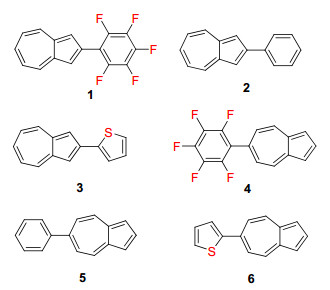
图式 1.
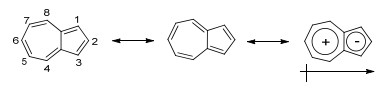
薁的共振结构
Scheme 1.
Numbering legend of azulene and its polarized resonance structure

薁(Azulene)又称甘菊蓝, 是一种青蓝色的具有特殊分子结构的化合物, 其成环原子具有10个π电子, 符合Hückel规则, 具有芳香性, 是典型的非苯芳香化合物(Scheme 1).薁是萘的同分异构体, 从分子结构上看, 薁是由带正电的环庚三烯与带负电的环戊二烯并合而成, 具有较大的分子偶极矩(约为1.08 D)[1].与萘相比, 薁具有非镜面对称的分子前线轨道(HOMO/LUMO), 具有较低的能隙, 这种独特的分子结构使薁表现出不同于其他传统芳香化合物的物理化学性质[2].薁的衍生物也存在于天然产物中, 表现出独特的生物活性[3], 并得到了相关应用, 例如薁类衍生物愈疮薁已应用于化妆品领域, 如防晒霜等[4]; 另外, 薁类衍生物还被用于眼睛和牙周炎的治疗[5].近年来, 薁及其衍生物因其独特的物理化学性质, 已经被应用于激光打印和静电复印[6], 分子开关[7], 液晶[8], 阴离子受体[9], 非线性光学材料[10]以及近红外材料[11]等研究.最近, 本课题组发展了一类基于薁的芳香二酰亚胺类化合物——联薁二酰亚胺(BAzDI), 并将其应用于有机场效应晶体管(OFET)与有机太阳能电池(OPV)研究[12].
薁的衍生物具有特殊的物理化学性质[13], 但由于该类化合物合成难度大, 大大限制了薁基功能分子材料的发展[13].薁类化合物的研究多局限于薁的1/3-位衍生物和4/7-位衍生物[14], 其2/6-位化学衍生物由于其合成难度较大, 故对于薁2/6-位化学衍生物研究报道相对较少[4, 15].薁的2/6-位化学衍生物具有C2V轴对称性, 相对于其他位置的衍生物, 具有更大的分子偶极矩和极化率, 易于形成强的分子间π-π相互作用, 有望应用于OFET和OPV等有机光电子领域[16].基于此, 我们合成了一系列薁2/6-位芳基取代的模型化合物1~6(图 1), 并对其吸收光谱、电化学、荧光光谱以及质子响应等物理化学性质进行了研究, 以期获得一些规律和认识, 为薁基功能分子的理性设计提供研究思路.
化合物1~3是薁2-位衍生物, 其取代基分别为五氟苯、苯和α-噻吩; 化合物4~6是薁6-位衍生物, 其取代基分别为五氟苯、苯和α-噻吩.化合物1~3的合成路线如Scheme 2所示.我们参照文献的合成方法[12, 17], 以环庚三烯酚酮(S1)为起始原料, 与氯化亚砜反应得到2-氯-2, 4, 6-环庚三烯-1-酮(S2), S2与1, 3-丙酮二羧酸二乙酯反应得到2-羟基-1, 3-薁二羧酸二乙酯(S3), S3脱去酯基得到2-羟基薁(S4), S4进一步与三溴化磷反应得到2-溴薁(S5), S5与联频哪醇硼酸酯反应得到2-硼酸酯薁(S6), 然后S6分别与五氟碘苯、碘苯和2-碘噻吩通过Suzuki-Miyaura偶联反应得到目标化合物1~3.
化合物4~6的合成路线如Scheme 2所示.我们参照文献的合成方法[8, 17], S2与氰基乙酸乙酯反应得到2-氨基-1, 3-薁二羧酸二乙酯(S7), S7进一步与液溴反应得到2-氨基-6-溴-1, 3-薁二羧酸二乙酯(S8), S8与联频哪醇硼酸酯在Pd催化下合成2-氨基-6-硼酸频哪醇酯-1, 3-薁二羧酸二乙酯(S9), 然后S9分别与五氟碘苯、碘苯和2-碘噻吩通过Suzuki-Miyaura偶联反应得到关键中间体2-氨基-6-五氟苯基-1, 3-薁二羧酸二乙酯(S10)、2-氨基-6-苯基-1, 3-薁二羧酸二乙酯(S11)和2-氨基-6-噻吩基-1, 3-薁二羧酸二乙酯(S12), 最后S10~S12进一步脱去氨基与酯基[12, 17], 得到目标化合物4~6.
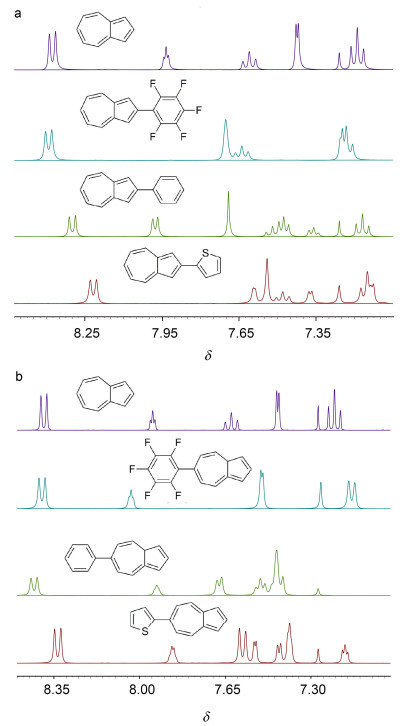
我们采用1H NMR、13C NMR和HRMS等手段表征确定了所合成的中间体以及目标化合物1~6的化学结构. 图 2是化合物1~6和薁的1H NMR图.化合物1~6和薁的1, 3-位化学位移分别为δ 7.70, 7.68, 7.54, 7.50, 7.43, 7.38和7.42; 4, 8-位化学位移分别为δ 8.39, 8.29, 8.22, 8.39, 8.42, 8.32和8.38; 5, 7-位的化学位移分别为δ 7.23, 7.16, 7.15, 7.13, 7.66, 7.57和7.19.研究发现, 在薁2/6-位引入给电子基团噻吩, 其化学位移向高场移动, 在薁2/6-位引入拉电子基团五氟苯, 其4, 5, 7, 8-位化学位移均向低场移动.
我们对化合物1~6的紫外可见吸收光谱进行了研究(图 3a).化合物1~6的紫外可见吸收光谱主要分为三个部分: 450 nm<λ<750 nm的吸收(分子S0→S1的跃迁吸收), 330 nm<λ<450 nm的吸收(分子S0→S2的跃迁吸收)和λ<330 nm的吸收(分子S0→S3或更高能级的跃迁吸收).
化合物1~6在450 nm<λ<750 nm区域中出现一个S0→S1的微弱吸收峰(图 3a), 与薁具有同样的末端吸收特征, 这是由于化合物1~6具有非镜面对称的分子前线轨道导致的[2].化合物1~6在330 nm<λ<450 nm出现较强的S0→S2吸收峰, 化合物1~6的吸收峰分别为374, 390, 407, 345, 368和387 nm, 相对于薁的吸收峰(339 nm), 分别红移了35, 51, 68, 5, 29和48 nm (图 3a, 表 1), 这是由于在薁2/6-位引入共轭基团, 分子π共轭体系增大, 电子离域范围增大造成的.在薁2/6-位引入供电子取代基团, 比引入相应的拉电子取代基团吸收光谱红移更加明显.薁单元1/3-位化学衍生物表现出同样的现象[18].对于相同的取代基, 在薁2-位引入相对于在薁6-位引入, 其吸收光谱会发生更明显的红移(1 vs 4, 2 vs 5和3 vs 6分别红移了30, 22和20 nm).化合物1~6在λ<330 nm的区域内表现出强的S0→S3或更高能级的跃迁吸收峰, 化合物1~6的吸收峰分别为292, 307, 315, 284, 298和322 nm, 相对于薁的吸收峰(275 nm), 分别红移17, 32, 40, 9, 23和47 nm(图 3a).
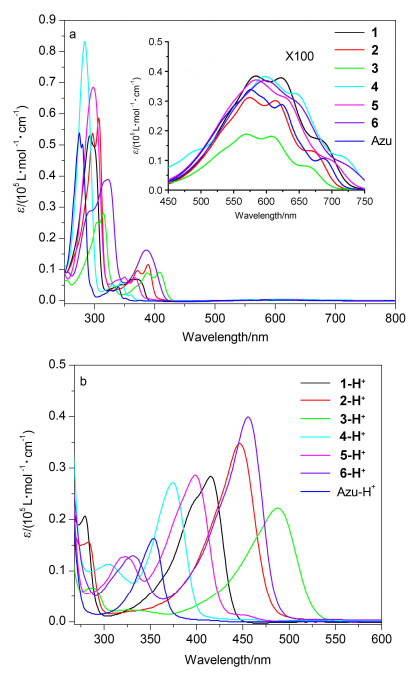
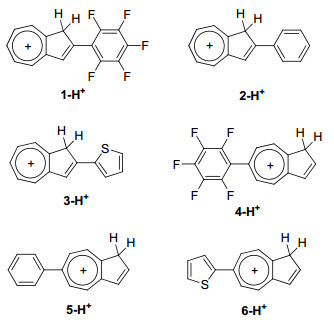
薁及其衍生物可以通过加入三氟乙酸(TFA)发生质子化, 从而改变其物理化学性质.质子化之后, 薁300 nm<λ<400 nm范围的吸收峰为S0→S1的跃迁[15b].加入30%的TFA对化合物1~6进行质子化, 然后对其吸收光谱进行测试(图 3b).结果表明, 化合物1~6质子化之后, 其吸收光谱相对于中性化合物均发生明显变化.这是由于薁单元质子化之后, 其富电子五元环形成环戊二烯, 其七元环形成一个稳定的环庚三烯阳离子(图 4)[14].化合物1-H+~6-H+最大吸收峰分别为415, 446, 488, 375, 399和456 nm, 相对于薁质子化的最大吸收峰354 nm, 分别红移了61, 92, 134, 21, 45和102 nm (图 3b, 表 1).质子化后, 化合物1-H+~6-H+的吸收峰变宽, 摩尔消光系数相对于Azulene-H+ (16305 L•mol-1•cm-1)有所增大(分别为28429, 34761, 22242, 27197, 28605和39936 L•mol-1•cm-1).以上结果表明, 化合物1-H+~6-H+的紫外可见吸收光谱在350~530 nm范围内, 归结于分子S0→S1的跃迁(表 1).结果表明, 质子化后, 在薁2/6-位引入供电子基团时相对于引入拉电子基团发生更大的红移(>100 nm), 另外, 在2-位引入共轭基团比6-位引入共轭基团发生更明显的红移.
 下载:
导出CSV
下载:
导出CSV
| Compd. | Unprotonated | Protonateda | |||||
| λmax(abs)/nm | λmax(em)/nm | ϕF | λmax(abs)/nm | λmax(em)/nm | ϕF | ||
| Azu | 340 | 375 | 0.054c | 354 | 413b | 0.005c | |
| 1 | 374 | 408b | 0.006c | 415 | 451 | 0.359c | |
| 2 | 390 | 424b | 0.003c | 446 | 503 | 0.095d | |
| 3 | 407 | 447b | 0.002c | 488 | 554b | 0.004d | |
| 4 | 345 | 386 | 0.082c | 375 | 421b | 0.002c | |
| 5 | 368 | 408 | 0.026c | 399 | 447b | 0.002c | |
| 6 | 387 | 435 | 0.001c | 456 | 500 | 0.029d | |
| a Treated with TFA; b extremely weak emission; c 9, 10-diphenylanthracene as standard subsure to measure; d rhodamine B as standand subsure to measure. | |||||||
薁是一种符合反Kasha规则的弱荧光物质, 对于薁的荧光测试, 通常测试薁S2→S0的光子辐射产生的荧光光谱[15].我们测试了化合物1~6及其质子化的化合物1-H+~6-H+的荧光光谱(图 5), 并采用等吸收点激发-荧光发射峰高对比法计算其荧光量子产率[19](表 1).结果表明, 化合物1~6和Azulene的荧光量子产率分别为0.006, 0.003, 0.002, 0.082, 0.026, 0.001和0.054, 化合物4相对于其他化合物显示出较强的荧光强度(ϕF=0.082).化合物1~6的荧光光谱表明, 在薁2/6-位引入不同取代基团通常会导致其荧光减弱, 这是由于共轭基团的引入导致分子刚性减弱, 分子振动增加, 进而导致荧光减弱[20].化合物4的荧光强度相对于薁增强, 原因可能是五氟苯中氟原子与薁七元环上5/7-位氢原子之间形成氢键, 分子刚性增强, 分子振动减弱, 荧光增强[20].加入TFA进行质子化后, 化合物1-H+~6-H+和Azulene-H+的荧光量子产率分别为0.359, 0.095, 0.004, 0.002, 0.002, 0.029和0.005, 化合物1-H+、2-H+和6-H+的荧光相对于Azu-H+(Y=0.005)有所增强(ϕF=0.359, 0.095和0.029), 其中化合物1-H+显示出最大的荧光量子产率(ϕF=0.359).化合物1-H+的荧光强度相对于中性化合物显著增强, 原因可能是存在较强的F—H氢键相互作用, 导致薁骨架与苯骨架存在一定夹角, 降低了分子内薁骨架与苯骨架之间的共轭效应, 有效地阻碍了分子内电荷复合(charge recombination)的过程, 从而造成了化合物1-H+的荧光量子产率大幅提升.

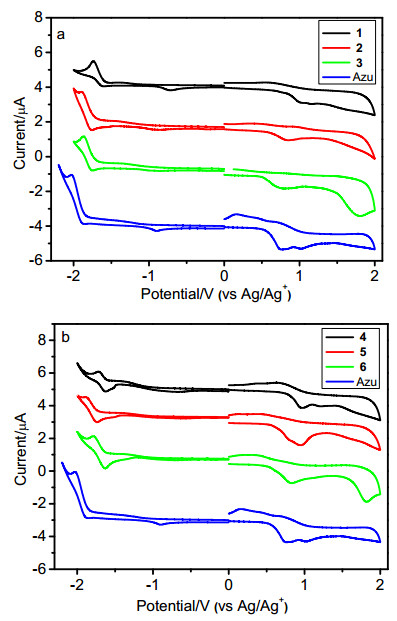
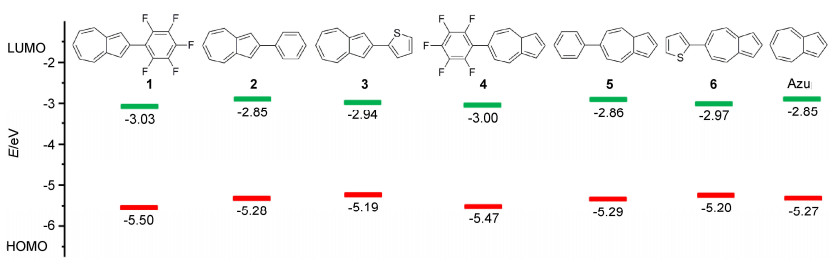
为了研究不同取代基团对分子能级的影响, 我们采用循环伏安法研究了化合物1~6在二氯甲烷溶液中的电化学性质. 图 6给出化合物1~6和薁的循环伏安曲线.如图 6a所示, 化合物1~3在正向和负向均表现出准可逆的氧化还原行为, 其起始还原电位(Eredonset)分别为-1.65, -1.82和-1.74 V, 其起始氧化电位(Eoxonset)分别为0.82, 0.60和0.51 V.由公式HOMO=-4.68-Eoxonset(经二茂铁校正的)计算出化合物1~3的HOMO能级分别为-5.50, -5.28和-5.19 eV; 由公式LUMO=-4.68-Eredonset(经二茂铁校正的)计算出化合物1~3的LUMO能级分别为-3.03, -2.86和-2.94 eV (图 7).如图 6b所示, 化合物4~6同样在正向和负向均表现出准可逆的氧化还原行为, 其起始还原电位(Eredonset)分别为-1.68, -1.82和-1.71 V, 其起始氧化电位(Eoxonset)分别为0.79, 0.61和0.52 V.由公式HOMO=-4.68-Eoxonset(经二茂铁校正的)计算出化合物4~6的HOMO能级分别为-5.47, -5.29和-5.20 eV; 由公式LUMO=-4.68-Eredonset(经二茂铁校正的)计算出化合物4~6的LUMO能级分别为-3.00, -2.86和-2.97 eV (图 7).薁的起始氧化还原电位分别为Eoxonset=0.59 V和Eredonset=-1.83 V(图 6), 计算出薁的HOMO和LUMO能级分别为-5.27 eV和-2.85 eV (图 7).电化学研究表明化合物1~6具有接受电子与给出电子的特性.化合物1~6与薁相比, 其HOMO能级变化分别为-0.23, -0.01, +0.08, -0.20, -0.02和+0.07 eV; 其LUMO能级变化分别为-0.18, 0, -0.09, -0.15, -0.01和-0.12 eV(图 7).化合物1和4, 2和5, 3和6具有两两相似的HOMO能级和LUMO能级, 表明在薁2/6-位引入同一共轭基团均可以有效调控分子的能级.整体来说, 在薁2/6-位引入共轭基团会导致分子的LUMO降低; 在薁2/6-位引入拉电子基团会导致分子HOMO有效降低(ΔEHOMO≈-0.2 eV), 而当引入给电子基团会导致分子HOMO能级略有升高.
0.1 mol/L Bu4+NPE6- as supporting electrolyte, Ag/Ag+ as reference electrode, scan rate of 100 mV/s
为了进一步理解不同取代基团对分子电子能级结构的影响, 我们采用密度泛函理论(DFT)在B3LYP/ 6-311+G(d, p)水平上对分子结构进行了优化, 计算了HOMO和LUMO能级, 并绘制了分子前线轨道轮廓图.在优化出的基态几何结构基础上, 进一步采用含时密度泛函理论(TDDFT)方法, 在B3LYP/6-311+G(d, p)水平上进行激发态计算, 拟合吸收光谱信息.
图 8给出了化合物1~6和薁及其质子化后化合物的分子结构.从图中可以看出, 化合物1~6总体表现出非平面的分子构型.在薁6-位引入共轭基团的分子扭转角大于在薁2-位引入同一共轭基团的分子扭转角, 削弱了分子内电荷转移行为[20], 这在一定程度上解释了相同取代基薁2-位衍生物吸收光谱相对于6-位衍生物发生明显红移; 在薁2/6-位引入噻吩基团的分子构型扭转角小于在薁2/6-位引入五氟苯基团的分子构型扭转角, 这与化合物1~6的紫外可见吸收光谱变化规律一致.质子化后, 化合物1-H+~3-H+表现出共平面的分子构型, 化合物4-H+~6-H+相对于中性化合物平面性变好[21], 这在一定程度上解释了化合物1-H+~3-H+的荧光强度比化合物1~3增强, 而化合物4-H+~6-H+的扭转角相对于中性分子变化较小, 质子化之后荧光强度减弱.
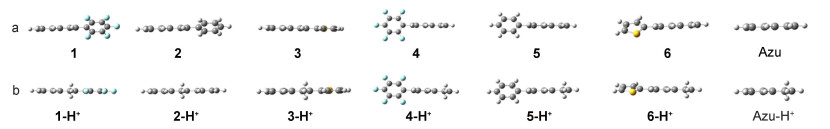
图 9列出了化合物1~6和薁及其质子化的衍生物1-H+~6-H+的分子前线轨道轮廓图与其对应的HOMO和LUMO能级.结果表明, 化合物1和4、2和5、3和6具有两两相似的电子云分布.对于HOMO轨道, 电子云集中分布在薁单元上; 而对于LUMO轨道, 电子云均匀分布在整个分子上.化合物1~6与薁相比, 其HOMO能级变化分别为-0.35, -0.03, -0.07, -0.31, +0.04和0 eV; 其LUMO能级变化分别为-0.52, -0.10, -0.16, -0.46, -0.08和-0.22 eV. DFT计算结果显示, 化合物1和4、2和5、3和6具有两两相近的HOMO和LUMO能级, 在薁2/6-位引入共轭基团会导致分子LUMO能级相对于薁降低, 其中引入拉电子基团大大降低分子LUMO能级(ΔELUMO≤-0.46 eV).在薁2/6-位引入拉电子基团同样会导致其HOMO能级有效降低(ΔEHOMO≤-0.31 eV), 而当引入给电子基团时对于分子HOMO能级变化不大.理论计算所得规律与实验结果一致.质子化后, 化合物1-H+~6-H+和Azulene-H+电子云表现出均匀分布的特点.在HOMO轨道上, 除了化合物4-H+电子云分布在五氟苯单元上, 其他化合物电子云均呈现均匀分布的特点; 而在LUMO轨道上, 所有化合物电子云均匀分布在整个分子上.化合物1-H+~6-H+与Azulene-H+相比, 其HOMO能级变化分别为+1.07, +1.61, +1.81, +0.86, +1.28和+1.52 eV; 其LUMO能级变化分别为+0.07, +0.46, +0.50, +0.01, +0.46和+0.50 eV. DFT计算结果显示, 化合物1-H+与4-H+、2-H+与5-H+、3-H+与6-H+具有两两相似的HOMO和LUMO能级, 在薁2/6-位引入共轭基团会导致其质子化合物的HOMO与LUMO能级升高; 在薁2/6-位引入拉电子基团对其质子化产物的HOMO与LUMO能级影响较小, 而当引入给电子基团使其质子化衍生物的HOMO能级与LUMO能级显著升高(ΔELUMO≈1.2 eV, ΔELUMO≈0.5 eV).
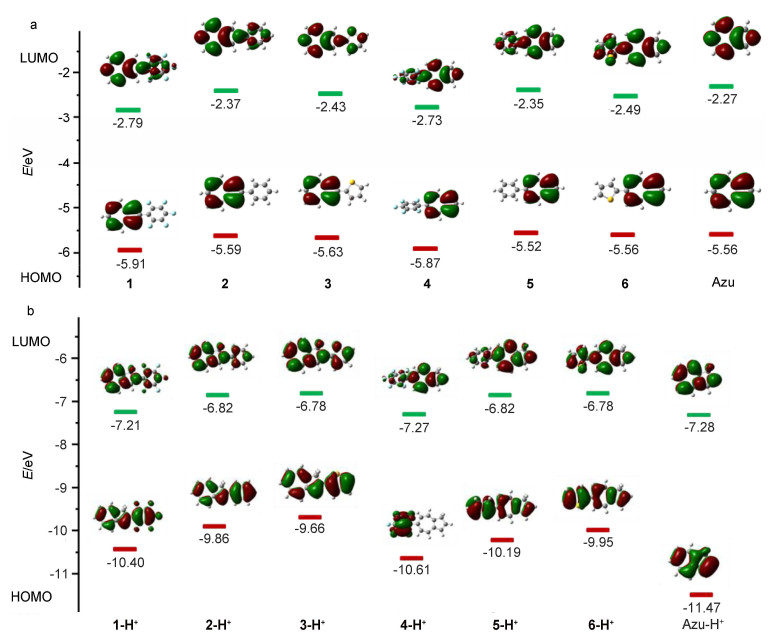
图 10a给出了化合物1~6和薁在TDDFT/B3LYP/ 6-311+G(d, p)水平上计算的吸收光谱.从图中可以看出, 吸收光谱同样分布在三个区域: 440 nm<λ<650 nm (S0→S1跃迁), 340 nm<λ<440 nm (S0→S2跃迁)和200 nm<λ<340 nm (S0→S3或更高能级跃迁).
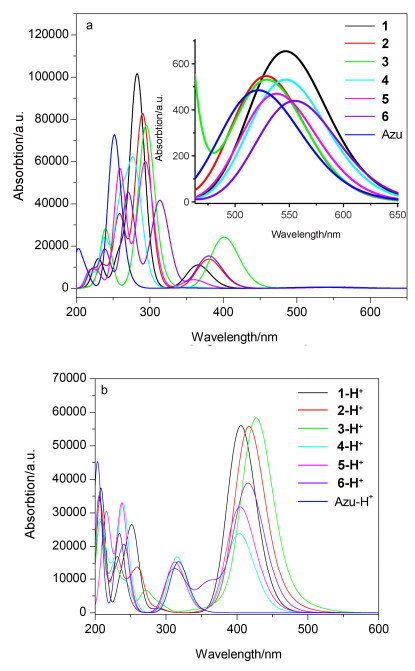
化合物1~6在440 nm<λ<650 nm的区域中均出现一个微弱的吸收峰(图 10a), 与薁具有同样的特征吸收, 这是由于化合物1~6具有非镜面对称的分子前线轨道导致的[2].化合物1~6在340 nm<λ<440 nm的区域具有较强的吸收, 吸收峰分别为365, 380, 401, 354, 359和380 nm, 相对于薁的吸收峰(344 nm), 分别红移了21, 36, 57, 10, 15和36 nm(图 10a), 与实验测得的吸收光谱变化规律相一致.这是由于在薁2/6-位引入共轭基团, 分子π共轭体系增大, 电子离域范围增大, 从而吸收光谱发生红移.结果表明, 在2/6-位引入供电子取代基团相比于拉电子取代基团, 其吸收光谱红移更加明显(可达57 nm).对于相同取代基, 在薁2-位引入相对于在薁6-位引入, 其吸收光谱会发生更明显的红移(1 vs 4, 2 vs 5, 3 vs 6分别红移了11 nm, 21 nm和21 nm), 与实验所得规律一致.化合物1~6在200 nm<λ<340 nm的区域内表现出强的吸收, 其吸收峰分别为282, 290, 295, 276, 293和313 nm, 相对于薁的吸收峰251 nm, 分别红移31, 39, 44, 25, 42和62 nm(图 10a).这同样与实验所得规律一致(图 10a).
图 10b给出了化合物1-H+~6-H+及Azulene-H+在TDDFT/B3LYP/6-311+G(d, p)水平上计算的吸收光谱.从图中可以看出, 质子化后在310 nm<λ<600 nm区域属于分子S0→S1的跃迁吸收峰[14].化合物1-H+~6-H+的最大吸收峰分别为405, 416, 427, 403, 404和415 nm, 相对于Azu-H+的最大吸收峰317 nm, 分别红移了88, 99, 110, 86, 87和98 nm(图 10b).结果表明在薁2/6-位引入共轭基团, 质子化后化合物1-H+~6-H+相对于Azu-H+发生明显的红移(Δλ=86~110 nm), 这与实验结果相一致.
我们设计合成了薁2/6-位芳基取代的六个模型化合物1~6, 并通过紫外吸收光谱、荧光光谱、电化学以及质子响应探究了薁2/6-位不同芳基取代对于分子物理化学性质的影响.紫外吸收光谱表明在薁2/6-位引入不同取代基团均可以使其S0→S2吸收峰相对于薁红移, 引入给电子噻吩基团红移现象更加明显(Δλ=68/48 nm), 有望通过在薁单元上引入给电子共轭基团拓展其光谱吸收.荧光光谱表明在薁单元6-位引入强拉电子基团五氟苯, 得到化合物4相对于其他化合物的荧光有明显增强(ϕF=0.082);质子化后, 同样引入五氟苯基化合物1-H+的荧光强度最强(ϕF=0.359).电化学表明, 在薁2/6-位引入不同取代芳基均具有较好的得失电子能力, 引入推拉电子基团均可以降低其LUMO能级, 其中在2/6-位引入五氟苯基可显著降低分子LUMO能级(1和4的ΔELUMO分别为-0.18和-0.15 eV); 引入拉电子基团同样可以降低其HOMO能级(1和4的ΔEHOMO分别为-0.23和-0.20 eV), 有望通过引入不同拉电子基团获得较低LUMO能级的功能材料分子.理论计算同样表明, 在薁单元2/6-位引入不同取代基团可以调控分子的能级, 在薁2/6-位引入共轭基团会导致LUMO能级降低, 在薁2/6-位引入拉电子基团会导致其HOMO/LUMO能级显著降低, 引入给电子基团对于分子HOMO能级影响不大.对于质子化后的化合物, 在薁2/6-位引入共轭基团会导致其HOMO与LUMO能级升高, 在薁2/6-位引入拉电子基团对其HOMO能级与LUMO能级影响不大, 而引入给电子基团使衍生物的HOMO能级与LUMO能级显著升高.这些研究结果为基于薁的有机功能分子的设计合成及性质研究提供了有效依据.
核磁共振(1H NMR, 13C NMR)由Bruker DRX 400, Varian Mercury 300或JEOL 400核磁共振波谱仪测定, 采用CDCl3或acetone-d6作为溶剂, TMS为内标.采用密度泛函理论(DFT)方法, 在B3LYP/6-31G(d, p)水平上进行分子结构优化和前线轨道能级进行计算.采用含时密度泛函理论(TDDFT)方法, 在B3LYP/6-311+G(d, p)水平上进行激发态计算, 拟合吸收光谱信息, 所有理论计算部分由Gaussian 09软件包完成.紫外-可见光谱(UV-Vis)由U-2910紫外可见光谱仪测得.荧光光谱由F-2700荧光分光光度计测得.电化学测试(CV)在计算机控制的CHI810D电化学分析仪上进行, 采用传统的三电极测试体系, 使用铂电极为工作电极, Ag/Ag+作为参比电极, 铂丝作为对电极, 样品溶于新蒸的二氯甲烷(浓度为1×10-3 mol/L), Bu4N+PF6- (0.1 mol/L)作为支持电解质, 扫描速度为100 mV/s.熔点在上海申光SGW X-4显微熔点仪测试.
参考文献[17a].将环庚三烯酚酮(S1) (1 g, 8.19 mmol)溶于25 mL甲苯中, 然后加入二氯亚砜(1.07 g, 9 mmol), 有白色固体析出, 将反应温度升至120 ℃回流1.5 h, 白色固体消失, 体系变成深红色, 减压旋掉过量的二氯亚砜和甲苯得棕色固体, 柱层析分离[V(石油醚):V(乙酸乙酯)=5:1]得白色固体1.03 g, 产率90%. m.p. 63~64 ℃ (lit.[17a] m.p. 64~65 ℃); 1H NMR (400 MHz, acetone-d6) δ: 7.90 (d, J=9.3 Hz, 1H), 7.34 (dd, J=12.1, 8.3 Hz, 1H), 7.19 (dd, J=10.6, 8.3Hz, 1H), 7.14~7.01 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ: 179.23, 148.12, 138.19, 135.97, 135.65, 134.38, 131.51.
参考文献[17b].将Na (4 g, 174 mmol)溶解于50 mL无水乙醇中得无色溶液, 1, 3-丙酮二羧酸二乙酯(4.34 g, 21.5 mmol)加入上述溶液中, 溶液变成乳白色. S2 (2.0 g, 14.0 mmol)溶于6 mL无水乙醇中, 快速加入到上述溶液, 体系变成砖红色, 体系在室温下搅拌反应12 h, 加入150 mL水, 有橙色固体析出, 过滤, 将所得的橙色固体溶于25 mL冰乙酸中, 加入70 mL水稀释, 用CHCl3萃取三次, 合并有机相, 浓缩后柱层析[[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体2.45 g, 产率58%. m.p. 93~94 ℃ (lit.[17b] m.p. 95~96 ℃); 1H NMR (400 MHz, CDCl3) δ: 11.76 (s, 1H), 9.36 (d, J=6.1 Hz, 2H), 7.84~7.52 (m, 3H), 4.51 (q, J=7.0 Hz, 4H), 1.48 (t, J=7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 172.19, 136.12, 134.65, 132.40, 60.50, 14.55.
参考文献[17e].将24 g五氧化二磷溶解于35 mL 85%磷酸中制得100%磷酸, 化合物S3 (2.3 g, 8.0 mmol)加入到上述新制的100%磷酸中, 玻璃棒搅拌下升温至130 ℃反应20 min, 反应液冷却至室温后倒入冰水, 用甲苯萃取三次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=10:1]分离得红色固体880 mg, 产率76%. m.p. 110~111 ℃ (lit.[17c] m.p. 115.5~116.5 ℃); 1H NMR (400MHz, acetone-d6) δ: 9.69 (s, 1H), 8.03 (d, J=10.0 Hz, 2H), 7.36 (t, J=9.8 Hz, 1H), 7.15 (dd, J=10.0, 9.8 Hz, 2H), 6.76 (s, 2H); 13C NMR (100 MHz, acetone-d6) δ: 167.64, 140.61, 131.49, 130.67, 123.98, 103.19.
参考文献[17e].化合物S4 (144 mg, 1.0 mmol)溶解于30 mL甲苯中, 加入三溴化磷(836 mg, 3.0 mmol), 溶液变成棕色, 有绿色固体析出, 升温至90 ℃搅拌反应2 h, 反应液冷却至室温后往体系中加入少量水中, 用甲苯萃取三次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=20:1]分离得紫色固体147 mg, 产率71%. m.p. 100~101 ℃ (lit.[17d]m.p. 104.2~104.8 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=9.6 Hz, 2H), 7.63 (t, J=9.9 Hz, 1H), 7.35 (s, 2H), 7.23 (dd, J=9.6, 9.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 139.84, 137.27, 135.24, 127.48, 124.55, 118.75.
参考文献[17d].向50 mL反应管中依次加入化合物S5 (411 mg, 2.0 mmol), Pd(dppf)2Cl2 (73 mg, 5 mol%), 联片呐醇硼酸酯(559 mg, 2.2 mmol), KOAc (588 mg, 3 mmol), 抽换N2三次, 在氮气保护下加入20 mL超干的DMSO, 然后反应体系升温至80 ℃搅拌反应6 h, 反应结束后将反应液倒入10% NH4Cl溶液中, 过滤, 所得固体柱层析[V(石油醚):V(二氯甲烷)=1:1]分离得蓝色固体260 mg, 产率51%. m.p. 88~91 ℃ (lit.[17d] m.p. 99~101 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.35 (d, J=9.3 Hz, 2H), 7.77 (s, 2H), 7.58 (t, J=9.9 Hz, 1H), 7.12 (dd, J=9.3, 9.9 Hz, 2H), 1.40 (s, 12H); 13C NMR (100 MHz, CDCl3) δ: 140.77, 138.89, 138.31, 125.16, 122.85, 83.84, 25.12, 25.04.
向25 mL反应管中依次加入化合物S6 (36 mg, 0.14 mmol), Pd(PPh3)4 (8 mg, 5 mmol%), Ag2CO3 (97 mg, 0.35 mmol), 抽换N2三次, 在氮气保护下加入五氟碘苯(45 mg, 0.21 mmol), 2 mL重蒸的THF, 然后升温至70 ℃, 搅拌回流24 h, 反应结束后体系倒入水中, 二氯甲烷萃取, 干燥, 浓缩柱层析(石油醚), 分离得蓝色固体36 mg, 产率87%. m.p. 202~205 ℃; 1H NMR (400 MHz, acetone-d6) δ: 8.60 (d, J=9.5 Hz, 2H), 7.96~7.73 (m, 3H), 7.40 (dd, J=9.5, 9.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 139.92, 138.66, 138.17, 124.12, 118.53; 19F NMR (376 MHz, CDCl3) δ: -142.98 (dd, J=22.2, 6.6 Hz, 2F), -157.86 (t, J=20.1 Hz, 1F), -164.40~-165.15 (m, 2F). HRMS (EI+) calcd for C16H7F5 294.0468, found 294.0475.
向25 mL反应管中依次加入化合物S6 (51 mg, 0.2 mmol), Pd(PPh3)4 (11 mg, 5 mmol%), Ag2CO3 (137 mg, 0.5 mmol), 抽换N2三次, 在氮气保护下加入碘苯(61 mg, 0.3 mmol), 2 mL重蒸的THF, 然后升温至70 ℃, 反应条件同化合物1, 得蓝色固体34 mg, 产率83%. m.p. 231~232 ℃ (lit.[22a] m.p. 228~230 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J=9.5 Hz, 2H), 7.97 (d, J=7.5 Hz, 2H), 7.68 (s, 2H), 7.58~7.42 (m, 3H), 7.35 (t, J=7.2 Hz, 2H), 7.16 (t, J=9.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 150.00, 141.37, 136.52, 136.07, 129.02, 128.33, 127.73, 123.84, 114.54. HRMS (EI+) calcd for C16H12 204.0939, found 204.0940.
向25 mL反应管中依次加入化合物S6 (51 mg, 0.2 mmol), Pd(PPh3)4 (11 mg, 5 mmol%), Ag2CO3 (137 mg, 0.5 mmol), 抽换N2三次, 在氮气保护下加入2-碘噻吩(62 mg, 0.3 mmol), 3 mL重蒸的THF, 然后升温至70 ℃, 反应条件同化合物1, 得到蓝色固体36 mg, 产率85%. 167~168 ℃ (lit.[22b] m.p. 143~144 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.22 (d, J=9.5 Hz, 2H), 7.59 (d, J=2.4 Hz, 1H), 7.54 (s, 2H), 7.48 (t, J=9.8 Hz, 2H), 7.37 (d, J=4.4 Hz, 1H), 7.13~7.18 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 143.35, 141.39, 141.02, 136.16, 135.52, 128.45, 126.58, 125.72, 124.21, 114.06. HRMS (EI+) calcd for C16H10S 210.0503, found 210.0509.
参考文献[17c].将Na (0.325 g, 14.1 mmol)溶解于15 mL无水乙醇中得无色溶液, 化合物氰基乙酸乙酯(1.78 g, 1.57 mmol)加入上述溶液中, S2 (1.0 g, 7.14 mmol)溶于5 mL无水乙醇中, 冰水浴冷却下滴加到上述溶液中并保持体系温度低于5 ℃, 室温下搅拌反应4 h, 反应结束后加入60 mL水, 用CHCl3萃取三次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体1.23 g, 产率61%. m.p. 91~92 ℃ (lit.[22e] m.p. 93~94 ℃); 1H NMR (300 MHz, CDCl3) δ: 9.15 (d, J=10.3 Hz, 2H), 7.80 (s, 2H), 7.56 (dd, J=10.3, 9.4 Hz, 2H), 7.44 (t, J=9.4 Hz, 1H), 4.47 (q, J=7.1 Hz, 4H), 1.48 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.52, 162.41, 146.05, 132.83, 132.58, 131.34, 99.68, 59.81, 14.65.
参考文献[17d].化合物S7 (2.87 g, 10.0 mmol)溶于70 mL氯仿中, 冰盐浴冷却下滴加稀释于10 mL氯仿中的Br2 (1.61 g, 10.1 mmol)到上述溶液中并保持体系温度低于0 ℃, 滴加结束后在冰浴下反应20 min, 撤去冰浴, 自然升至室温搅拌反应30 min, 反应结束后向体系中加入200 mL水, 用CHCl3萃取三次, 合并有机相, 旋干后柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体3.6 g, 产率98%. m.p. 158~159 ℃ (lit.[22c] m.p. 160~161 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J=11.7 Hz, 2H), 7.78 (s, 2H), 7.77 (d, J=11.7 Hz, 2H), 4.44 (q, J=7.1 Hz, 4H), 1.45 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.14, 162.23, 144.14, 135.17, 129.28, 128.27, 100.83, 60.04, 14.59.
参考文献[22c].在150 mL反应管中加入化合物S8 (5 g, 13.62 mmol), 联频哪醇硼酸酯(3.8 g, 14.98 mmol), Pd(dppf)2Cl2 (298.7 mg, 0.409 mmol), KOAc (4.0 g, 40.89 mmol)溶于60 mL无水DMSO中, 抽换N2三次, 升温至80 ℃搅拌6 h, 反应结束后反应液倒入10% NH4Cl溶液中, 过滤, 所得固体柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体4.65 g, 产率82%. m.p. 154~157 ℃ (lit.[22c] m.p. 167~170 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.08 (d, J=10.8 Hz, 2H), 8.05 (d, J=10.8 Hz, 2H), 7.93 (s, 2H), 4.45 (q, J=7.1 Hz, 4H), 1.47 (t, J=7.1 Hz, 6H), 1.38 (s, 12H); 13C NMR (100 MHz, CDCl3) δ: 166.73, 147.45, 138.76, 130.37, 99.79, 84.65, 59.96, 29.80, 25.02, 14.68.
在25 mL反应管中依次加入化合物S9 (82.64 mg, 0.2 mmol), 五氟碘苯(70.55 mg, 0.24 mmol), Pd(PPh3)4 (34.7 mg, 0.03 mmol), Ag2CO3 (137.87 mg, 0.5 mmol)溶于3 mL重蒸THF中, 抽换N2三次, 升温至70 ℃搅拌24 h, 反应结束后向体系中加入20 mL水, 用二氯甲烷(DCM)萃取三次, 合并有机相, 柱层析[V(石油醚):V(二氯甲烷)=1:2]分离得橙黄色固体46 mg, 产率51%. m.p. 177~181 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.15 (d, J=11.4 Hz, 2H), 7.95 (s, 2H), 7.51 (d, J=11.4 Hz, 2H), 4.50 (q, J=7.1 Hz, 4H), 1.50 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.50, 163.45, 145.92, 134.44, 129.88, 100.96, 60.27, 14.79; 19F NMR (376 MHz, CDCl3) δ: -142.33 (dd, J=24.5, 9.4 Hz, 2F), -154.26 (t, J=21.6 Hz, 1F), -161.46 (td, J=24.3, 9.3 Hz, 2F). HRMS (EI+) calcd for C22H16F5NO4 453.0999, found 453.0997.
在25 mL反应管中依次加入化合物S9 (82.64 mg, 0.2 mmol), 碘苯(70.55 mg, 0.24 mmol), Pd(PPh3)4 (34.7 mg, 0.03 mmol), Ag2CO3 (137.87 mg, 0.5 mmol)溶于3 mL重蒸THF中, 抽换N2三次, 升温至70 ℃搅拌24 h.反应条件同化合物S10得到橙黄色固体59 mg, 产率81%. m.p. 146~147 ℃ (lit.[12c] m.p. 148~149 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.17 (d, J=11.1 Hz, 2H), 7.78 (m, 4H), 7.62 (d, J=7.6 Hz, 2H), 7.48 (dd, J=10.8, 7.2 Hz, 2H), 7.41 (t, J=7.2 Hz, 1H), 4.48 (q, J=7.1 Hz, 4H), 1.49 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.72, 162.58, 146.56, 145.01, 144.01, 132.95, 131.03, 129.01, 128.43, 128.07, 100.14, 60.00, 14.84.
在25 mL反应管中依次加入化合物S9 (82.64 mg, 0.2 mmol), 2-碘噻吩(70.55 mg, 0.24 mmol), Pd(PPh3)4 (34.7 mg, 0.03 mmol), Ag2CO3 (137.87 mg, 0.5 mmol)溶于3 mL重蒸THF中, 抽换N2三次, 升温至70 ℃搅拌24 h, 反应条件同化合物S10, 得到橙黄色固体44 mg, 产率: 59%. m.p. 136~138 ℃ (lit.[12c] m.p. 127~128 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.08 (d, J=11.1 Hz, 2H), 7.89 (d, J=11.1 Hz, 2H), 7.79 (s, 2H), 7.46 (d, J=3.3 Hz, 1H), 7.39 (d, J=5.0 Hz, 1H), 7.18~7.10 (m, 1H), 4.47 (q, J=7.1 Hz, 4H), 1.49 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.63, 162.42, 146.81, 144.65, 138.79, 130.75, 130.67, 128.78, 127.16, 125.51, 100.40, 60.01, 14.81.
参考文献[17e].将化合物S10 (148 mg, 0.325 mmol)溶于10 mL 1, 4-二氧六环中, 加入对苯二酚(35.7 mg, 0.325 mmol), 浓硫酸(0.325 mmol), 随后同时滴加溶于5 mL 1, 4-二氧六环和亚硝酸异戊酯(0.44 mL, 3.25 mmol)溶液和溶于5 mL 1, 4-二氧六环的对苯二酚(357 mg, 3.25 mmol)溶液, 搅拌过夜, 反应结束后加入20 mL水, 用DCM萃取三次, 合并有机相, 柱层析[V(石油醚):V(二氯甲烷)=8:1], 分离得紫红色固体82 mg, 产率57.6%. m.p. 120~123 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.83 (d, J=10.9 Hz, 2H), 8.93 (s, 1H), 7.70 (d, J=10.8 Hz, 2H), 4.45 (q, J=7.1 Hz, 4H), 1.47 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 164.54, 144.84, 143.38, 138.22, 137.80, 132.12, 117.17, 60.15, 14.36; 19F NMR (376 MHz, CDCl3) δ: -141.98 (dd, J=22.4, 7.4 Hz, 2F), -152.58 (t, J=20.9 Hz, 1F), -160.66 (td, J=21.4, 6.8 Hz, 2F). HRMS (EI+) calcd for C22H15F5O4 438.0890, found 438.0893.
参考文献[17e].将化合物S11 (82 mg, 0.224 mmol)溶于16 mL 1, 4-二氧六环中, 加入对苯二酚(24.7 mg, 0.224 mmol), 浓硫酸(0.224 mmol), 随后同时滴加溶于3 mL 1, 4-二氧六环的亚硝酸异戊酯(0.3 mL, 2.24 mmol)溶液与溶于3 mL 1, 4-二氧六环对苯二酚(246 mg, 2.24 mmol)溶液, 搅拌过夜, 反应结束后加入20 mL水, 用DCM萃取三次, 合并有机相, 柱层析[V(石油醚):V(二氯甲烷)=8:1]分离得红色固体32 mg, 产率68%. m.p. 148~149 ℃ (lit.[22d] m.p. 150~152 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.82 (d, J=10.8 Hz, 2H), 8.82 (s, 1H), 7.96 (d, J=10.8 Hz, 1H), 7.69 (d, J=7.2 Hz, 2H), 7.60~7.45 (m, 3H), 4.45 (q, J=7.1 Hz, 4H), 1.47 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 165.22, 154.71, 143.71, 143.35, 142.88, 138.62, 131.00, 129.23, 129.08, 128.93, 116.60, 60.20, 14.73. HRMS (EI+) calcd for C22H20O4 348.1362, found 348.1358.
参考文献[17e].将化合物S12 (738 mg, 2 mmol)溶于130 mL 1, 4-二氧六环中, 加入对苯二酚(225 mg, 2 mmol), 浓硫酸(2 mmol), 随后同时滴加溶于70 mL 1, 4-二氧六环亚硝酸异戊酯(5.3 mL, 39.2 mmol)溶液与溶于70 mL 1, 4-二氧六环对苯二酚(4.3 g, 39.2 mmol)溶液, 搅拌过夜, 反应结束后加入100 mL水, 用DCM萃取三次, 合并有机相, 柱层析[V(石油醚):V(二氯甲烷)=8:1]分离得粉红色固体354 mg, 产率50%. m.p. 160~161 ℃ (lit.[22d] m.p. 142~150 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.68 (d, J=10.1 Hz, 2H), 8.73 (s, 1H), 8.05 (d, J=10.1 Hz, 2H), 7.63 (d, J=3.2 Hz, 1H), 7.52 (d, J=4.9 Hz, 1H), 7.19 (dd, J=3.2 Hz, 4.9 Hz, 1H), 4.43 (q, J=6.9 Hz, 4H), 1.45 (t, J=6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 164.98, 146.46, 146.08, 142.80, 142.37, 138.27, 129.40, 129.11, 128.25, 127.67, 116.79, 60.02, 14.55. HRMS (EI+) calcd for C20H18O4S 354.0926, found 354.0929.
将6.35 g五氧化二磷溶解于9.5 mL 85%磷酸中制得100%磷酸, 化合物S13 (82 mg, 0.187 mmol)加入到上述新制的100%磷酸中, 玻璃棒搅拌下升温至130 ℃反应30 min, 反应液冷却至室温后倒入冰水中, 乙酸乙酯萃取三次, 合并有机相, 浓缩后柱层析(石油醚)分离得蓝色固体42 mg, 产率76%. m.p. 193~194 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.39 (d, J=10.2 Hz, 2H), 8.03 (t, J=3.7 Hz, 1H), 7.50 (d, J=3.7 Hz, 2H), 7.13 (d, J=10.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 139.87, 139.01, 135.17, 134.54, 124.70, 119.32; 19F NMR (376 MHz, CDCl3) δ: -142.05 (dd, J=22.9, 7.6 Hz, 2F), -154.69 (t, J=20.9 Hz, 1F), -161.72 (td, J=22.4, 7.6 Hz, 2F). HRMS (EI+) calcd for C16H7F5 294.0468, found 294.0473.
将6.35 g五氧化二磷溶解于9.5 mL 85%磷酸中制得100%磷酸, 化合物S14 (32 mg, 0.09 mmol)加入到上述新制的100%磷酸中, 反应条件同化合物4, 得到蓝色固体17 mg, 产率90%. m.p. 163~164 ℃ (lit.[22e] m.p. 158~159 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.42 (d, J=10.0 Hz, 2H), 7.92 (br, 1H), 7.66 (d, J=7.3 Hz, 2H), 7.50 (t, J=7.2 Hz, 2H), 7.41~7.45 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 151.06, 145.45, 139.01, 137.03, 136.00, 128.80, 128.69, 128.03, 123.55, 118.44. HRMS (EI+) calcd for C16H12 204.0939, found 204.0940.
将6.35 g五氧化二磷溶解于9.5 mL 85%磷酸中制得100%磷酸, 化合物S15 (354 mg, 1 mmol)加入到上述新制的100%磷酸中, 反应条件同化合物4, 得到蓝色固体120 mg, 产率57%. m.p. 174~177 ℃ (lit.[22f] m.p. 160~161 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.32 (d, J=10.2 Hz, 2H), 7.85 (d, J=3.4 Hz, 1H), 7.57 (d, J=10.2 Hz, 2H), 7.52 (d, J=3.4 Hz, 1H), 7.42 (d, J=4.9 Hz, 1H), 7.36~7.38 (br, 2H), 7.15 (t, J=4.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 148.12, 142.95, 138.92, 136.94, 135.82, 128.63, 127.49, 126.08, 121.47, 118.93. HRMS (EI+) calcd for C16H10S 210.0503, found 210.0507.
辅助材料(Supporting Information) 核磁谱图和高分辨质谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Lemal, D. M.; Goldman, G. D. J. Chem. Educ. 1988, 65, 923. doi: 10.1021/ed065p923
(a) Michl, J.; Thulstrup, E. W. Tetrahedron 1976, 32, 205.
(b) Dong J.; Zhang H. Chin. Chem. Lett. 2016, 27, 1097.
(a) Rekka, E.; Chrysselis, M.; Siskou, I.; Kourounakis, A. Chem. Pharm. Bull. 2002, 50, 904.
(b) Ishihara, M.; Wakabayashi, H.; Motohashi, N.; Sakagami, H. Anticancer Res. 2011, 31, 515.
Saka, M.; Tsuchikawa, H. JP 04020312, 1992.
Kitamura, Y.; Nakaguchi, T. JP 11125497, 1999.
Yasushige, N.; Shinichi, Y.; Yoshimichi, K.; Seijiro, I.; Satoshi, T. US 20030129516, 2003
(a) Wang, X.; Ng, J. K.-P.; Jia, P.; Lin, T.; Cho, C. M.; Xu, J.; Lu, X.; He, C. Macromolecules 2009, 42, 5534.
(b) Amir, E.; Amir, R. J.; Campos, L. M.; Hawker, C. J. J. Am. Chem. Soc. 2011, 133, 10046.
(c) Koch, M.; Blacque, O.; Venkatesan, K. Org. Lett. 2012, 14, 1580.
(d) Murai, M.; Amir, E.; Amir, R. J.; Hawker, C. J. Chem. Sci. 2012, 3, 2721.
(e) Ghazvini Zadeh, E. H.; Tang, S.; Woodward, A. W.; Liu, T.; Bondar, M. V.; Belfield, K. D. J. Mater. Chem. C 2015, 3, 8495.
(f) Murai, M.; Takami, K.; Takeshima, H.; Takai, K. Org. Lett. 2015, 17, 1798.
Ito, S.; Inabe, H.; Morita, N.; Ohta, K.; Kitamura, T.; Imafuku, K. J. Am. Chem. Soc. 2003, 125, 1669. doi: 10.1021/ja0209262
(a) Salman, H.; Abraham, Y.; Tal, S.; Meltzman, S.; Kapon, M.; Tessler, N.; Speiser, S.; Eichen, Y. Eur. J. Org. Chem. 2005, 2207.
(b) Zielinski, T.; Kedziorek, M. J.; Jurczak, J. Chem.-Eur. J. 2008, 14, 838.
Cristian, L.; Sasaki, I.; Lacroix, P. G.; Donnadieu, B.; Asselberghs, I.; Clays, K.; Razus, A. C. Chem. Mater. 2004, 16, 3543. doi: 10.1021/cm0492989
(a) Kurotobi, K.; Kim, K. S.; Noh, S. B.; Kim, D.; Osuka, A. Angew. Chem., Int. Ed. 2006, 45, 3944.
(b) Wang, F. K.; Lin, T. T.; He, C. B.; Chi, H.; Tang, T.; Lai, Y. H. J. Mater. Chem. 2012, 22, 10448.
(c) Ince, M.; Bartelmess, J.; Kiessling, D.; Dirian, K.; Martinez-Diaz, M. V.; Torres, T.; Guldi, D. M. Chem. Sci. 2012, 3, 1472.
(a) Xin, H.; Ge, C.; Gao, H.; Yang, X.; Gao, X. Chem. Sci. 2016, 7, 6701.
(b) Xin, H.; Ge, C.; Jiao, X.; Yang, X.; Rundel, K.; McNeill, C. R.; Gao, X. Angew. Chem., Int. Ed. 2018, 57, 1322.
(c) Xin, H.; Li, J.; Ge, C.; Yang, X.; Xue, T.; Gao, X. Mater. Chem. Front. 2018, 2, 975.
(a) Dutta, S.; Lakshmi, S.; Pati, S. K. Bull. Mater. Sci. 2008, 31, 353.
(b) Dias, J. R. J. Phys. Org. Chem. 2007, 20, 395.
(a) Nozoe, T.; Asao, T.; Oda, M. Bull. Chem. Soc. Jpn. 1974, 47, 681.
(b) Nozoe, T.; Takase, K.; Shimazaki, N., Bull. Chem. Soc. Jpn. 1964, 37, 1644.
(a) Xin, H.; Gao, X. ChemPlusChem 2017, 82, 945.
(b) Koch, M.; Blacque, O.; Venkatesan, K. J. Mater. Chem. C 2013, 1, 7400.
Kim, H.; Schulte, N.; Zhou, G.; Müllen, K.; Laquai, F. Adv. Mater. 2011, 23, 894. doi: 10.1002/adma.v23.7
(a) Zhang, J.; Petoud, S. Chem. Eur. J. 2008, 14, 1264.
(b) Nozoe, T.; Takase, K.; Shimazaki, N. Bull. Chem. Soc. Jpn. 1964, 37, 1644.
(c) Nozoe, T.; Seto, S.; Matsumura, S.; Murase, Y. Bull. Chem. Soc. Jpn. 1962, 35, 1179.
(d) McDonald, R. N.; Richmond, J. M.; Curtis, J. R.; Petty, H. E.; Hoskins, T. L. J. Org. Chem. 1976, 41, 1811.
(e)Xin, H.; Ge, C.; Fu, L.; Yang, X.; Gao, X. Chin. J. Org. Chem. 2017, 37, 711(in Chinese).
(辛涵申, 葛从武, 傅丽娜, 杨笑迪, 高希珂, 有机化学, 2017, 37, 711.)
Robert, S. H. Liu, J. Chem. Educ. 2002, 79, 183. doi: 10.1021/ed079p183
(a) Li, L.; Zhang, M. Anal. Chem. 1988, 16, 732(in Chinese).
(李隆弟, 张满, 分析化学, 1988, 16, 732.)
(b) Yang, X.; Pan, Z.; Ma, Y. J. Analy. Sci. 2003, 19, 588(in Chinese).
(杨洗, 潘祖亭, 马勇, 分析化学, 2003, 19, 588.)
韩亮, 吴靓, 童永正, 祖晓燕, 蒋绍亮, 有机化学, 2017, 37, 2940. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346195.shtmlHan, L.; Wu, L.; Tong, Y.; Zu, X.; Jiang, S. Chin. J. Org. Chem. 2017, 37, 2940(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346195.shtml
林丹燕, 宋森川, 陈智勇, 郭鹏然, 陈江韩, 史华红, 麦裕良, 宋化灿, 有机化学, 2018, 38, 103. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346366.shtmlLin, D.; Song, S.; Chen, Z.; Guo, P.; Chen, J.; Shi, H.; Mai, Y.; Song, H. Chin. J. Org. Chem. 2018, 38, 103(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346366.shtml
(a) Shoji, T.; Maruyama, A.; Ito, S.; Okujima, T; ; Yasunami, M.; Higashi, J.; Moritae, N. Heterocyles 2014, 89, 2588.
(b) Shoji, T.; Kikuchi, S.; Ito, S.; Morita, N. Heterocyles 2005, 1, 91.
(c) Chen, A.; Chiu, S.; Kuo, Y. Synth. Commun. 2003, 15, 2701.
(d) Kurotobi, K.; Tabata, H.; Miyauchi, M.; Murafuji, T.; Sugihara, Y. Synthesis 2002, 1013.
(e) Jutz, C.; Schweiger, E. Chem. Ber. 1974, 107, 2383.
(f) Shoji, T.; Ito, S.; Toyota, K.; Iwamoto, T.; Yasunami, M.; Morita, N. Eur. J. Org. Chem. 2009, 25, 4307.

图式 1 薁的共振结构
Scheme 1 Numbering legend of azulene and its polarized resonance structure

图 2 (a) 化合物1~3和Azulene的1H NMR以及(b)化合物4~6和Azulene的1H NMR
Figure 2 (a) 1H NMR of compounds 1~3 and azulene, and (b) 1H NMR of compounds 4~6 and azulene

图 3 (a) 化合物1~6和Azulene的紫外吸收光谱, 以及(b)化合物1-H+~6H+和Azulene-H+的紫外吸收光谱
Figure 3 (a) UV-vis absorption spectra of compounds 1~6 and azulene, (b) UV-vis absorption spectra of compounds 1-H+~ 6H+ and azulene-H+

图 5 (a) 化合物1~6和Azulene的荧光光谱以及(b)化合物1-H+~6-H+和Azulene-H+的荧光光谱
Figure 5 (a) Fluorescence spectra of compounds 1~6 and azulene; and (b) fluorescence spectra of compounds 1-H+~6-H+ and azulene-H+

图 6 (a) 化合物1~3和Azulene在二氯甲烷溶液中的循环伏安曲线, 以及(b)化合物4~6和Azulene在二氯甲烷溶液中循环伏安曲线
Figure 6 (a) Cyclic voltammograms of compounds 1~3 and azulene in dichloromethane, and (b) cyclic voltammograms of compounds 4~6 and azulene in dichloromethane
0.1 mol/L Bu4+NPE6- as supporting electrolyte, Ag/Ag+ as reference electrode, scan rate of 100 mV/s

图 7 化合物1~6和Azulene的HOMO能级和LUMO能级
Figure 7 HOMO and LUMO energy levels of compounds 1~6 and azulene

图 8 (a) 化合物1~6和Azulene优化后的分子构型示意图以及(b)化合物1-H+~6-H+和Azulene-H+优化后的分子构型示意图
Figure 8 (a) Optimized geometries of compounds 1~6 and azulene, (b) optimized geometries of compounds 1-H+~6-H+ and azulene-H+

图 9 (a) 化合物1~6和Azulene的前线分子轨道的分子轨道能量和电子密度等势图, 以及(b)化合物1-H+~6-H+和Azulene-H+的前线分子轨道的分子轨道能量和电子密度等势图
Figure 9 (a) Molecular orbital energy diagrams and isodensity surface plots of the frontier orbitals of compounds 1~6 and azulene, and (b) Molecular orbital energy diagrams and isodensity surface plots of the frontier orbitals of compounds 1-H+~6-H+ and azulene-H+

图 10 (a) 化合物1~6和Azulene计算拟合的电子吸收光谱, 以及(b)化合物1-H+~6-H+及Azulene-H+计算拟合的电子吸收光谱
Figure 10 (a) UV-Vis absorption spectra of compounds 1~6 and azulene by TDDFT, and (b) UV-Vis absorption spectra of compounds 1-H+~6-H+ and azulene-H+ by TDDFT
表 1 化合物1~6和Azulene在氯仿溶液中紫外可见吸收光谱、荧光光谱及量子产率数据及其质子化合物的相关数据
Table 1. Absorption, emission, fluorescence quantum yield data of compounds 1~6 and azulene, and the corresponding data of 1-H+~6-H+ and azulene-H+species in CHCl3 at r.t.
| Compd. | Unprotonated | Protonateda | |||||
| λmax(abs)/nm | λmax(em)/nm | ϕF | λmax(abs)/nm | λmax(em)/nm | ϕF | ||
| Azu | 340 | 375 | 0.054c | 354 | 413b | 0.005c | |
| 1 | 374 | 408b | 0.006c | 415 | 451 | 0.359c | |
| 2 | 390 | 424b | 0.003c | 446 | 503 | 0.095d | |
| 3 | 407 | 447b | 0.002c | 488 | 554b | 0.004d | |
| 4 | 345 | 386 | 0.082c | 375 | 421b | 0.002c | |
| 5 | 368 | 408 | 0.026c | 399 | 447b | 0.002c | |
| 6 | 387 | 435 | 0.001c | 456 | 500 | 0.029d | |
| a Treated with TFA; b extremely weak emission; c 9, 10-diphenylanthracene as standard subsure to measure; d rhodamine B as standand subsure to measure. | |||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们