bcage-C were not observed; 19F{1H} NMR (376 MHz, CDCl3) δ: -112.7; 11B NMR (128 MHz, CDCl3) δ: -1.8 (d, JBH=150 Hz, 2B, BcageH), -4.2 (s, 2B, ArBcage), -10.8 (d, JBH=152 Hz, 2B), -12.6 (d, JBH=163 Hz, 4B, BcageH). Anal. calcd for C14H18B10F2: C 50.59, H 5.46; found C 50.57, H 5.49.
3, 6-(3', 5'-C6H3F2)2-1, 2-C2B10H10 (4q): White solid, yield 74%. m.p. 133~135 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.11 (m, 4H, ArH), 6.87 (tt, J=8.8, 2.4 Hz, 2H, ArH) 3.81 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 163.1 (dd, JCF=253, 12 Hz), 116.0 (dd, JCF=18, 6 Hz), 105.7 (t, JCF=25 Hz, aromatic C), 58.9 (cage CH), the Bcage-C were not observed; 19F{1H} NMR (376 MHz, CDCl3) δ: -108.9; 11B NMR (128 MHz, CDCl3) δ: -1.5 (d, JBH=150 Hz, 2B, BcageH), -4.7 (s, 2B, ArBcage), -10.6 (d, JBH=150 Hz, 2B), -12.6 (d, JBH=165 Hz, 4B) (BcageH). Anal. calcd for C14H16B10F4: C 45.65, H 4.38; found C 45.73, H 4.44.
3, 6-(p-CF3C6H4)2-1, 2-C2B10H10 (4s): White solid, yield 42%. m.p. 141~142 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.75 (d, J=8.0 Hz, 4H, ArH), 7.63 (d, J=8.0 Hz, 4H, ArH), 3.88 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 133.7, 132.1 (q, 2JCF=32 Hz), 125.2 (q, 3JCF=4 Hz, aromatic C), 124.0 (q, 1JCF=274 Hz, CF3), 58.9 (cage CH), the Bcage-C were not observed; 19F{1H} NMR (376 MHz, CDCl3) δ: -63.1; 11B NMR (128 MHz, CDCl3) δ: -1.6 (d, JBH=151 Hz, 2B, BcageH), -4.3 (s, 2B, ArBcage), -10.6 (d, JBH=145 Hz, 2B), -12.6 (d, JBH=169 Hz, 4B, BcageH). Anal. calcd for C16H18B10F6: C 44.44, H 4.20; found C 44.30, H 4.31.
3, 6-(m-CF3C6H4)2-1, 2-C2B10H10 (4t): White solid, yield: 61%. m.p. 125~126 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.84 (sm, 4H, ArH), 7.70 (d, J=8.0 Hz, 2H, ArH), 7.52 (t, J=8.0 Hz, 2H, ArH), 3.89 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 136.8, 130.9 (q, 2JCF=32 Hz), 129.7 (q, 3JCF=4 Hz), 129.0, 126.9 (q, 3JCF=4 Hz, aromatic C), 124.1 (q, 1JCF=274 Hz) (CF3), 59.0 (cage CH), the Bcage-C were not observed; 19F{1H} NMR (376 MHz, CDCl3) δ: -62.6; 11B NMR (128 MHz, CDCl3) δ: -1.5 (d, JBH=150 Hz, 2B, BcageH), -4.1 (s, 2B, ArBcage), -10.5 (d, JBH=151 Hz, 2B), -12.6 (d, JBH=164 Hz, 4B, BcageH). Anal. calcd for C16H18B10F6: C 44.44, H 4.20; found C 44.41, H 4.24.
3, 6-(p-C6H5C6H4)2-1, 2-C2B10H10 (4v): White solid, yield 65%. m.p. 216~217 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.72 (m, 4H, ArH), 7.61 (m, 8H, ArH), 7.47 (m, 4H, ArH), 7.39 (m, 2H, ArH), 3.93 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 142.9, 140.6, 133.8, 129.0, 127.9, 127.3, 127.1 (aromatic C), 59.3 (cage CH), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.1 (d, JBH=157 Hz, 2B, BcageH), -3.6 (s, 2B, ArBcage), -11.1 (d, JBH=214 Hz, 2B), -12.7 (d, JBH=184 Hz, 4B, BcageH). Anal. calcd for C26H28B10: C 69.61, H 6.29; found C 69.27, H 6.24.
3, 6-(m-C6H5-C6H4)2-1, 2-C2B10H10 (4w): White solid, yield 47%. m.p. 235~237 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.86 (s, 2H, ArH), 7.64 (m, 8H, ArH), 7.48 (m, 6H, ArH), 7.40 (m, 2H, ArH), 3.96 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 141.4, 140.8, 132.2, 132.1, 129.0, 128.9, 128.8, 127.7, 127.4 (aromatic C), 59.2 (cage CH), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -0.8 (d, JBH=157 Hz, 2B, BcageH), -2.6 (s, 2B, ArBcage), -9.8 (d, JBH=214 Hz, 2B), -11.5 (d, JBH=184 Hz, 4B, BcageH). HRMS (DART-nagative mode) calcd for C26H2710B211B8: 447.3121, found 447.3134.
3, 6-(2'-C10H7)2-1, 2-C2B10H10 (4x): White solid, yield 57%. m.p. 154~156 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.15 (s, 2H, ArH), 7.86 (m, 6H, ArH), 7.71 (d, J=8.4 Hz, 2H), 7.54 (m, 4H, ArH), 4.03 (s, 2H, cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 133.9, 132.9, 129.5, 128.2, 127.9, 127.8, 127.1, 126.7 (aromatic C), 59.4 (cage CH), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.2 (d, JBH=195 Hz, 2B, BcageH), -3.6 (s, 2B, ArBcage), -11.2 (d, JBH=177 Hz, 2B), -12.7 (d, JBH=180 Hz, 4B, BcageH). Anal. calcd for C22H24B10: C 66.64, H 6.10; found C 66.30, H 6.04.
An oven-dried Schlenk flask equipped with a stir bar was charged with 1 (198 mg, 0.5 mmol), 4-bromoanisole (281 mg, 1.5 mmol), Pd(OAc)2 (22 mg, 0.1 mmol), PPh3 (52 mg, 0.2 mmol) and Cs2CO3 (490 mg, 1.5 mmol), followed by dry cyclohexane (5 mL). The flask was closed under an atmosphere of nitrogen and stirred at 130 ℃ (bath temperature) for 12 h. After cooled to room temperature, water (10 mL) and 30% H2O2 (5 mL) aqueous solution were successively added and the mixture was stirred at room temperature overnight. After extraction with diethyl ether (10 mL× 3), the ether solutions were combined and concentrated to dryness in vacuo. The residue was subjected to flash column chromatography on silica gel (230~400 mesh) using n-hexane and ethyl acetate (V/V=100/1) as eluent to give 4i (86 mg, 48%) and 5i (43 mg, 26%). 5i: White solid, m.p. 136~137 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.62 (d, J=7.2 Hz, 2H), 7.55 (d, J=8.4 Hz, 2H), 7.40 (m, 3H), 6.91 (d, J=8.4 Hz, 2H) (aromatic CH), 3.83 (m, 5H) (OCH3 & cage CH); 13C{1H} NMR (101 MHz, CDCl3) δ: 161.2, 134.7, 133.3, 129.9, 128.4, 114.1 (aromatic C), 59.3 (cage CH), 55.4 (s, OCH3), the Bcage-C were not observed; 11B NMR (128 MHz, CDCl3) δ: -2.3 (d, JBH=152 Hz, 2B, BcageH), -3.5 (s, 2B, ArBcage), -11.3 (d, JBH=188 Hz, 2B), -12.9 (d, JBH=174 Hz, 4B, BcageH). Anal. calcd for C15H22B10O: C 55.19, H 6.79; found C 54.91, H 6.90.
Data were collected at 293 K on a Bruker SMART 1000 CCD diffractometer using Mo-Kα radiation. An empirical absorption correction was applied using the SADABS program.[17] The structure was solved by direct methods and subsequent Fourier difference techniques and refined anisotropically for all non-hydrogen atoms by full-matrix least squares calculations on F2 using the SHELXTL program package.[18] All hydrogen atoms were geometrically fixed using the riding model.
Supporting Information NMR spectra of B(3, 6)-dia-rylated-o-carboranes 4 and 5i. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/. CCDC 1827267 for 5i contain the supplementary crystallographic data. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html(or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk).
(a) Hawthorne, M. F. Angew. Chem., Int. Ed. 1993, 32, 950.
(b) Armstrong, A. F.; Valliant, J. F. Dalton Trans. 2007, 4240.
(c) Issa, F.; Kassiou, M.; Rendina, L. M. Chem. Rev. 2011, 111, 5701.
(d) Scholz, M.; Hey-Hawkins, E. Chem. Rev. 2011, 111, 7035.
(e) Leśnikowski, Z. J. J. Med. Chem. 2016, 59, 7738.
(a) Xie, Z. Coord. Chem. Rev. 2002, 231, 23.
(b) Hosmane, N. S.; Maguire, J. A. In Comprehensive Organometallic Chemistry Ⅲ, Eds.: Mingos, D. M. P.; Crabtree, R. H., Elsevier, Oxford, 2007, Vol. 3.
(c) Lee, J.-D.; Co, T. T.; Kim, T.-J.; Kang, S. O. Synlett 2009, 771.
(d) Himmelspach, A.; Finze, M.; Raub, S. Angew. Chem., Int. Ed. 2011, 50, 2628.
(e) Spokoyny, A. M.; Machan, C. W.; Clingerman, D. J.; Rosen, M. S.; Wiester, M. J.; Kennedy, R. D.; Stern, C. L.; Sarjeant, A. A.; Mirkin, C. A. Nat. Chem. 2011, 3, 590.
(f) El-Hellani, A.; Lavallo, V. Angew. Chem., Int. Ed. 2014, 53, 4489.
(g) Joost, M.; Zeineddine, A.; Estévez, L.; Mallet-Ladeira, S.; Miqueu, K.; Amgoune, A.; Bourissou, D. J. Am. Chem. Soc. 2014, 136, 14654.
(h) Adams, R. D.; Kiprotich, J.; Peryshkov, D. V.; Wong, Y. O. Chem. Eur. J. 2016, 22, 6501.
(i) Axtell, J. C.; Kirlikovali, K. O.; Djurovich, P. I.; Jung, D.; Nguyen, V. T.; Munekiyo, B.; Royappa, A. T.; Rheingold, A. L.; Spokoyny, A. M. J. Am. Chem. Soc. 2016, 138, 15758.
(j) Hailmann, M.; Wolf, N.; Renner, R.; Schä fer, T. C.; Hupp, B.; Steffen, A.; Finze, M. Angew. Chem., Int. Ed. 2016, 55, 10507.
(k) Zhou, Y.-P.; Raoufmoghaddam, S.; Szilvási, T.; Driess, M. Angew. Chem., Int. Ed. 2016, 55, 12868.
(a) Tsuboya, N.; Lamrani, M.; Hamasaki, R.; Ito, M.; Mitsuishi, M.; Miyashita, T.; Yamamoto, Y. J. Mater. Chem. 2002, 12, 2701.
(b) Guo, J.; Liu, D.; Zhang, J.; Zhang, J.; Miao, Q.; Xie, Z. Chem. Commun. 2015, 51, 12004.
(c) Li, X.; Yan, H.; Zhao, Q. Chem. Eur. J. 2016, 22, 1888.
(d) Mukherjee, S.; Thilagar, P. Chem. Commun. 2016, 52, 1070.
(e) Núñez, R.; Tarrés, M.; Ferrer-Ugalde, A.; de Biani, F. F.; Teixidor, F. Chem. Rev. 2016, 116, 14307.
(a) Yang, X.; Jiang, W.; Knobler, C. B.; Hawthorne, M. F. J. Am. Chem. Soc. 1992, 114, 9719.
(b) Jude, H.; Disteldorf, H.; Fischer, S.; Wedge, T.; Hawkridge, A. M.; Arif, A. M.; Hawthorne, M. F.; Muddiman, D. C.; Stang, P. J. J. Am. Chem. Soc. 2005, 127, 12131.
(c) Koshino, M.; Tanaka, T.; Solin, N.; Suenaga, K.; Isobe, H.; Nakamura, E. Science 2007, 316, 853.
(d) Sasaki, T.; Guerrero, J. M.; Leonard, A. D.; Tour, J. M. Nano Res. 2008, 1, 412.
(e) Dash, B. P.; Satapathy, R.; Gaillard, E. R.; Maguire, J. A.; Hosmane, N. S. J. Am. Chem. Soc. 2010, 132, 6578.
(f) Bauduin, P.; Prevost, S.; Farràs, P.; Teixidor, F.; Diat, O.; Zemb, T. Angew. Chem., Int. Ed. 2011, 50, 5298.
(g) Cioran, A. M.; Musteti, A. D.; Teixidor, F.; Krpetić, Ž.; Prior, I. A.; He, Q.; Kiely, C. J.; Brust, M.; Viñ as, C. J. Am. Chem. Soc. 2012, 134, 212.
(h) Grimes, R. N. Dalton Trans. 2015, 44, 5939.
(i) Schwartz, J. J.; Mendoza, A. M.; Wattanatorn, N.; Zhao, Y.; Nguyen, V. T.; Spokoyny, A. M.; Mirkin, C. A.; Baše, T.; Weiss, P. S. J. Am. Chem. Soc. 2016, 138, 5957.
(j) Qian, E. A.; Wixtrom, A. I.; Axtell, J. C.; Saebi, A.; Jung, D.; Rehak, P.; Han, Y.; Moully, E. H.; Mosallaei, D.; Chow, S.; Messina, M. S.; Wang, J. Y.; Royappa, A. T.; Rheingold, A. L.; Maynard, H. D.; Král, P.; Spokoyny, A. M. Nat. Chem. 2017, 9, 333.
(a) Kokado, K.; Chujo, Y. J. Org. Chem. 2011, 76, 316.
(b) Ferrer-Ugalde, A.; Juárez-Pérez, E. J.; Teixidor, F.; Viñ as, C.; Sillanpä ä, R.; Pérez-Inestrosa, E.; Núñ ez, R. Chem. Eur. J. 2012, 18, 544.
(c) Wee, K.-R.; Cho, Y.-J.; Jeong, S.; Kwon, S.; Lee, J.-D.; Suh, I.-H.; Kang, S. O. J. Am. Chem. Soc. 2012, 134, 17982.
(d) Wee, K.-R.; Han, W.-S.; Cho, D. W.; Kwon, S.; Pac, C.; Kang, S. O. Angew. Chem., Int. Ed. 2012, 51, 2677.
(e) Shi, C.; Sun, H.; Jiang, Q.; Zhao, Q.; Wang, J.; Huang, W.; Yan, H. Chem. Commun. 2013, 49, 4746.
(f) Shi, C.; Sun, H.; Tang, X.; Lv, W.; Yan, H.; Zhao, Q.; Wang, J.; Huang, W. Angew. Chem., Int. Ed. 2013, 52, 13434.
(g) Wee, K.-R.; Cho, Y.-J.; Song, J. K.; Kang, S. O. Angew. Chem., Int. Ed. 2013, 52, 9682.
(h) Bae, H. J.; Chung, J.; Kim, H.; Park, J.; Lee, K. M.; Koh, T.-W.; Lee, Y. S.; Yoo, S.; Do, Y.; Lee, M. H. Inorg. Chem. 2014, 53, 128.
(i) Naito, H.; Morisaki, Y.; Chujo, Y. Angew. Chem., Int. Ed. 2015, 54, 5084.
(j) Furue, R.; Nishimoto, T.; Park, I. S.; Lee, J.; Yasuda, T. Angew. Chem., Int. Ed. 2016, 55, 7171.
(k) Kirlikovali, K. O.; Axtell, J. C.; Gonzalez, A.; Phung, A. C.; Khan, S. I.; Spokoyny, A. M. Chem. Sci. 2016, 7, 5132.
(l) Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Angew. Chem., Int. Ed. 2017, 56, 254.
(m) Tu, D.; Leong, P.; Guo, S.; Yan, H.; Lu, C.; Zhao, Q. Angew. Chem., Int. Ed. 2017, 56, 11370.
(a) Tang, C.; Xie, Z. Angew. Chem., Int. Ed. 2015, 54, 7662.
(b) Lu, J.-Y.; Wan, H.; Zhang, J.; Wang, Z.; Li, Y.; Du, Y.; Li, C.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. Chem.-Eur. J. 2016, 22, 17542.
(a) Heying, T. L.; Ager, J. W.; Clark, S. L.; Mangold, D. J.; Goldstein, H. L.; Hillman, M.; Polak, R. J.; Szymanski, J. W. Inorg. Chem. 1963, 2, 1089.
(b) Kusari, U.; Li, Y.; Bradley, M. G.; Sneddon, L. G. J. Am. Chem. Soc. 2004, 126, 8662.
(c) Li, Y.; Carroll, P. J.; Sneddon, L. G. Inorg. Chem. 2008, 47, 9193.
Cao, K.; Huang, Y.; Yang, J.; Wu, J. Chem. Commun. 2015, 51, 7257. doi: 10.1039/C5CC01331C
(a) Jiang, W.; Knobler, C. B.; Curtis, C. E.; Mortimer, M. D.; Hawthorne, M. F. Inorg. Chem. 1995, 34, 3491.
(b) Tominaga, M.; Morisaki, Y.; Chujo, Y. Macromol. Rapid Commun. 2013, 34, 1357.
(a) Quan, Y.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 1295.
(b) Zhang, X.; Zheng, H.; Li, J.; Xu, F.; Zhao, J.; Yan, H. J. Am. Chem. Soc. 2017, 139, 14511.
(c) Zhang, X.; Yan, H. Chem. Sci. 2018, 9, 3964.
(a) Qiu, Z.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2013, 135, 12192.
(b) Quan, Y.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 7599.
(c) Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 15513.
(d) Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem., Int. Ed. 2015, 54, 10623.
(e) Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2015, 137, 3502.
(f) Lyu, H.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2016, 138, 12727.
(g) Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 11840.
(h) Quan, Y.; Tang, C.; Xie, Z. Chem. Sci. 2016, 7, 5838.
(i) Zhang, Y.; Sun, Y.; Lin, F.; Liu, J.; Duttwyler, S. Angew. Chem., Int. Ed. 2016, 55, 15609.
(j) Zhao, D.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 3166.
(k) Quan, Y.; Lyu, H.; Xie, Z. Chem. Commun. 2017, 53, 4818.
(l) Shen, Y.; Pan, Y.; Zhang, K.; Liang, X.; Liu, J.; Spingler, B.; Duttwyler, S. Dalton Trans. 2017, 46, 3135.
(m) Tang, C.; Zhang, J.; Xie, Z. Angew. Chem., Int. Ed. 2017, 56, 8642.
(n) Zhang, X.; Zheng, H.; Li, J.; Xu, F.; Zhao, J.; Yan, H. J. Am. Chem. Soc. 2017, 139, 14511.
(a) Teixidor, F.; Barberà, G.; Vaca, A.; Kivekäs, R.; Sillanpää, R.; Oliva, J.; Viñas, C. J. Am. Chem. Soc. 2005, 127, 10158.
(b) Yamazaki, H.; Ohta, K.; Endo, Y. Tetrahedron Lett. 2005, 46, 3119.
(a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(b) Prokopcová, H.; Kappe, C. O. Angew. Chem., Int. Ed. 2009, 48, 2276.
(c) Guo, L.; Chatupheeraphat, A.; Rueping, M. Angew. Chem., Int. Ed. 2016, 55, 11810.
(d) Ben Halima, T.; Zhang, W.; Yalaoui, I.; Hong, X.; Yang, Y.-F.; Houk, K. N.; Newman, S. G. J. Am. Chem. Soc. 2017, 139, 1311.
(e) Yadav, M. R.; Nagaoka, M.; Kashihara, M.; Zhong, R.-L.; Miyazaki, T.; Sakaki, S.; Nakao, Y. J. Am. Chem. Soc. 2017, 139, 9423.
(f) Huo, P.; Li, J.; Liu, W.; Mei, G. Chin. J. Chem. 2017, 35, 363.
(g) Ortega-Jiménez, F.; Penieres-Carrillo, J. G.; López-Cortés, J. G.; Ortega-Alfaro, M. C.; Lagunas-Rivera, S. Chin. J. Chem. 2017, 35, 1881.
Cheng, R.; Qiu, Z.; Xie, Z. Nat. Commun. 2017, 8, 14827. doi: 10.1038/ncomms14827
(a) Fahey, D. R.; Mahan, J. E. J. Am. Chem. Soc. 1976, 98, 4499.
(b) Garrou, P. E. Chem. Rev. 1985, 85, 171.
(c) Kong, K. C.; Cheng, C. H. J. Am. Chem. Soc. 1991, 113, 6313.
(d) O'Keefe, D. F.; Dannock, M. C.; Marcuccio, S. M. Tetrahedron Lett. 1992, 33, 6679.
(e) Baranano, D.; Hartwig, J. F. J. Am. Chem. Soc. 1995, 117, 2937.
(f) Morita, D. K.; Stille, J. K.; Norton, J. R. J. Am. Chem. Soc. 1995, 117, 8576.
(g) Segelstein, B. E.; Butler, T. W.; Chenard, B. L. J. Org. Chem. 1995, 60, 12.
(h) Goodson, F. E.; Wallow, T. I.; Novak, B. M. J. Am. Chem. Soc. 1997, 119, 12441.
(i) Grushin, V. V. Organometallics 2000, 19, 1888.
(j) Kwong, F. Y.; Chan, K. S. Organometallics 2000, 19, 2058.
(k) Kwong, F. Y.; Chan, K. S. Chem. Commun. 2000, 1069.
(l) Kwong, F. Y.; Lai, C. W.; Chan, K. S. J. Am. Chem. Soc. 2001, 123, 8864.
(m) Inoue, A.; Shinokubo, H.; Oshima, K. J. Am. Chem. Soc. 2003, 125, 1484.
(n) Baba, K.; Tobisu, M.; Chatani, N. Org. Lett. 2015, 17, 70.
(a) Amatore, C.; Azzabi, M.; Jutand, A. J. Am. Chem. Soc. 1991, 113, 8375.
(b) Csákai, Z.; Skoda-Földes, R.; Kollár, L. Inorg. Chim. Acta 1999, 286, 93.
(c) Wei, C. S.; Davies, G. H. M.; Soltani, O.; Albrecht, J.; Gao, Q.; Pathirana, C.; Hsiao, Y.; Tummala, S., T.; Eastgate, M. D. Angew. Chem., Int. Ed. 2013, 52, 5822.
Sheldrick, G. M. SADABS:Program for Empirical Absorption Correction of Area Detector Data, University of G ttingen, Germany, 1996.
Sheldrick, G. M. SHELXTL 5.10 for Windows NT:Structure Determination Software Programs, Bruker Analytical X-ray systems, Inc., Madison, Wisconsin, USA, 1997.
Table 1. Optimization of reaction conditions with phenylbromidea
|
|||||||
| Entry | Cat. | L | Base | Solvent | Yieldb/% | ||
| 1 | 3a | 4a | |||||
| 1 | Pd(PPh3)4 | — | Cs2CO3 | THF | 10 | — | 90 |
| 2 | Pd(PPh3)4 | — | Cs2CO3 | Dioxane | 9 | 16 | 75 |
| 3 | Pd(PPh3)4 | — | Cs2CO3 | DCE | 93 | 7 | — |
| 4 | Pd(PPh3)4 | — | Cs2CO3 | Cyclohexane | — | — | 100 |
| 5 | Pd(PPh3)4 | — | K2CO3 | Cyclohexane | 92 | 8 | — |
| 6 | Pd(PPh3)4 | — | AgOAc | Cyclohexane | — | — | — |
| 7 | Pd(PPh3)4 | — | K2HPO3 | Cyclohexane | 100 | — | — |
| 8 | Pd(PPh3)4 | — | KOAc | Cyclohexane | 100 | — | — |
| 9 | Pd2(dba)3 | — | Cs2CO3 | Cyclohexane | 100 | — | — |
| 10 | Pd2(dba)3 | PPh3 | Cs2CO3 | Cyclohexane | 7 | 7 | 86 |
| 11 | Pd(acac)2 | PPh3 | Cs2CO3 | Cyclohexane | 100 | — | — |
| 12 | Pd(MeCN)4(BF4)2 | PPh3 | Cs2CO3 | Cyclohexane | 72 | 20 | 8 |
| 13 | [PdCl(C3H5)]2 | PPh3 | Cs2CO3 | Cyclohexane | 28 | 21 | 51 |
| 14 | PdCl2(cod) | PPh3 | Cs2CO3 | Cyclohexane | 2 | 1 | 97 |
| 15 | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | — | — | 100 |
| 16c | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | 21 | 17 | 62 |
| 17d | Pd(OAc)2 | PPh3 | Cs2CO3 | Cyclohexane | 20 | 18 | 62 |
| 18 | Pd(OAc)2 | P(C6H4OMe-o)3 | Cs2CO3 | Cyclohexane | 39 | 22 | 39 |
| 19 | Pd(OAc)2 | PCy3 | Cs2CO3 | Cyclohexane | 8 | 8 | 84 |
| 20 | PdCl2(cod) | PCy3 | Cs2CO3 | Cyclohexane | 7 | 10 | 83 |
| a Reaction condition: 1 (0.1 mmol), PhBr (0.25 mmol), 20 mol% cat., 40 mol% L, base (0.3 mmol), 110 ℃, 8 h; DCE=dichloroethane; dba=dibenzylideneacetone, cod=1, 5-cyclooctadiene; Cy=cyclohexyl. b GC yield. c10 mol% Pd(OAc)2, 20 mol% PPh3. d 80 ℃. | |||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Initial study of the reaction of 1 with arylbromide under different conditionsa
|
|||
| Entry | Ar | L | Yieldb/% |
| 1 | C6H5 | PPh3 | 84 (4) |
| 2 | 4-MeC6H4 | PPh3 | 85 (4:5=64:21) |
| 3 | 3-MeC6H4 | PPh3 | 87 (4:5=73:14) |
| 4c | 4-MeOC6H4 | PPh3 | 48 (4), 26 (5) |
| 5 | 4-ClC6H4 | PPh3 | 69 (4) |
| 6 | 4-CF3C6H4 | PPh3 | 60 (4) |
| 7 | 4-MeC6H4 | PCy3 | 61 (4:6=49:12) |
| 8 | 3-MeC6H4 | PCy3 | 51 (4:6=48:3) |
| 9c | 4-MeOC6H4 | PCy3 | 25 (4:6=22:3) |
| 10 | 4-FC6H4 | PCy3 | 65 (4:6=62:3) |
| a Reaction condition: 1 (0.5 mmol), ArBr (1.25 mmol), Cs2CO3 (1.5 mmol), 20 mol% Pd(OAc)2, 40 mol% L, 110 ℃, 24 h. b Isolated yield, ratio determined by GC. c130 ℃. | |||
下载: 导出CSV
Table 3. Optimization of reaction conditions with p-tolyl halidesa
|
||||||
| Entry | Cat. | L | Yieldb/% | |||
| 1 | 3b | 4b | 6b | |||
| 1 | Pd(OAc)2 | PnBu3 | 75 | 20 | 5 | — |
| 2 | Pd(OAc)2 | PCy3 | 8 | 9 | 65 | 16 |
| 3 | Pd2(dba)3 | PCy3 | 16 | 19 | 62 | 3 |
| 4 | PdCl2(cod) | PCy3 | 7 | 12 | 78 | 3 |
| 5 | PdCl2(PCy3)2 | — | 51 | 31 | 18 | 0 |
| a Reaction condition: 1 (0.1 mmol), p-tolyl halide (0.25 mmol), 20 mol% cat., 40 mol% L, Cs2CO3 (0.3 mmol), 110 ℃, 24 h. b GC yield. | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们