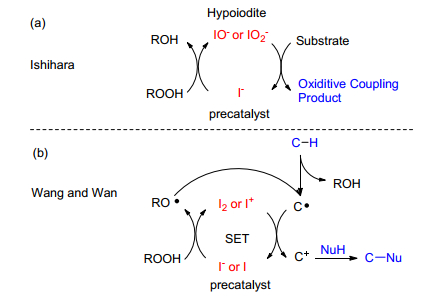
图式 1.
低价碘催化氧化偶联的催化机理
Scheme 1.
Catalytic mechanism for oxidative coupling by the low-valence iodine

碘元素化学性质较活泼, 非金属性强.在传统的有机合成中, 低价碘(碘化物和单质碘)经常被用于碘代反应以及催化环化反应中.近些年来, 过渡金属催化的交叉脱氢偶联(即氧化偶联)已经成为了一种构建C—C、C—O、C—N、C—S、C—P键的有效合成方法[1].反应中两个不同的亲核试剂在过渡金属和氧化剂的作用下, 通过氧化脱氢的方式直接偶联构建新的化学键.反应失去两个氢原子, 原子经济性高; 副产物为水, 绿色无毒.研究反应机理发现, 反应一般都经历了低价金属到高价金属的催化循环.碘化学价态丰富, 可以从-1价一直变化到+7价, 具有很好的氧化还原能力.因此就有可能代替过渡金属实现非金属催化的氧化偶联反应, 从而解决过渡金属催化剂的高成本和毒性问题.
受此启发, Ishihara[2]、汪志勇[3]以及万小兵[4]小组分别发现了低价碘催化的氧化偶联反应, 构建了分子内或者分子间的C—O键和C—N键.从那以后的短短几年时间, 低价碘催化的氧化偶联反应发展快速.通过这种方法, 能有效地构建一系列分子间或者分子内的碳杂键(C—X, X=C、O、N、S、P)及其他化学键.反应无需过渡金属、条件温和且副产物一般为水和醇, 符合绿色化学可持续发展的要求.
综合目前已经发表的低价碘催化氧化偶联反应, 主要有两种占主导地位的反应机理. 2010年, 日本的Ishihara小组[2]首次报道了负一价的碘化物催化的分子内氧化C—O键偶联反应, 并在随后又发展了分子间的氧化C—O键偶联反应.作者提出的反应机理是负一价的碘化物(I-)在氧化剂(如过氧叔丁醇或者过氧化氢)的作用下原位生成一价的次碘酸盐或亚碘酸盐(IO-或者IO2-), 然后这种活性催化剂再去氧化底物, 最终得到偶联产物(Scheme 1a).
随后汪志勇小组[3]和万小兵小组[4]都提出了一种单电子转移(SET)的自由基机理.首先, 负一价的碘化物或者零价的单质碘在过氧叔丁醇的作用下, 原位生成零价碘或者正一价碘, 同时氧化剂自身产生叔丁基氧自由基和羟基负离子(也可以生成过氧叔丁基自由基); 叔丁基氧自由基可以夺取活泼碳氢键的氢自由基, 生成碳自由基, 自身生成叔丁醇副产物; 而产生的碳自由基则在零价碘或者一价碘的作用经过一个单电子转移过程, 生成碳正离子.然后, 各种不同的亲核试剂就可以进攻碳正离子, 最后生成偶联产物(Scheme 1b).
这两种机理已被大家广泛接受, 然而目前所有的氧化偶联反应并不都能用以上两种机理解释.比如最近报道的碘/氧气或碘/二甲基亚砜(DMSO)等催化体系中, 反应一般经历零价碘到负一价碘的催化循环, 且非自由基历程, 下文详细介绍.
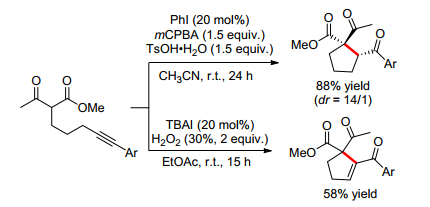
2011年, Moran小组[5]首次报道了低价碘催化的δ-炔基取代的β-酮酯的五元环化反应(Scheme 2).开始当他们以20 mol%的碘苯作为催化剂、1.5 equiv.的间氯过苯甲酸(mCPBA)作为氧化剂、1.5 equiv.的对甲苯磺酸一水化合物(TsOH·H2O)作为添加剂及乙腈作溶剂时, 室温条件下24 h就可以以88%的优秀产率得到多取代环戊烷.产物同时含有一个季碳中心和叔碳中心, 且非对映选择性(dr)达到14/1.有趣的是, 当他们以20 mol%的四丁基碘化铵(TBAI)作为催化剂, 2 equiv.的30%的过氧化氢水溶液作为氧化剂及乙酸乙酯作溶剂时, 室温条件下15 h就可以58%的产率得到多取代环戊烯.这个结果表明了TBAI/H2O2催化体系的特殊催化性能.然而, 作者对于生成环戊烯的机理并没有详细解释, 只是推测次碘酸钾可能是活性催化剂.
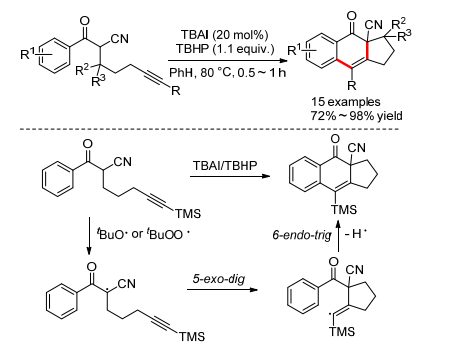
2012年, Shia小组[6]报道了一个α-氰基-α-炔基芳酮的串联环化反应(Scheme 3).该研究小组以20 mol% TBAI作为催化剂, 1.1 equiv.的叔丁基过氧化氢(TBHP)作为氧化剂, 80 ℃下就可以得到[6, 6, 5]三环结构.作者提出的机理与Scheme 1b基本类似, 原位生成的叔丁氧或叔丁过氧自由基首先夺取底物中α位的氢得到烷基自由基, 然后经过5-exo-dig环化得到烯基自由基中间体, 最后经过6-endo-trig加成得到目标产物.
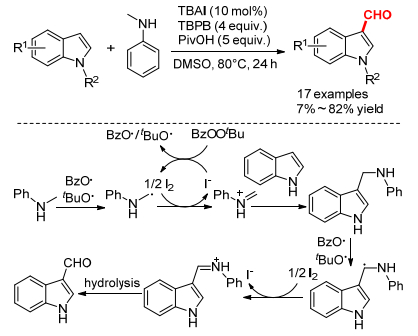
2012年, 王斌小组[7]报道了TBAI催化的N-甲基苯胺作为酰化试剂的吲哚甲酰化反应, 对于N保护的或者不保护的吲哚都能得到相应的3-吲哚醛(Scheme 4).与传统的Vilsmeier方法[POCl3/N, N-二甲基甲酰胺(DMF)]和已报道的过渡金属催化的吲哚甲酰化反应相比, 该方法条件温和、毒性小且选择性高.反应在当量n-Bu4NOH和碘存在下, 没有甲酰化产物生成, 这说明n-Bu4NIO不是活性催化剂.反应机理也与Scheme 1b基本类似, 具体为:首先原位生成的苯甲酰氧或叔丁氧自由基夺取N-甲基苯胺的氢原子生成碳自由基, 接着在原位生成的零价碘条件下经过SET得到亚胺中间体, 然后吲哚对其加成得到碳碳氧化偶联产物, 重复上述过程水解得到甲酰化产物.
同年, 该小组[8]又报道了碘化钾催化的吲哚和N, N-二甲基苯胺的氧化偶联反应(Eq. 1).催化剂由TBAI变为KI, 氧化剂使用量增加到6 equiv., 其他条件基本相同, 但是得到了不同的产物, 反应仅对未保护的吲哚和4位有取代基的N, N-二甲基苯胺适用.机理研究显示, 在亲核试剂吲哚对亚胺中间体加成时, 同时得到碳碳键和碳氮键偶联产物.
|
|
(1) |
近来, 蔡虎小组[9]也报道了单质碘催化的吲哚的甲酰化反应(Eq. 2).作者使用N, N, N', N'-四甲基乙二胺(TMEDA)作为一种新型的甲酰化试剂, 采用101 kPa的氧气作为氧化剂, 以高产率得到了3-吲哚醛.
|
|
(2) |
2013年, Nagano和Li小组[10]共同报道了TBAI催化的N-芳基烯胺的分子内氧化偶联反应, 能以高产率得到多取代的吲哚化合物(Eq. 3).其中R1可以是不同的吸电子或给电子基团, 而R2只能是吸电子取代的芳环, R3只能是酯基、酰胺或者酰基.作者随后对机理进行了研究, 通过筛选一系列碘催化体系, 最终发现次碘酸盐或亚碘酸盐可能是反应的活性催化剂.
|
|
(3) |
2013年, Itoh小组[11]报道了单质碘催化的N-芳基四氢异喹啉和碳亲核试剂的氧化偶联反应, 40 ℃下就可以得到偶联产物(Scheme 5).然而, 亲核试剂仅限于与吸电子取代基相连的sp3碳氢键(例如硝基烷烃、丙二酸酯及脂肪酮).加入自由基抑制剂2, 6-二叔丁基-4-甲基苯酚(BHT)后, 反应产率没有明显降低, 这就排除了自由基机理.作者提出了一个可能的反应机理, 原位产生的活性催化剂次碘酸(HIO)能氧化N-芳基四氢异喹啉生成亚胺碘化物, 然后再发生亲核加成得到偶联产物, 同时产生的HI再氧化循环成单质碘.
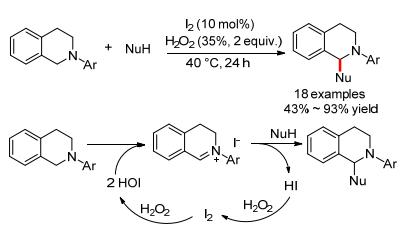
同年, Prabhu小组[12]也报道了类似的反应(Eq. 4).不同的是, 他们使用更绿色的氧气作为氧化剂, 室温下就可以得到偶联产物.反应经历了和Itoh小组相同的中间体, 除了适用于含有C(sp2)—H键或活泼C(sp3)—H键的亲核试剂(例如香豆素、苯酚、吲哚, 硝基烷烃, TMSCN和酮等)外, 还适用于过氧叔丁醇, 酰胺和亚磷酸酯等底物.
|
|
(4) |
同年, 万小兵小组[13]也报道了TBAI催化的N-甲基苯胺的α-氰基化反应(Eq. 5).作者使用了苯乙腈代替氰化物作为安全的氰化试剂, 并且首次采用DFT计算, 给出了一个和上述两个反应类似的自由基机理.
|
|
(5) |
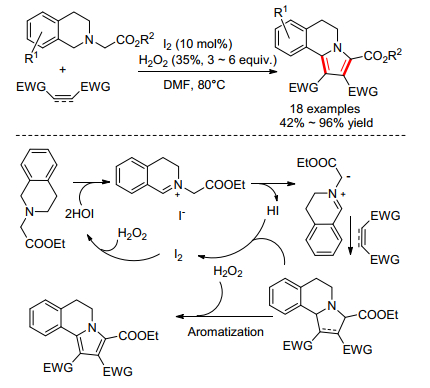
2014年, 高建荣小组[14]报道了单质碘催化的N-取代四氢异喹啉和活化烯烃、炔烃的[3+2]氧化环加成反应(Scheme 6).活化烯烃为1, 4-萘醌、丙烯酸酯、丁二酰亚胺等, 活化炔烃为丁炔二酸酯, 都能以高产率得到吡咯并异喹啉化合物.作者提出了可能的反应机理:原位产生的HIO能氧化异喹啉成亚胺碘化物, 然后再脱去HI生成氮叶立德, 接着和炔键或者烯键发生环加成和氧化芳构基化生成吡咯并异喹啉环.
2015年, 雷爱文小组[15]报道了碘催化的β-酮酯或2-吡啶酯和取代乙烯的氧化环化反应, 分别合成了二氢呋喃和吲嗪(Scheme 7).反应仅适用于芳基乙烯, 1, 1-二苯乙烯和1-甲基苯乙烯, 对脂肪烯烃不适用. β-酮酯的反应产率明显大于2-吡啶酯, 一方面因为其活性较高, 另一方面因为二氢呋喃没有经过进一步氧化得到呋喃.作者加入自由基捕获剂2, 2, 6, 6-四甲基哌啶氧化物(TEMPO)后发现, 并没有得到目标产物, 这说明反应经历了自由基历程.具体机理如下:在原位生成的一价碘存在下, β-酮酯转变为次甲基自由基, 其对烯键进行自由基加成得到苄基自由基, 其对羰基进行分子内自由基加成得到四氢呋喃, 最后被原位产生的一价碘或叔丁氧自由基氧化得到最终产物二氢呋喃.吲嗪的产生也经历了一个类似的过程, 唯一的区别在于多了一步氧化芳构化过程.
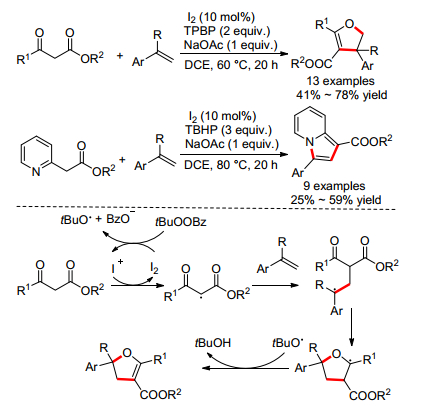
接着作者[16]又报道了碘催化的β-酮酯或2-吡啶酯和取代乙炔的氧化环化反应, 分别合成了呋喃和吲嗪(Eqs. 6, 7).前后两种方法都能得到吲嗪, 而后者产率显著提高.反应对于芳基取代的乙炔产率较高, 而脂肪基取代乙炔较低.作者提出的机理基本和上述相同, 不同在于经历了自由基对炔键的加成过程.
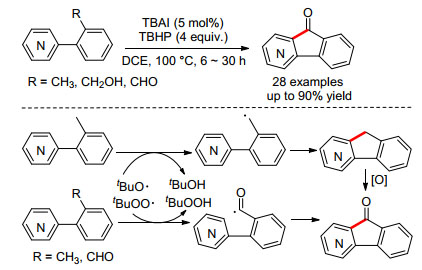
2016年, Patel小组[17]报道了TBAI催化的芳烃或杂芳烃分子内的氧化酰基化反应, 可以合成一系列芴酮化合物(Scheme 8).其中R基团可以为甲基、羟甲基和醛基.当R为甲基时, 反应首先生成苄基自由基; 当R为羟甲基或醛基时, 反应首先生成酰基自由基, 随后经由自由基加成消除得到最终的产物, 而产物可进一步发生氧化反应生成.
|
|
(6) |
|
|
(7) |
随后, 杨罗小组[18]报道了碘催化的异喹啉和甲基芳烃的选择性氧化偶联反应, 以中等产率得到了1-苄基异喹啉化合物(Eq. 8).同样地, 由甲基芳烃失去一个苄位H原子产生的苄基自由基为反应的活性中间体.
|
|
(8) |
2017年, Itoh小组[19]报道了碘催化可见光促进的噻吩和丙二酸酯的氧化偶联反应(Scheme 9).反应对不同取代基的噻吩和噻唑都适用, 然而仅α位吸电子取代的丙二酸酯可作为偶联试剂.控制实验证明碘、氧气和光照对反应缺一不可, 使用碘代三酯基甲烷也能得到相应产物, 表明反应可能首先发生了碘化.具体机理如下:首先丙二酸酯和碘生成碘代丙二酸酯, 接着在光照下发生C—I键断裂生成碳自由基和碘自由基, 最终经由碳自由基对噻吩中双键的加成、消除生成最终产物.体系中生成两分子HI可被氧气重新氧化成单质碘, 实现催化循环.
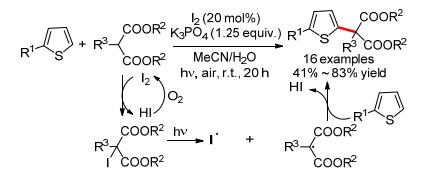
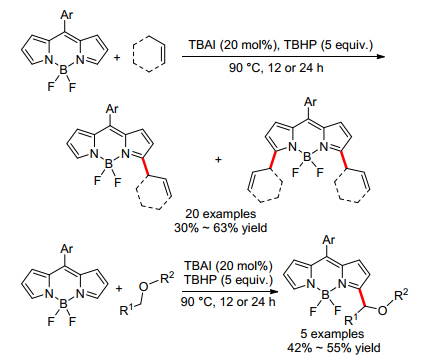
同年, Boens小组[20]报道了TBAI催化的硼-二吡咯亚甲基(BODIPY)和烯丙基烯烃(或醚)的氧化偶联反应, 以中等产率得到了烯丙基或氧烷基取代的BODIPY (Scheme 10).反应对含烯丙基C—H键的不同烯烃都适用, 通过控制反应时间可以分别得到单取代或双取代的偶联产物.反应对不同环状或脂肪醚也适用, 得到醚的α-芳基化产物.机理研究表明烯丙基自由基或α-氧烷基自由基为反应的中间体.反应机理也与Scheme 1b基本类似.
同年, 杜云飞小组[21]也报道了TBAI催化的香豆素和三级胺的氧化偶联反应, 以满意的产率得到了3-胺烷基取代香豆素, 反应对N, N-二甲基芳胺、N-甲基吡咯烷酮和N-甲基二苯胺都适用(Eq. 9).研究表明α-胺烷基自由基为反应的中间体, 反应机理也与Scheme 1b基本类似.
|
|
(9) |
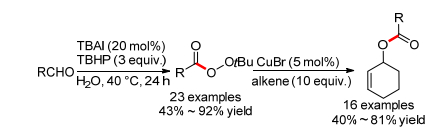
2011年, 万小兵小组[22]报道了首例TBAI催化的醛和过氧叔丁醇的直接氧化偶联反应, 能以中等到优秀的产率得到过氧化羧酸叔丁酯, 芳香醛和脂肪醛都适用于这个反应(Scheme 11).反应可能经历了一个酰基自由基和过氧叔丁基自由基的直接偶联过程.在铜催化下, 所得过氧化羧酸叔丁酯可以和烯丙基碳氢键反应生成一系列烯丙酯.
2010年, Ishihara小组[2]报道了首例TBAI催化的邻羰乙基苯酚的分子内氧化醚化反应, 室温就可以以几乎当量得到2-羰基二氢苯并呋喃化合物(Scheme 12a).当作者将TBAI换成手性碘化季铵盐后, 通过优化导向基团, 可以形成手性的碳氧键, ee值高达96%, 而且具有叔碳或季碳中心.机理研究发现, 反应经历第一种催化机理(Scheme 1a), 原位产生的次碘酸季铵盐可能为反应的活性催化剂. 2014年该小组[23]又将该体系扩展到邻羰丙基苯酚的分子内氧化醚化反应中, 获得了手性的2-羰基二氢苯并吡喃化合物(Scheme 12b).
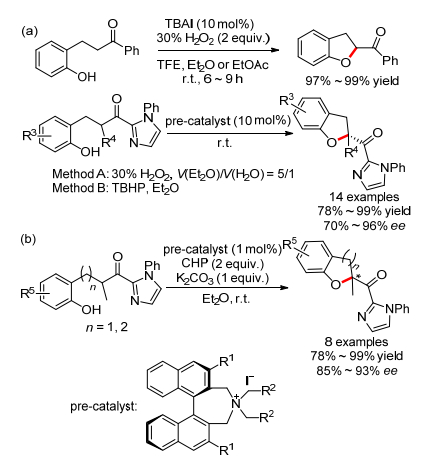
2014年, Wang小组[24]报道了一例类似的TBAI催化的酰基相邻C—H键的分子内氧化醚化反应, 得到2-酰基苯并噁唑啉, 在当量K2CO3促进下进一步脱去Ts就可得到芳构化产物2-酰基苯并噁唑(Eq. 10). R1无论是芳香取代基, 还是脂肪取代基, 反应都能顺利进行且产率高达94%, 反应机理与上述报道相似.
2011年, Ishihara小组[25]又报道了TBAI催化的与羰基相邻C—H键和羧酸的分子内或分子间的氧化氧酰基化反应, 得到环内酯或者脂肪酯(Eqs. 11~13).在分子间反应中, 酮和脂肪醛底物都适用.除此之外, 芳香羧酸和脂肪酸也都适用于这个反应.
|
|
(10) |
|
|
(11) |
|
|
(12) |
|
|
(13) |
2014年, 成江小组[26]报道了TBAI催化的酮α-C—H键和取代甲醇的氧化氧酰基化反应, 得到了α-酰氧基酮化合物(Eq. 14).芳酮和脂肪酮都适用于这个反应, 苄醇、肉桂醇以及脂肪醇也都适用.
|
|
(14) |
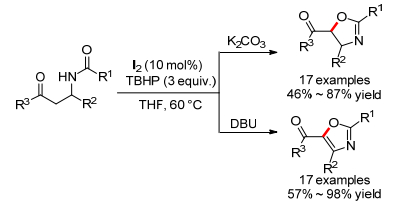
2015年, 高文超小组[27]报道了单质碘催化的β-乙酰氨基苯酮的分子内C—O键偶联反应, 且通过碱可以调节反应选择性(Scheme 13).当碳酸钾作为碱时, 可以得到多取代噁唑啉, 而当DBU作为碱时得到了进一步氧化的产物多取代噁唑.
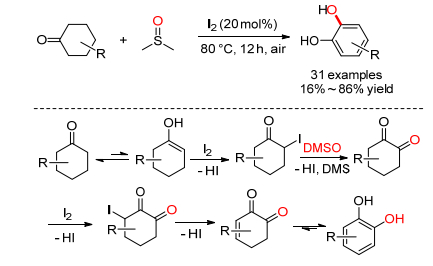
2016年, 焦宁小组[28]报道了碘催化的环己酮氧化生成邻苯二酚的反应, 反应中DMSO既作为溶剂又作氧化剂(Scheme 14).机理研究发现, 碘代环己酮和环己二酮可能都是反应的中间体; 18O标记实验证明DMSO提供了邻苯二酚的氧源.具体机理为:环己酮首先发生碘代生成碘代环己酮, 接着发生Kornblum氧化生成1, 2-环己二酮, 继续发生碘代、消除生成中间体, 最后重排得到最终产物.反应中产生的HI被DMSO或空气重新氧化生成单质碘, 实现催化循环.
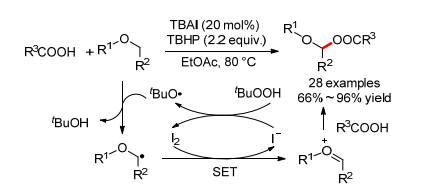
2011年, 万小兵小组[4]报道了首例TBAI催化的醚类化合物中与氧原子相邻碳氢键和羧酸的分子间氧化偶联反应, 能选择性得到α-酰氧基醚化合物, 芳香羧酸和脂肪酸都适用于这个反应(Scheme 15).反应经历了第二种催化机理, 产生的α-烷基自由基和氧鎓离子是活性的中间体.值得注意的是, 作者通过动力学同位素效应, 指出碳氢键断裂即第一步的自由基夺氢过程是反应的决速步骤.
2013年, 段兴华小组[29]报道了TBAI催化的与氧原子或氮原子相邻碳氢键和α-酮酸的脱羰氧化偶联反应, 能选择性得到α-酰氧基醚或α-酰氧基酰胺(Eq. 15).然而, 只有芳香羧酸适用于这个反应.
|
|
(15) |
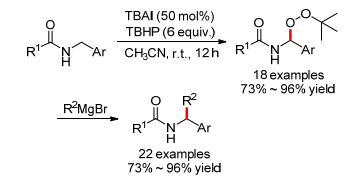
2014年, 于辉小组[30]报道了TBAI催化的与氮原子相邻碳氢键和过氧叔丁醇的氧化偶联反应, 得到α-叔丁过氧酰胺, 在室温可以和格式试剂进一步生成α-芳基或烷基取代酰胺(Scheme 16).然而, 只有芳香羧酸适用于这个反应.
2012年, 余孝其小组[31]报道了首例TBAI催化的甲基芳烃的苄位C—H键和羧酸的氧化偶联反应, 获得了一系列羧酸苄酯, 其中芳香羧酸、脂肪酸甚至N保护氨基酸都适用于这个反应(Eq. 16).反应同样经历了第二种催化机理, 产生的苄基自由基和正离子是活性的中间体.作者通过动力学同位素效应, 指出苄基碳氢键断裂是反应的决速步骤.此外, 作者发现NaI代替TBAI时反应几乎不发生, 这表明季铵离子起到了十分重要的作用.这可能是因为在过量的甲基芳烃中TBAI溶解性远大于NaI, 故其催化活性也明显高于NaI.
|
|
(16) |
同年, 王斌小组[32]报道了TBAI催化的2-甲基喹啉或甲基芳烃的苄位C—H键和芳香醛的氧化偶联反应, 分别在水中或者无溶剂条件下得到了一系列苯甲酸苄酯.遗憾的是仅芳香羧酸适用于这个反应(Eq. 17).作者研究机理发现, 原位产生的过氧化苯甲酸叔丁酯起到了重要的作用.
|
|
(17) |
2013年, 付雪峰小组[33]也报道了类似的TBAI催化的甲基芳烃的苄位C—H键和苄醇的氧化偶联反应, 同样获得了苄酯(Eq. 18).
|
|
(18) |
同年, Patel小组[34]报道了TBAI催化的甲基芳烃苄位C—H键的自身偶联和交叉偶联反应, 在无溶剂条件下得到了羧酸苄酯(Eq. 19).
|
|
(19) |
2014年, 唐果小组[35]报道了TBAI催化的甲基芳烃苄位C—H键和二苯基磷氧或亚磷酸酯的氧化偶联反应, 得到了亚磷酸苄酯(Eq. 20).反应同样经历了自由基历程, 区别在于P—H键先发生了氧化生成P—OH, 再去进攻苄基正离子形成C—O键, 而不是直接进攻构建C—P键.基于上述工作, 近来唐课文小组[36]报道了TBAI催化的甲基芳烃苄位C—H键和二芳基磷酸的氧化偶联反应得到了同样的产物.反应同样经历了自由基历程, 苄基自由基为反应的关键中间体(Eq. 21).
|
|
(20) |
|
|
(21) |
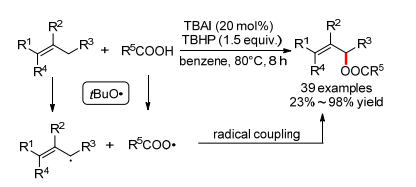
2012年, 万小兵小组[37]报道了首例TBAI催化的烯丙位C—H键和羧酸的分子间自由基偶联反应, 可以得到羧酸烯丙酯(Scheme 17).反应底物范围广, 对不同类型的芳香羧酸和脂肪酸都适用.然而值得注意的是, 当作者使用苯甲酸钠代替苯甲酸作底物时, 反应无法顺利进行, 且自由基抑制实验证明反应经历一个自由基历程.因此作者认为生成的烯丙基自由基并不会进一步生成烯丙基正离子, 然后羧基负离子进攻形成C—O键, 而是直接发生一个烯丙基自由基和羧基自由基的直接自由基偶联反应.
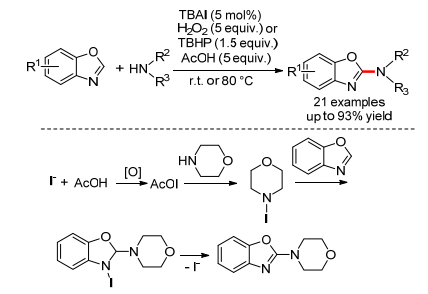
2011年, Nachtsheim小组[38]报道了首例TBAI催化的苯并噁唑和脂肪胺的氧化C—N偶联反应(Scheme 18).反应可以使用5 equiv.的过氧化氢或者1.5 equiv.的叔丁基过氧化氢作为氧化剂, 乙酸作为添加剂, 能得到2-氨基取代的苯并噁唑.通过控制实验, 作者认为原位生成的乙酸碘起到了关键的作用.具体机理为: I-首先在H2O2或TBHP及乙酸存在下生成AcOI, 接着和吗啉反应生成N-碘代吗啉, 然后和苯并噁唑反应生成中间体, 最终生成产物, 同时生成I-.
2013年, 纪顺俊小组[39]报道了碘催化的吲哚和芳胺的氧化偶联反应, 可以获得2, 3-二氨基取代的吲哚(Eq. 22).不同取代基的吲哚都适用且产率较高, 而对于给电子如甲基和甲氧基取代的苯胺产率较高, 吸电子基团取代的苯胺产率较低.遗憾的是, 作者对机理没有进行详细研究.
|
|
(22) |
2015年, Yotphan小组[40]报道了碘催化的吲哚和唑类化合物的氧化偶联反应, 能选择性地得到2-取代吲哚(Scheme 19).吡唑、咪唑、1, 2, 3-三氮唑、1, 2, 4-三氮唑、苯并咪唑和苯并三氮唑都适用于这个反应.随后作者对机理进行了研究, 发现加入TEMPO或BHT并没有抑制反应, 这说明反应不是一般的自由基机理; 当HI水溶液代替单质碘时, 也能以85%产率得到相应产物, 这说明可能存在着单质碘到负一价碘的催化循环.综合以上实验结果, 作者提出了一个可能的机理:首先吲哚发生碘代反应生成碘鎓离子, 随后吡唑亲核进攻生成中间体, 最后消除HI生成产物, 而HI被重新氧化成单质碘.
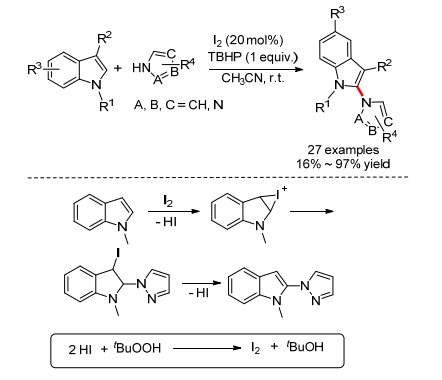
2017年, 王斌小组[41]报道了碘催化的色氨酸酯的分子内氧化偶联反应, 能得到吲哚并吡咯稠杂环化合物(Eq. 23).反应经历了和上述反应相似的机理, 经由碘鎓离子生成了中间体, 最后氧化芳构化得到最终产物.
|
|
(23) |
2012年, 万小兵小组[42]报道了首例TBAI催化的芳香醛和甲酰胺的氧化酰胺键形成反应, 反应通过脱去甲酰胺中的CO, 得到了N, N-二取代的苯甲酰胺(Scheme 20).反应仅对不饱和的共轭醛适用, 脂肪醛不适用.作者将DMF中羰基碳进行13C标记, 发现产物中并没有检测到13C, 这说明羰基可能以CO的形式释放.基于控制实验作者认为反应是一个自由基偶联过程.原位产生的叔丁氧基自由基或叔丁过氧自由基能夺取醛基氢生成醛基自由基, 同时使甲酰胺生成氨基自由基, 最后醛基自由基和氨基自由基发生自由基偶联直接形成C—N键.
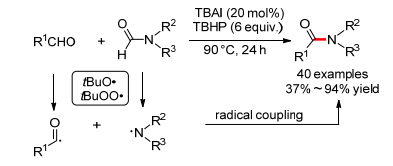
紧接着, 汪志勇小组[43](Eq. 24)和朱成建小组[44] (Eq. 25)几乎同时报道了碘催化的苄醇和甲酰胺的氧化酰胺键形成反应, 反应中苄醇首先被氧化为苯甲醛, 随后发生上述的自由基偶联反应. 2014年, 汪志勇小组[45]又报道了碘催化的苄胺和甲酰胺的氧化酰胺键形成反应, 反应中苄胺首先被氧化为苯甲醛, 随后也发生上述的自由基偶联反应(Eq. 26).
|
|
(24) |
|
|
(25) |
|
|
(26) |
2012年, Barbas Ⅲ小组[46]也报道了TBAI催化的芳醛和亲核试剂的氧化酯化或者酰胺化反应, 可以得到一系列酯或者酰胺(Scheme 21a).随后余孝其小组[47]也报道了低价碘催化的苄醇或者芳香醛和铵盐的氧化酰胺键形成反应.反应以四乙基碘化铵作为催化剂, 碳酸氢胺作为氮源, 可以得到酰胺化合物(Scheme 21b). 2014年, Li小组[48]也报道了碘化钾催化的芳香醛和不同唑类化合物的氧化酰胺键形成反应(Scheme 21c).反应机理与上述报道不用, 首先亲核试剂(氧或氮)对醛基进行加成生成醇, 然后叔丁氧基自由基夺取与羟基相邻α氢生成自由基中间体, 最后过氧叔丁醇氧化脱氢得到产物, 同时生成叔丁氧基自由基和水.
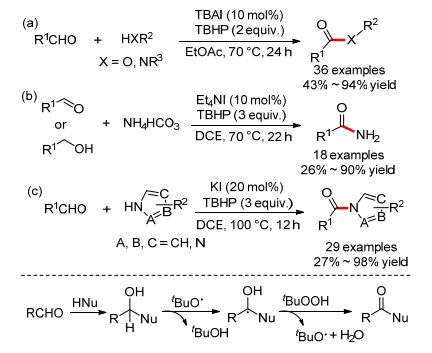
2012年, 王磊小组[49]报道了首例碘催化的苯乙酮和二级胺的氧化偶联反应, 可以得到α-羰基酰胺(Scheme 22a).通过控制实验, 作者认为该反应经历了一个碘化、氨基化再氧化的串联过程.几乎同时, 万小兵小组[50]也报道了一个相似的碘催化的苯乙酮和胺的氧化偶联反应, 不但二级胺适用, 一级胺也同样适用于这个反应(Scheme 22b).而且作者通过18O标记水证明了新产生的氧原子来源于水分子.
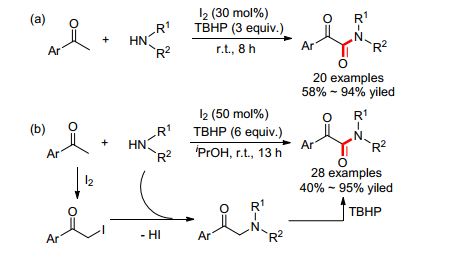
同年, 屈凌波[51] (Eq. 27)和王磊小组[52] (Eq. 28)分别报道了低价碘催化的苯乙酮和甲酰胺的氧化偶联反应.值得注意的是, 酰胺的碳来自于苯乙酮, 而甲酰胺仅提供氮源.
|
|
(27) |
|
|
(28) |
2012年, Prabhu小组[53]报道了NIS催化的苯乙酮或苯丙酮和二级胺的分子间氧化偶联反应, 可以分别得到α-羰基酰胺和α-氨基苯丙酮(Eqs. 29, 30).反应同样经历了一个先碘化、再亲核胺化的串联过程.
|
|
(29) |
|
|
(30) |
同年, Loh小组[54]报道了碘催化的脂肪醛和二级胺的氧化偶联反应(Scheme 24).作者采用单质碘作为催化剂, 碳酸钠过氧化氢作为氧化剂, 可以得到氨基化产物.通过控制实验, 作者证明了碘代亚胺和烯胺都是反应的中间体.基于实验结果作者提出了一个可能的反应机理:首先脂肪醛和二级胺脱水生成烯胺中间体, 接着在原位生成次碘酸存在下生成碘代亚胺, 然后甲醇加成、分子内亲核取代生成氮杂三元环, 最终碳氮消除、甲醇加成生成产物.
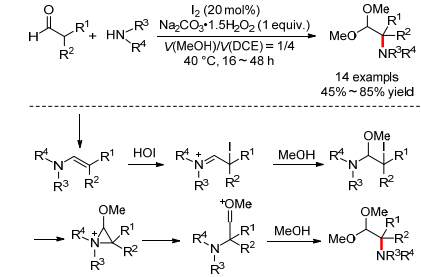
2011年, 韩丙小组[55]报道了TBAI催化的β-酮酯和2-氨基吡啶的氧化偶联反应, 可以得到[1, 2-a]咪唑并吡啶(Eq. 31).作者认为该反应同样经历了一个先碘化, 后吡啶氮亲核取代, 再吡啶2位的氨基和酮羰基缩合关环的串联过程.
2012年, 朱成建小组[56]报道了TBAI催化的β-酮酯和苄胺的氧化偶联反应, 可以得到多取代的噁唑(Eq. 32).反应经历了一个串联的过程, 首先β-酮酯的α位C—H键和苄基的氨基发生分子间氧化C—N偶联, 然后酰基上的氧再和苄胺的苄位碳氢键发生分子内碳氧键偶联, 一步实现了C—N键和C—O键的构建.
|
|
(31) |
|
|
(32) |
2013年, 汪志勇小组[57]又报道了首例NIS催化的2-吡啶乙酸乙酯和苄胺的氧化偶联反应, 在室温下就能得到[1, 5-a]咪唑并吡啶(Eq. 33).首先2-吡啶乙酸乙酯的α-C—H键和苄胺氨基发生氧化C—N键偶联, 然后吡啶氮和苄胺的苄基C—H键再发生氧化C—N键偶联, 一步反应实现了两个C—N键构建.
|
|
(33) |
2014年, 于辉小组[58]报道了低价碘催化的邻羟基苯酮和二级胺的氧化偶联反应, 可以得到2-氨基苯并呋喃酮(Eqs. 34, 35).作者认为该反应经历了一个碘化、羟基和氨基分别亲核取代的串联过程.
|
|
(34) |
|
|
(35) |
同年, Ilangovan小组[59]报道了碘催化的邻氨基苯乙酮的分子内氧化偶联反应, 可以得到一系列N保护的靛红, 反应机理和以前报道类似(Eq. 36).
|
|
(36) |
2012年, 汪志勇小组[60]报道了低价碘催化的与杂原子相邻C—H键和邻氨基酮中氨基的氧化C—N偶联反应, 在额外氮源存在下缩合氧化可以得到4-取代喹唑啉(Eq. 37).不同溶剂例如酰胺类[DMF、N, N-二甲基乙酰胺(DMA)、N-甲基吡咯烷酮(NMP)等]、醚(甲基叔丁醚、乙醚、乙二醇二甲醚、乙二醇单甲醚等)和醇(甲醇、乙醇、乙二醇)都适用, 这也是首例非金属催化的一级胺和与杂原子相连的C—H键的氧化偶联反应.通过动力学同位素效应, 指出C—H键断裂是反应的决速步骤.当使用高价碘试剂代替低价碘催化体系时, 反应并不能发生, 也证明了高价碘试剂不是反应的活性催化剂.自由基抑制实验也证明了反应经历了自由基历程.因此, 基于以上实验首次提出了一个零价碘到正一价碘的催化循环机理(详见Scheme 1b).
|
|
(37) |
同年, 李中军小组[61]又报道了KI催化的N-甲基酰胺和酰亚胺的分子间氧化偶联反应, 在无溶剂条件下就可以得到酰亚胺化产物(Eq. 38).但是遗憾的是, 仅有酰亚胺作为底物, 一般的胺和酰胺都不适用, 反应机理与Scheme 1b相同.
|
|
(38) |
2015年, 毛金成小组[62]报道了NaI催化的N, N-二甲基苯胺和磺酰胺的氧化偶联反应(Scheme 25).和上述报道不同之处在于, 反应首先经由自由基历程生成C—N键, 然后在氧化剂存在下能够进一步氧化生成C=N键.反应中只有磺酰胺产率较高, 而苯甲酰胺产率极低.
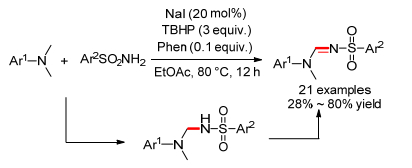
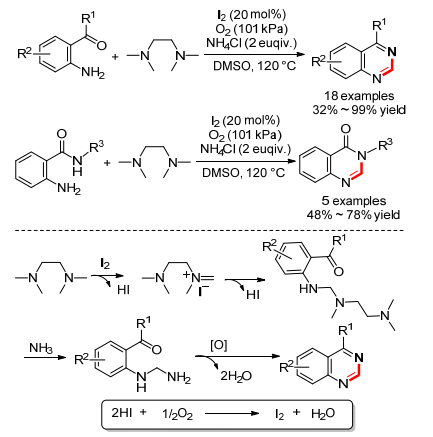
2015年, 我们课题组[63]报道了碘催化的TMEDA和邻氨基酮(或邻氨基苯甲酰胺)的氧化偶联反应, 在氧气做氧化剂条件下可以得到喹唑啉和喹唑啉酮(Scheme 26).当反应加入自由基抑制剂TEMPO时, 反应产率反而增加, 这说明反应为非自由基机理.根据实验结果我们给出了一个可能的机理, 首先TMEDA和碘反应生成亚胺碘化物, 接着邻位氨基进攻双键得到碳氮偶联中间体, 随后和氨发生交换得到中间体, 最后脱水缩合、氧化芳构化得到目标产物.
2014年, 孙凯小组[64]报道了首例TBAI催化的醚和糖精的分子间氧化偶联反应, 可以得到醚的α-酰亚胺化产物(Eq. 39).不同取代基的苯甲醚、乙二醇二甲醚、四氢呋喃、1, 4-二氧六环都适用.通过动力学同位素效应, 也证明了与氧相邻C—H键断裂为反应的决速步骤.随后Patel小组[65]也报道了一例TBAI催化的苯甲醚和四氮唑的分子间偶联反应(Eq. 40).两者机理均与Scheme 1b中相同.
|
|
(39) |
|
|
(40) |
2017年, 我们小组[66]又报道了KI催化的醚和脒盐酸盐的氧化偶联反应, 并通过随后的亲核取代、脱氨缩合及氧化芳构化的串联过程合成了1, 3, 5-三嗪化合物(Eq. 41).反应对于芳基、吡啶和烷基取代的脒都适用, 也对不同脂肪醚也适用.值得注意的是, 当两分子相同的脒作底物时, 可合成结构对称的2, 4-二取代-1, 3, 5-三嗪; 而当两个不同脒作底物时, 在生成两种对称2, 4-二取代-1, 3, 5-三嗪的同时, 也生成了不对称的2, 4-二取代-1, 3, 5-三嗪化合物.机理研究表明, 反应可能经历了一个负一价碘到零价碘的催化循环机理(与Scheme 1b相同), 该催化循环在醚的C—H氨基化和C—O键断裂中发挥了重大的作用.
|
|
(41) |
近来, 林东恩小组[67]报道了TBAI催化的嘌呤和醚的氧化偶联反应, 合成得到了N-氧烷基修饰的嘌呤衍生物(Eq. 42).四氢呋喃、1, 4-二氧六环、乙二醇二甲醚、甲基叔丁基醚和四氢吡喃都适用于这个反应, 反应机理与Scheme 1b相同.
|
|
(42) |
2013年, 朱成建小组[68]报道了TBAI催化的甲基芳烃苄位C—H键和与咪唑、三氮唑或者酰亚胺的分子间氧化C—N偶联反应, 可以得到苄位氨基化产物(Eq. 43).反应同样经历了第二种催化机理(详见Scheme 1b), 产生的苄基自由基和苄基正离子是活性中间体.
|
|
(43) |
2014年, 李建新小组[69]报道了TBAI催化的4-甲基喹唑啉和苄胺的氧化偶联反应, 在乙酸作添加剂条件下可以得到[1, 5-a]咪唑并喹唑啉(Eq. 44).反应经历了一个串联过程, 首先4-甲基喹唑啉的苄位C—H键和苄胺的氨基发生氧化C—N偶联, 随后喹唑啉3位氮和苄胺的苄位C—H键再发生氧化C—N键偶联, 最后芳构化得到产物. 2015年, 该小组[70]又发展了KI催化的邻氨基苯酮、醋酸铵和甲苯的三组分氧化偶联反应.甲苯的苄位C—H键连续发生了两次氧化C—N偶联, 最后芳构化形成2, 4-二取代喹唑啉(Eq. 45).
|
|
(44) |
|
|
(45) |
2015年, 李剑小组[71]也报道了类似的KI催化的二苯甲烷和亚胺的氧化偶联反应, 后续经过简单的酸解就可以得到苄胺(Eqs. 46, 47).反应对对称取代的二苯甲烷和非对称取代的二苯甲烷都适用, 该体系同样适用于3-取代吲哚酮, 可以用来合成3-氨基吲哚酮.
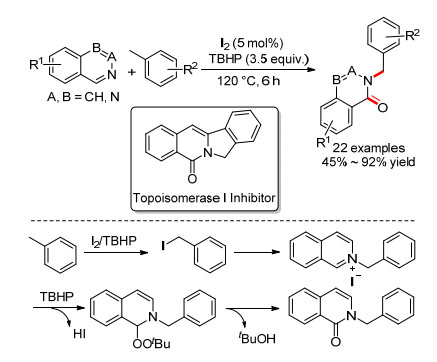
2016年, 杨罗小组[72]报道了碘催化的甲基芳烃苄位C—H键和喹啉类化合物的氧化偶联反应, 合成了N-取代喹啉酮化合物(Scheme 27).反应对异喹啉、喹啉、喹唑啉和酞嗪都适用, 而且作者也将此方法用于了Topoisomerase Ⅰ抑制剂的合成.机理研究发现, 碘代甲苯可能是反应中间体.通过18O标记实验, 证明了羰基氧可能不是来自于水, 而是来自于氧化剂过氧叔丁醇.具体机理为:甲苯先碘代生成碘代甲苯, 接着异喹啉亲核取代生成异喹啉碘化物, 随后TBHP对碳氮双键加成得到过氧中间体, 最后消除一分子叔丁醇得到产物.
|
|
(46) |
|
|
(47) |
2014年, 王磊小组[73]报道了首例TBAI催化的烯丙位C—H键和芳胺的氧化偶联反应, 获得了一系列N-芳基取代的烯丙胺, 其中不同取代基苯胺、N-甲基苯胺和邻氨基吡啶都适用于这个反应(Eqs. 48, 49).作者也尝试了烷基苯和芳胺的氧化偶联反应, 反应也能顺利进行.该反应同样经历了第二种催化机理, 产生的烯丙基自由基和烯丙基正离子是反应的中间体.
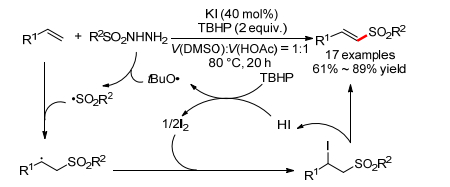
2014年, 雷爱文小组[74]报道了KI催化的取代乙烯和磺酰肼的氧化偶联反应, 可以得到β-磺酰化苯乙烯(Scheme 28).反应对于芳香磺酰肼和脂肪磺酰肼都适用.作者提出反应可能经历了一个类似与氧化Heck反应的自由基机理:由磺酰肼原位产生的磺酰基自由基对烯烃进行加成得到β-磺酰烷基自由基, 然后和产生的碘生成碘代产物, 最终消除HI得到产物, HI重生被氧化单质碘实现催化循环.
|
|
(48) |
|
|
(49) |
2014年, 邓国军小组[75]报道了碘催化的吲哚和苯亚磺酸钠的氧化磺酰化反应, 可以得到2-磺酰化吲哚(Eq. 50).反应对于芳香亚磺酸钠和亚磺酸钠都适用.反应同样经历了一个和上述反应类似的串联自由基机理:由苯亚磺酸钠原位产生的磺酰基自由基对吲哚2位进行加成, 再生成碘代产物, 最后消除一分子HI得到最终产物.
|
|
(50) |
近来, Prabhu小组[76]报道了碘催化的烯胺酮和硫酚的氧化偶联反应, 可以得到硫醚化烯胺酮(Eq. 51).反应对于芳香硫酚和杂环硫酚都适用, 溶剂DMSO作为反应的氧化剂.当加入自由基抑制剂TEMPO时, 反应正常进行, 故反应可能不涉及自由基历程.当使用相应的二硫化物代替硫酚时, 目标产物也能获得, 说明二硫化物可能是反应的中间体.
|
|
(51) |
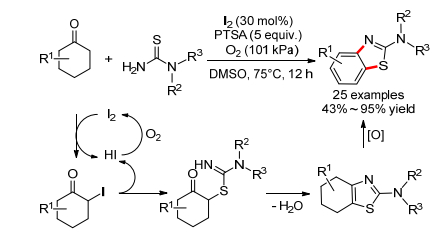
2013年, 江焕峰小组[77]报道了碘催化的环己酮和硫脲的氧化偶联反应, 在氧气作为氧化剂条件下可以得到2-氨基苯并噻唑, 不同取代基的环己酮和苯并环己酮都适用(Scheme 29).作者认为环己酮首先发生碘代生成碘代环己酮, 接着硫脲亲核取代、脱水缩合得到2-氨基噻唑, 最后氧化芳基化得到产物.
同年, 邓国军小组[78]报道了碘催化的环己酮和硫醇的氧化C—S偶联反应, 在需氧条件下能得到邻羟基硫醚, 反应对不同取代的环己酮都适用, 芳基硫酚和硫醇也都适用(Scheme 30).反应机理与上述反应略有不同, 硫酚在氧气存在下自身氧化偶联成二硫化物, 接着和碘反应生成亲电硫试剂, 然后和环己酮偶联生成中间体, 最后氧化脱氢得到邻羟基硫醚.
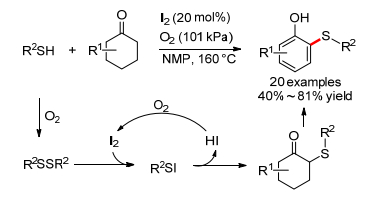
2015年, 雷爱文小组[79]报道了碘催化的1, 3-二羰基化合物和芳基硫酚的氧化偶联反应, 可以到α-硫醚化产物(Eq. 52).遗憾的是, 反应底物范围不是很广, 仅乙酰丙酮、3, 5-庚烷二酮、芳基硫酚适合.随后, 王正小组[80]也报道了一个类似的KI催化的1, 3-二羰基化合物和芳基硫酚的需氧氧化偶联反应, 可得到相同的产物(Eq. 53).反应使用绿色的氧气作氧化剂, 且底物范围进一步扩大.机理研究表明, 反应不是自由基历程, 二硫化物和碘代硫酚均可能是反应的中间体.
|
|
(52) |
|
|
(53) |
2014年, 雷爱文小组[81]报道了碘催化的甲基芳烃和芳基硫酚的氧化偶联反应, 在氮气条件下能得到苄硫醚(Eq. 54).机理研究发现, 苄基自由基和二硫化物可能是反应的中间体, 随后发生偶联生成C—S键.
|
|
(54) |
同年, 邓国军小组[82]报道了KI催化的2-甲基喹啉和亚磺酸钠的氧化偶联反应, 可以得到2-磺酰甲基喹啉(Scheme 31).芳基亚磺酸钠和脂肪亚磺酸钠都适用于这个反应, 2-甲基喹啉和2-甲基喹喔啉也都适用.反应可能经历了一个磺酰自由基对2-甲基喹啉重排结构加成、随后消除的历程.
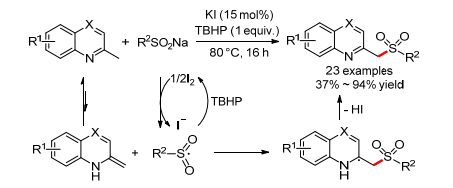
2012年, 李小青小组[83]报道了TBAI催化的α-烷基苯乙烯和磺酰肼的磺酰化反应, 可以得到β-磺酰化的烯烃(Scheme 32).反应对于大多数α-甲基芳基乙烯都适用, 对不同烷基取代的α-苯乙烯也适用.遗憾的是, 作者对磺酰肼的底物范围没有详细研究.反应同样经历了一个自由基机理, 原位由磺酰肼产生的磺酰基自由基对烯烃进行加成, 然后脱氢得到最终的产物.
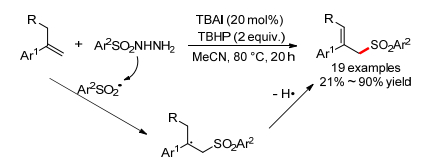
2013年, 王倩小组[84]报道了TBAI催化的N-芳基四氢异喹啉和亚磷酸酯的氧化C—P键偶联反应, 可以高产率地得到α-氨基磷酸酯(Eq. 55).亚磷酸二甲酯、亚磷酸二乙酯和亚磷酸二异丙酯都适用于这个反应, 然而作者对N-芳基四氢异喹啉仅研究了不同N-芳基对产率的影响, 底物范围有限.作者对机理没有进行详细研究, 初步推测亚胺为反应中间体.
|
|
(55) |
2017年, 孙凯小组[85]报道了KI催化的芳酮和硒氰酸钾的氧化偶联反应, 可以得到α-氰硒化苯酮(Eq. 56).反应对于苯乙酮、苯丙酮及其杂环酮都适用, 反应可能经历了一个先碘代后亲核取代的串联过程.
|
|
(56) |
2013年, Prabhu小组[86]报道了碘催化的亚磷酸酯和其他亲核试剂的氧化偶联反应, 可以高产率的得到磷酰胺或者磷酸酯(Scheme 33).其中亲核试剂可以是胺、醇和亚砜胺, 亚磷酸酯可以是亚磷酸二乙酯、亚磷酸二甲酯、亚磷酸二异丙酯和亚磷酸二苯酯.作者认为首先亚磷酸酯和碘生成磷酰碘中间体, 然后发生亲核取代得到最终产物, HI重新被氧化成单质碘.
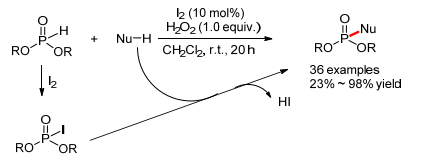
2015年, 潘远江小组[87]也报道了相似的KI催化的二取代磷氧和硫醇(或芳基硫酚)的氧化偶联反应(Eq. 57).然而, 作者认为反应机理与上述反应不同, 可能经历了一个磷氧自由基和硫自由基的直接偶联机理.
|
|
(57) |
2015年, 雷爱文小组[88]报道了NIS催化的芳基硫酚和硫醇的交叉氧化S—S偶联反应, 能以高产率得到非对称的二硫化合物, 同时也有自身二硫化物生成(Eq. 58).
|
|
(58) |
2013年, 万小兵小组[89]报道了KI催化的二级胺和硝基甲烷的氧化氮氮键偶联, 可以合成N-亚硝胺化合物(Eq. 59). 2016年, 我们课题组[90]利用这一思想, 报道了KI催化的邻氨基苯甲酰胺和硝基甲烷的氧化氮氮键偶联反应, 能以高产率得到1, 2, 3-苯并三嗪-4-酮化合物(Eq. 60).研究机理发现, HNO和HNO2可能为反应的中间体.
|
|
(59) |
|
|
(60) |
2016年, 吴小峰小组[91]报道了单质碘催化的芳基硫酚(或硫醇)和氨的氧化S-N偶联反应, 可以得到芳基磺酰胺, 反应对于不同取代基的苯硫酚、2-萘硫酚、杂环硫酚和硫醇都适用(Eq. 61).
|
|
(61) |
综上所述, 低价碘催化的脱氢氧化偶联反应因其原子经济性高、低毒且绿色高效已被广泛应用于碳碳、碳氧、碳氮、碳硫、碳磷键以及其他化学键构建中.尽管如此, 目前仍然有很多挑战性的问题亟待解决.首先, 低价碘并不能完全代替金属, 目前仅适用于键能相对较低的X—H (X=C、O、N、S、P)键的官能化反应中.因此, 成功实现键能更高的惰性化学键(如C—C、C—O、C—N键)官能化将是今后的发展方向.此外, 大多数氧化偶联反应中都需要过量的过氧化试剂作为氧化剂, 这些试剂潜在的爆炸性限制了其大规模的工业应用.因此, 需要我们发展更绿色安全的氧化条件去实现氧化官能化, 比如目前发展迅速的电化学氧化和光催化氧化.除此之外, 更多类似于碘性质的非金属催化剂亟待开发(比如单质硫等), 其很有可能作为潜在的非金属催化剂应用氧化偶联反应中.
Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2013, 52, 2.
Uyanik, M.; Okamoto, H.; Yasui, T.; Ishihara, K. Science 2010, 328, 1376. doi: 10.1126/science.1188217
Zhang, J.; Zhu, D.; Yu, C.; Wan, C.; Wang, Z. Org. Lett. 2010, 12, 2841. doi: 10.1021/ol100954x
Chen, L.; Shi, E.; Liu, Z.; Chen, S.; Wei, W.; Li, H.; Xu, K.; Wan, X. Chem.-Eur. J. 2011, 17, 4085. doi: 10.1002/chem.201100192
Rodríguez, A.; Moran, W. J. Org. Lett. 2011, 13, 2220.
Wong, Y.-C.; Tseng, C.-T.; Kao, T.-T.; Yeh, Y.-C.; Shia, K.-S. Org. Lett. 2012, 14, 6024. doi: 10.1021/ol3028972
Li, L.-T.; Huang, J.; Li, H.-Y.; Wen, L.-J.; Wang, P.; Wang, B. Chem. Commun. 2012, 48, 5187. doi: 10.1039/c2cc31578e
Li, L.-T.; Li, H.-Y.; Xing, L.-J.; Wen, L.-J.; Wang, P.; Wang, B. Org. Biomol. Chem. 2012, 10, 9519. doi: 10.1039/c2ob26636a
Lu, L.; Xiong, Q.; Guo, S.; He, T.; Xu, F.; Gong, J.; Zhu, Z.; Cai, H. Tetrahedron 2015, 71, 3637. doi: 10.1016/j.tet.2014.11.069
Jia, Z.; Nagano, T.; Li, X.; Chan, A. S. C. Eur. J. Org. Chem. 2013, 858.
Nobuta, T.; Tada, N.; Fujiya, A.; Kariya, A.; Miura, T.; Itoh, A. Org. Lett. 2013, 15, 574. doi: 10.1021/ol303389t
Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K. R. Org. Lett. 2013, 15, 1092. doi: 10.1021/ol4001153
Zhang, C.; Liu, C.; Shao, Y.; Bao, X.; Wan, X. Chem.-Eur. J. 2013, 19, 17917. doi: 10.1002/chem.201303296
Huang, H.-M.; Li, Y.-J.; Ye, Q.; Yu, W.-B.; Han, L.; Jia, J.-H.; Gao, J.-R. J. Org. Chem. 2014, 79, 1084. doi: 10.1021/jo402540j
Tang, S.; Liu, K.; Long, Y.; Gao, X.; Gao, M.; Lei A. Org. Lett. 2015, 17, 2404. doi: 10.1021/acs.orglett.5b00912
Tang, S.; Liu, K.; Long, Y.; Qi, X.; Lan, Y.; Lei, A. Chem. Commun. 2015, 51, 8769. doi: 10.1039/C5CC01825K
Laha, J. K.; Jethava, K. P.; Patel, S.; Patel, K. V. J. Org. Chem. 2017, 82, 76. doi: 10.1021/acs.joc.6b02065
Shi, X.; Zhang, F.; Luo, W.-K.; Yang, L. Synlett 2016, 28, 494. doi: 10.1055/s-0036-1588331
Sudo, Y.; Yamaguchi, E.; Itoh, A. Org. Lett. 2017, 19, 1610. doi: 10.1021/acs.orglett.7b00428
Yu, Y.; Jiao, L.; Wang, J.; Wang, H.; Yu, C.; Hao, E.; Boens, N. Chem. Commun. 2017, 53, 581. doi: 10.1039/C6CC08098G
Dian, L.; Zhang-Negrerie, D.; Du, Y. Adv. Synth. Catal. 2017, 359, 3090. doi: 10.1002/adsc.201700521
Wei, W.; Zhang, C.; Xu, Y.; Wan, X. Chem. Commun. 2011, 47, 10827. doi: 10.1039/c1cc14602e
Uyanik, M.; Hayashi, H.; Ishihara, K. Science 2014, 345, 291. doi: 10.1126/science.1254976
Boominathan, S. S. K.; Hu, W.-P.; Senadi, G. C.; Vandavasi, J. K.; Wang, J.-J. Chem. Commun. 2014, 50, 6726. doi: 10.1039/C4CC02425G
Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331. doi: 10.1002/anie.201101522
Guo, S.; Yu, J.-T.; Dai, Q.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 6240. doi: 10.1039/c4cc01652a
Gao, W.-C.; Hu, F.; Huo, Y.-M.; Chang, H.-H.; Li, X.; Wei, W.-L. Org. Lett. 2015, 17, 3914. doi: 10.1021/acs.orglett.5b01933
Liang, Y.-F.; Li, X.; Wang, X.; Zou, M.; Tang, C.; Liang, Y.; Song, S.; Jiao, N. J. Am. Chem. Soc. 2016, 138, 12271. doi: 10.1021/jacs.6b07269
Zhang, S.; Guo, L.-N.; Wang, H.; Duan, X.-H. Org. Biomol. Chem. 2013, 11, 4308. doi: 10.1039/c3ob40748a
Yu, H.; Shen, J. Org. Lett. 2014, 16, 3204. doi: 10.1021/ol5012168
Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287. doi: 10.1002/adsc.201100920
Huang, J.; Li, L.-T.; Li, H.-Y.; Husan, E.; Wang P.; Wang, B. Chem. Commun. 2012, 48, 10204. doi: 10.1039/c2cc35450k
Liu, L.; Yun, L.; Wang, Z.; Fu, X.; Yan, C.-H. Tetrahedron Lett. 2013, 54, 5383. doi: 10.1016/j.tetlet.2013.07.114
Majji, G.; Guin, S.; Gogoi, A.; Rout, S. K.; Patel, B. K. Chem. Commun. 2013, 49, 3031. doi: 10.1039/c3cc40832a
Xu, J.; Zhang, P.; Li, X.; Gao, Y.; Wu, J.; Tang, G.; Zhao, Y. Adv. Synth. Catal. 2014, 356, 3331. doi: 10.1002/adsc.201400436
Xiong, B.; Wang, G.; Zhou, C.; Liu, Y.; Zhang, P.; Tang, K. J. Org. Chem. 2018, 83, 993. doi: 10.1021/acs.joc.7b02422
Shi, E.; Shao, Y.; Chen, S.; Hu, H.; Liu, Z.; Zhang, J.; Wan, X. Org. Lett. 2012, 14, 3384. doi: 10.1021/ol3013606
Froehr, T.; Sindlinger, C. P.; Kloeckner, U.; Finkbeiner, P.; Nachtsheim, B; J. Org. Lett. 2011, 13, 3754. doi: 10.1021/ol201439t
Cai, Z.-J.; Wang, S.-Y.; Ji, S.-J. Org. Lett. 2013, 15, 5226. doi: 10.1021/ol4023936
Beukeaw, D.; Udomsasporn, K.; Yotphan, S. J. Org. Chem. 2015, 80, 3447. doi: 10.1021/jo502933e
Yang, Z.-Y.; Tian, T.; Du, Y.-F.; Li, S.-Y.; Chu, C.-C.; Chen, L.-Y.; Li, D.; Liu, J.-Y.; Wang, B. Chem. Commun. 2017, 53, 8050. doi: 10.1039/C7CC03983B
Liu, Z.; Zhang, J.; Chen, S.; Shi, E.; Xu, Y.; Wan, X. Angew. Chem., Int. Ed. 2012, 51, 3231. doi: 10.1002/anie.v51.13
Xu, K.; Hu, Y.; Zhang, S.; Zha, Z.; Wang, Z. Chem.-Eur. J. 2012, 18, 9793. doi: 10.1002/chem.v18.32
Li, H.; Xie, J.; Xue, Q.; Cheng, Y.; Zhu, C. Tetrahedron Lett. 2012, 53, 6479. doi: 10.1016/j.tetlet.2012.09.039
Gao, L.; Tang, H.; Wang, Z. Chem. Commun. 2014, 50, 4085. doi: 10.1039/C4CC00621F
Tan, B.; Toda, N.; Barbas Ⅲ, C. F. Angew. Chem., Int. Ed. 2012, 51, 12538. doi: 10.1002/anie.201205921
Wang, G.; Yu, Q.-Y.; Chen, S.-Y.; Yu, X.-Q. Org. Biomol. Chem. 2014, 12, 414. doi: 10.1039/C3OB42037J
Zhao, J.; Li, P.; Xia, C.; Li, F. Chem. Commun. 2014, 50, 4751. doi: 10.1039/c4cc01587h
Zhang, X.; Wang, L. Green Chem. 2012, 14, 2141. doi: 10.1039/c2gc35489f
Wei, W.; Shao, Y.; Hu, H.; Zhang, F.; Zhang, C.; Xu, Y.; Wan, X. J. Org. Chem. 2012, 77, 7157. doi: 10.1021/jo301117b
Mai, W.-P.; Wang, H.-H.; Li, Z.-C.; Yuan, J.-W.; Xiao, Y.-M.; Yang, L.-R.; Mao, P.; Qu, L.-B. Chem. Commun. 2012, 48, 10117. doi: 10.1039/c2cc35279f
Zhao, Q.; Miao, T.; Zhang, X.; Zhou, W.; Wang, L. Org. Biomol. Chem. 2013, 11, 1867. doi: 10.1039/c3ob27433k
Lamani, M.; Prabhu, K. R. Chem.-Eur. J. 2012, 18, 14638. doi: 10.1002/chem.201202703
Tian, J.-S.; Jeffrey Ng, K. W.; Wong, J.-R.; Loh, T.-P. Angew. Chem., Int. Ed. 2012, 51, 9105. doi: 10.1002/anie.201204215
Ma, L.; Wang, X.; Yu, W.; Han, B. Chem. Commun. 2011, 47, 11333. doi: 10.1039/c1cc13568f
Xie, J.; Jiang, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2012, 48, 979. doi: 10.1039/C2CC15813B
Yan, Y.; Zhang, Y.; Zha, Z.; Wang, Z. Org. Lett. 2013, 15, 2274. doi: 10.1021/ol4008487
Yu, H.; Huang, W.; Zhang, F. Eur. J. Org. Chem. 2014, 3156.
Ilangovan, A.; Satish, G. J. Org. Chem. 2014, 79, 4984. doi: 10.1021/jo500550d
Yan, Y.; Zhang, Y.; Feng, C.; Zha, Z.; Wang, Z. Angew. Chem., Int. Ed. 2012, 51, 8077. doi: 10.1002/anie.v51.32
Lao, Z.-Q.; Zhong, W.-H.; Lou, Q.-H.; Li, Z.-J.; Meng, X.-B. Org. Biomol. Chem. 2012, 10, 7869. doi: 10.1039/c2ob26430g
Zheng, Y.; Mao, J.; Chen, J.; Rong, G.; Liu, D.; Yan, H.; Chi, Y.; Xu, X. RSC Adv. 2015, 5, 50113. doi: 10.1039/C5RA06773A
Yan, Y.; Xu, Y.; Niu, B.; Xie, H.; Liu, Y. J. Org. Chem. 2015, 80, 5581. doi: 10.1021/acs.joc.5b00474
Sun, K.; Wang, X.; Li, G.; Zhu, Z.; Jiang, Y.; Xiao, B. Chem. Commun. 2014, 50, 12880. doi: 10.1039/C4CC06003B
Rajamanickam, S.; Majji, G.; Santra, S. K.; Patel, B. K. Org. Lett. 2015, 17, 5586. doi: 10.1021/acs.orglett.5b02749
Yan, Y.; Li, Z.; Li, H.; Cui, C.; Shi, M.; Liu, Y. Org. Lett. 2017, 19, 6228. doi: 10.1021/acs.orglett.7b03171
Luo, Z.; Jiang, Z.; Jiang, W.; Lin, D. J. Org. Chem. 2018, 83, 3710. doi: 10.1021/acs.joc.8b00066
Xue, Q.; Xie, J.; Li, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2013, 49, 3700. doi: 10.1039/c3cc41558a
Zhao, D.; Wang, T.; Shen, Q.; Li, J.-X. Chem. Commun. 2014, 50, 4302. doi: 10.1039/C4CC01444H
Zhao, D.; Shen, Q.; Li, J.-X. Adv. Synth. Catal. 2015, 357, 339. doi: 10.1002/adsc.201400827
Huang, H.; Chen, W.; Xu, Y.; Li, J. Green Chem. 2015, 17, 4715. doi: 10.1039/C5GC01056J
Luo, W.-K.; Shi, X.; Zhou, W.; Yang, L. Org. Lett. 2016, 18, 2036. doi: 10.1021/acs.orglett.6b00646
Zhang, X.; Wang, M.; Li, P.; Wang, L. Chem. Commun. 2014, 50, 8006. doi: 10.1039/C4CC01189A
Tang, S.; Wu, Y.; Liao, W.; Bai, R.; Liu, C.; Lei, A. Chem. Commun. 2014, 50, 4496. doi: 10.1039/C4CC00644E
Xiao, F.; Chen, H.; Xie, H.; Chen, S.; Yang, L.; Deng, G.-J. Org. Lett. 2014, 16, 50. doi: 10.1021/ol402987u
Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2017, 82, 3084. doi: 10.1021/acs.joc.7b00073
Zhao, J.; Huang, H.; Wu, W.; Chen, H.; Jiang, H. Org. Lett. 2013, 15, 2604. doi: 10.1021/ol400773k
Liao, Y.; Jiang, P.; Chen, S.; Qi, H.; Deng, G.-J. Green Chem. 2013, 15, 3302. doi: 10.1039/c3gc41671b
Cao, H.; Yuan, J.; Liu, C.; Hu, X.; Lei, A. RSC Adv. 2015, 5, 41493. doi: 10.1039/C5RA04906G
Jiang, Y.; Zou, J.-X.; Huang, L.-T.; Peng, X.; Deng, J.-D.; Zhu, L.-Q.; Yang, Y.-H.; Feng, Y.-Y.; Zhang, X.-Y.; Wang, Z. Org. Biomol. Chem. 2018, 16, 1641. doi: 10.1039/C8OB00080H
Yuan, J.; Ma, X.; Yi, H.; Liu, C.; Lei, A. Chem. Commun. 2014, 50, 14386. doi: 10.1039/C4CC05661B
Xiao, F.; Chen, S.; Chen, Y.; Huang, H.; Deng, G.-J. Chem. Commun. 2015, 51, 652. doi: 10.1039/C4CC07546C
Li, X.; Xu, X.; Zhou, C. Chem. Commun. 2012, 48, 12240. doi: 10.1039/c2cc36960e
王倩, 徐洲, 有机化学, 2013, 33, 2430. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343474.shtmlWang, Q.; Xu, Z. Chin. J. Org. Chem. 2013, 33, 2430(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract343474.shtml
Sun, K.; Lv, Y.; Chen, Y.; Zhou, T.; Xing, Y.; Wang, X. Org. Biomol. Chem. 2017, 15, 4464. doi: 10.1039/C7OB00958E
Dhineshkumar, J.; Prabhu, K. R. Org. Lett. 2013, 15, 6062. doi: 10.1021/ol402956b
Wang, J.; Huang, X.; Ni, Z.; Wang, S.; Wu, J.; Pan, Y. Green Chem. 2015, 17, 314. doi: 10.1039/C4GC00944D
Yuan, J.; Liu, C.; Lei, A. Org. Chem. Front. 2015, 2, 677. doi: 10.1039/C5QO00027K
Zhang, J.; Jiang, J.; Li, Y.; Wan, X. J. Org. Chem. 2013, 78, 11366. doi: 10.1021/jo401915t
Yan, Y.; Niu, B.; Xu, K.; Yu, J.; Zhi, H.; Liu, Y. Adv. Synth. Catal. 2016, 358, 212. doi: 10.1002/adsc.201500619
Feng, J.-B.; Wu, X.-F. Org. Biomol. Chem. 2016, 14, 6951. doi: 10.1039/C6OB01301E

图式 1 低价碘催化氧化偶联的催化机理
Scheme 1 Catalytic mechanism for oxidative coupling by the low-valence iodine

图式 2 TBAI催化的δ-炔基β-酮酯的分子内氧化环化反应
Scheme 2 TBAI-catalyzed intramolecular oxidative cyclization reactions of δ-alkynyl β-ketoesters

图式 3 TBAI催化的α-氰基-α-炔基芳酮的分子内氧化环化反应
Scheme 3 TBAI-catalyzed intramolecular oxidative cyclization reactions of α-cyano-α-alkynyl aryl ketones

图式 4 TBAI催化的N-甲基苯胺作为酰化试剂的吲哚C(3)甲酰化反应
Scheme 4 TBAI-catalyzed C(3)-formylation of indoles with N-methylaniline as a formaylation reagent

图式 5 碘催化的N-芳基四氢异喹啉和碳亲核试剂的氧化偶联反应
Scheme 5 Iodine-catalyzed oxidative coupling of N-aryltetrahy-droisoquinolines with carbon nucleophile

图式 6 碘催化的四氢异喹啉和不饱和键的氧化环加成反应
Scheme 6 Iodine-catalyzed oxidative cycloaddition of tetrahydroisoquinolines with unsaturated bonds

图式 7 碘催化的β-酮酯(或2-吡啶酯)和烯烃的氧化环化反应
Scheme 7 Iodine-catalyzed oxidative annulation of β-keto esters or 2-pyridinyl-β-ester with alkenes

图式 8 TBAI催化的芳烃或杂芳烃分子内的氧化酰基化反应
Scheme 8 TBAI-catalyzed intramolecular oxidative acylation of arenes or heteroarenes

图式 9 碘催化的异喹啉和甲基芳烃的选择性氧化偶联反应
Scheme 9 Iodine-catalyzed oxidative coupling of isoquinolines with methylarenes

图式 10 TBAI催化的BODIPY和烯丙基烯烃(或醚)的氧化偶联反应
Scheme 10 TBAI-catalyzed oxidative coupling of BODIPY with allylic alkenes or ethers

图式 11 TBAI催化的醛和叔丁基过氧化氢的直接氧化偶联反应
Scheme 11 TBAI-catalyzed oxidative coupling of aldehydes with tert-butyl hydroperoxide

图式 12 碘化季铵盐催化的邻羰烷基苯酚的分子内氧化醚化反应
Scheme 12 Quaternary ammonium iodide-catalyzed intramolecular oxidative etherification of ketophenols

图式 13 碘催化的β-乙酰氨基苯酮的分子内氧化C—O键偶联反应
Scheme 13 Iodine-catalyzed intramolecular oxidative C—O coupling of β-acylamino ketones

图式 14 碘催化的环己酮氧化生成邻苯二酚
Scheme 14 Iodine-catalyzed conversion of simple cyclohexanones into catechols

图式 15 TBAI催化的醚的与氧相邻碳氢键和羧酸的氧化偶联反应
Scheme 15 TBAI-catalyzed oxidative coupling of C—H bond adjacent to oxygen in ethers with carboxylic acids

图式 16 TBAI催化的与氮原子相邻碳氢键和过氧叔丁醇的氧化偶联反应
Scheme 16 TBAI-catalyzed oxidative coupling of C—H bond adjacent to nitrogen atom with TBHP

图式 17 TBAI催化的烯丙基碳氢键和羧酸的氧化偶联反应
Scheme 17 TBAI-catalyzed oxidative coupling of allylic C—H bonds with carboxylic acids

图式 18 TBAI催化的苯并噁唑和羧酸的氧化偶联反应
Scheme 18 TBAI-catalyzed oxidative coupling of benzoxazoles with carboxylic acids

图式 19 碘催化的吲哚和唑的氧化偶联反应
Scheme 19 Iodine-catalyzed oxidative coupling of indoles with azoles

图式 20 TBAI催化的芳香醛和甲酰胺的氧化酰胺键形成反应
Scheme 20 TBAI-catalyzed oxidative formation of amides from aromatic aldehydes and formamides

图式 21 碘化物催化的醛和亲核试剂的氧化偶联反应
Scheme 21 Iodide-catalyzed intramolecular oxidative coupling of aldehydes with nucleophile

图式 22 碘催化的苯乙酮和胺的氧化偶联反应
Scheme 22 Iodine-catalyzed oxidative coupling of acetophenones with amines

图式 24 碘催化的脂肪醛和胺的氧化偶联反应
Scheme 24 Iodine-catalyzed oxidative coupling of aliphatic aldehydes with amines

图式 25 碘化钠催化的N, N-二甲基苯胺和磺酰胺的氧化偶联反应
Scheme 25 NaI-catalyzed oxidative coupling of N, N-dimethyl anilines with sulfamides

图式 26 碘催化的TMEDA和邻氨基酮(或邻氨基苯甲酰胺)的氧化偶联反应
Scheme 26 Iodine-catalyzed oxidative coupling of TMEDA with o-aminoketones or o-aminobenzamides

图式 27 碘催化的甲基芳烃和喹啉的氧化偶联反应
Scheme 27 Iodine-catalyzed oxidative coupling of methylarenes with quinolones

图式 28 碘化钾催化的取代乙烯和磺酰肼的氧化偶联反应
Scheme 28 KI-catalyzed oxidative coupling of ethylenes with sulfonohydrazides

图式 29 碘催化的环己酮和硫酚的需氧氧化偶联反应
Scheme 29 Iodine-catalyzed aerobic oxidative coupling of cyclohexanones with thioureas

图式 30 碘催化的环己酮和硫酚的需氧氧化偶联反应
Scheme 30 Iodine-catalyzed aerobic oxidative coupling of cyclohexanones with thiols

图式 31 碘催化的2-甲基喹啉和亚磺酸钠的氧化偶联反应
Scheme 31 Iodine-catalyzed oxidative coupling of 2-methyl-quinolines with sodium sulfinates

图式 32 TBAI催化的α-烷基苯乙烯和磺酰肼的氧化偶联反应
Scheme 32 TBAI-catalyzed oxidative coupling of α-alkylstyr-enes with sulfonylhydrazides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





























































 下载:
下载:
