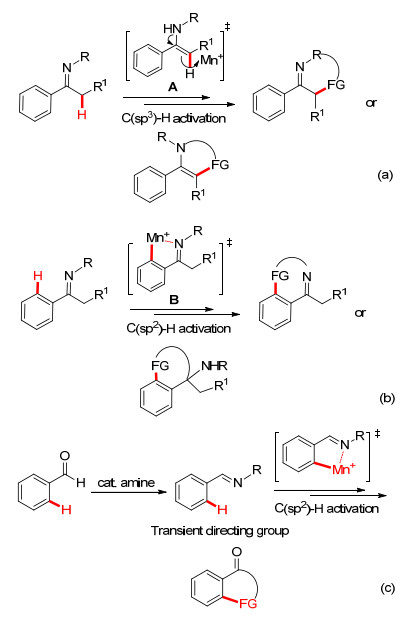
图式 1.
亚胺参与的C—H键官能团化/环化反应
Scheme 1.
C—H bond functionalization/cyclization involving imines

众所周知, 含氮化合物广泛存在于药物分子、天然产物和材料分子之中[1].因此, 发展高效合成策略来构建这类化合物不仅是有机化学的重要研究内容, 也已经成为当今化学家的研究热点领域之一[2].在众多合成方法中, 过渡金属催化的C—H键官能团化策略因具有独特的优势而成为合成这类骨架化合物的重要手段之一[3].与传统的交叉偶联相比, 直接的C—H官能化反应因避免了预官能团化(如卤化等)步骤而提高了反应的原子经济性, 符合低碳绿色化学的理念.另一方面, 鉴于环化反应类型的多样化以及环化产物是许多生物活性分子的核心骨架, 开展经由C—H键官能团化引起的串联环化反应研究一直成为化学家感兴趣的研究课题.
亚胺合成子因其反应活性的多样性而作为合成化学中常见的关键砌块[4].我们课题组先后报道了过渡金属催化亚胺的烷基化、酰胺化、芳基化等反应, 为构建多样性的含氮化合物提供了新的途径[5].此外, 近年来亚胺参与的C—H键官能团化反应为制备结构新颖的氮杂环提供了简捷的合成方法.从其参与的C—H键反应的类型来看, 通常分为C(sp3)—H键和C(sp2)—H键的功能化反应:一方面, 亚胺可互变异构形成烯胺, 继而被金属亲电进攻形成金属物种(A), 再与各种偶联试剂发生反应实现亚胺α位的C—H键官能团化, 最后引入的功能基与N原子或者取代基(R)发生分子内的环化反应(Scheme 1a); 另一方面, 直接通过亚胺功能基中N原子与过渡金属螯合经由五元环金属中间体(B)活化芳环C—H键, 从而实现惰性C—H键的官能团化/环化反应(Scheme 1b), 其中还包括催化剂量的胺类化合物与醛或者酮直接形成瞬态的亚胺功能基, 通过无痕导向的环金属化策略实现C—H键官能团化/环化反应(Scheme 1c).在此, 本文将系统地总结了通过上述反应模式实现亚胺关联的C—H官能团化反应/环化反应的最新研究进展.
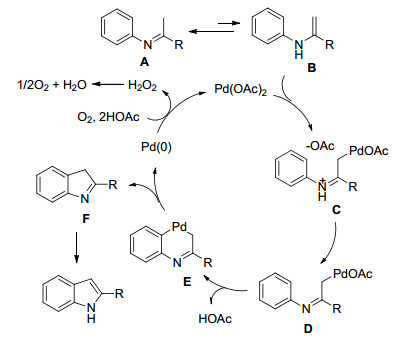
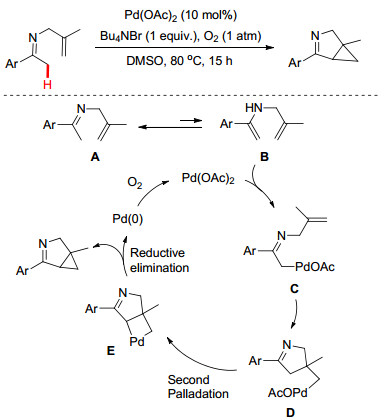
吲哚及其衍生物属于天然生物碱、药物分子以及功能材料的核心结构单元[6], 因此吲哚骨架的高效制备一直成为合成工作者的研究重点. 2012年, Yoshikai小组[7]首次报道了Pd(OAc)2-催化亚胺α位的C—H键活化及随后的分子内环化反应高效地合成了吲哚衍生物(Scheme 2).该反应条件比较温和, 并且使用分子氧作为氧化剂.苯环上无论是被吸电子基还是给电子基取代时, 都能够获得良好的产率.作者进一步通过控制实验和H/D交换证实了该反应涉及亚胺/烯胺异构及环钯中间体E的形成, 最后通过还原消除得到吲哚产物.
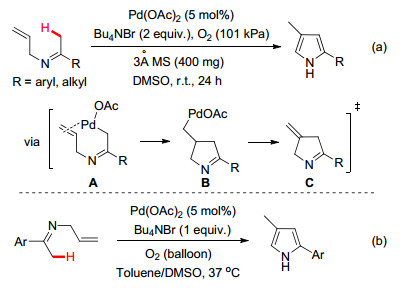
随后, Yoshikai小组[8]于2013年进一步将亚胺N原子上的芳基替换成烯丙基, 同样利用亚胺/烯胺异构形成的烷基钯亚胺中间体A (Scheme 3a)对烯烃的迁移插入和β-H消除等反应历程, 最后通过芳构化得到目标的吡咯化合物(Scheme 3a).该方法底物容忍性广, 不仅适用于芳基酮亚胺, 对于烷基酮亚胺也能兼容.需要指出的是, 几乎在同一时间里, 雷爱文小组[9]也报道了类似的工作(Scheme 3b), 并进一步通过动力学同位素效应研究证实了亚胺α位C(sp3)—H键的亲钯化反应是整个反应过程的决速步骤.
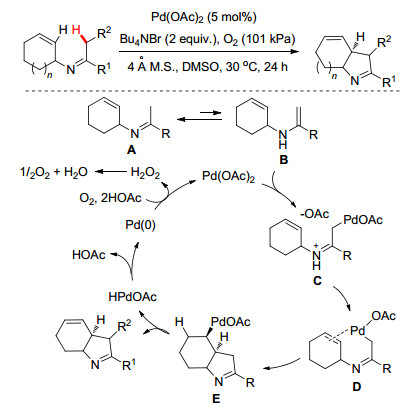
与此同时, Glorius小组[10]在2013年也报道了Pd催化N-环烯基酮亚胺分子内的C—H键活化/环化串联反应(Scheme 4).其不同之处在于作者通过巧妙地在N原子上引入环烯基, 利用中间体E单键难以自由旋转的特点, 高选择性地实现中间体E上定点氢原子的β-H消除, 从而生成二氢吡咯化合物(Scheme 4).该方法能够快速构建五元环并大环核心骨架.
鉴于过去报道的N-烯丙基酮亚胺的环化反应, 都需要经过β-H消除生成相应的目标产物. 2017年雷爱文小组[11]利用N-(2-甲基)-烯丙基亚胺为底物分子, 致使烷基钯物种D (Scheme 5)的β-C上没有氢原子而无法经历β-H消除, 而是烷基钯再次通过亲电钯化活化邻近的C(sp3)—H键生成四元环钯中间体E, 最后通过还原消除实现了钯催化的[2+1]环化反应(Scheme 5), 其深入的原位红外研究表明第二次的C(sp3)—H键活化可能是反应历程中的决速步骤.
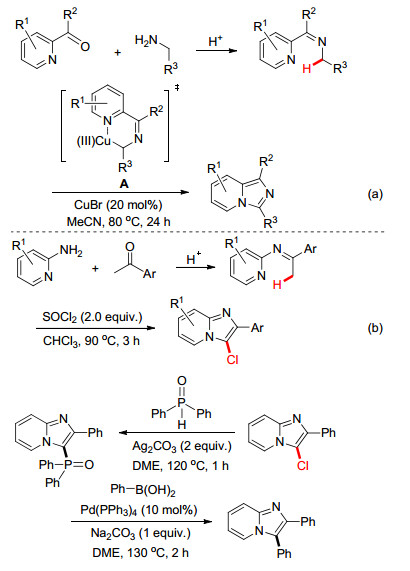
发展吡啶并咪唑[12]核心骨架的有效合成方法同样是合成化学家所关注的研究领域.我们[13]于2013年则尝试了在CuBr催化体系下芳香酮和伯胺直接通过一锅法构建咪唑[1, 5-a]并吡啶类化合物的可能性(Scheme 6a).利用芳香酮和伯胺在酸性条件下易于快速生成亚胺的特点, 吡啶N可协助Cu催化活化亚胺α位的C—H键并形成六元环铜中间体A, 中间体A再经由还原消除及芳构化反应生成目标产物.该反应底物范围适用性广, 各种功能基取代的苄胺和烷基胺都能转化为目标产物.在此基础上, 我们[14]继续发展了无金属催化合成吡啶并咪唑化合物的反应体系, 仅通过芳基胺、芳基甲酮和SOCl2一锅法反应便可高效构建3-氯取代的咪唑[1, 2-a]并吡啶化合物(Scheme 6b), 该化合物还通过与二苯基氧膦直接偶联衍生成为结构更为复杂三芳基氧膦.
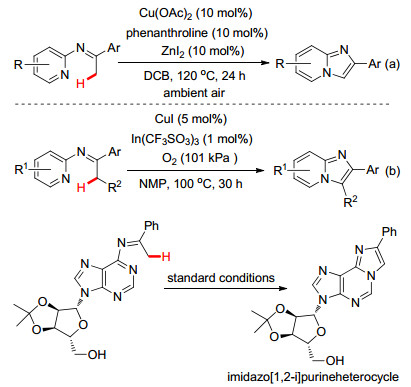
此外, Hajra小组[15]也报道了Cu(Ⅱ)-催化N-吡啶亚胺经由分子内的C—H键胺化反应合成了咪唑[1, 2-a]并吡啶衍生物的工作, 但该方法的底物比较受限, 只能实现亚胺α-甲基的C—H键活化, 而对于α-亚甲基的C—H键功能化反应并不适用(Scheme 7a).随后苏伟平小组[16]同样报道了类似的反应(Scheme 7b), 并进一步利用该方法学策略完成了具有生物活性的咪唑[1, 2-i]嘌呤的合成.
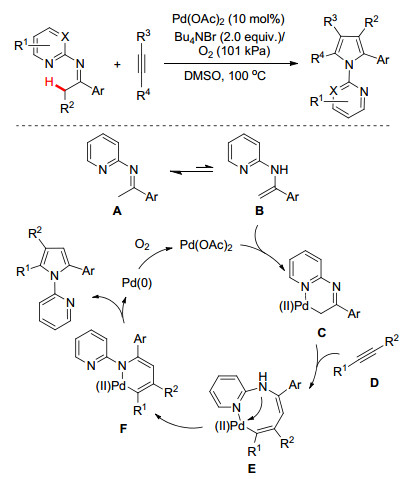
除了亚胺功能基参与的分子内C—H键官能团化反应外, 亚胺关联的分子间的C—H键官能团化/环化反应在构建氮杂环方面也取得了不错的进展.在这方面, 我们[17]最近报道了Pd(OAc)2催化N-吡啶酮亚胺与炔烃分子间的[3+2]环化反应快速构建多取代吡咯化合物(Scheme 8).机理研究证实:亚胺/烯胺异构并经由C(sp3)—H键活化形成六元环钯金属中间体C是实现本反应的关键步骤.
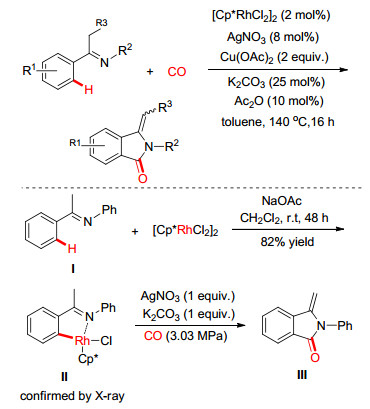
鉴于CO与炔烃一样都属于三键亲电体, 同时又是廉价易得C1羰基源, 我们[18]继续探索了N-吡啶酮亚胺在Pd(Ⅱ)催化作用下与CO发生羰化/环化反应的可能性.结果发现Pd(Ⅱ)催化剂可有效实现酮亚胺与CO的羰化/环反应(Eq. 1).该反应体系具有较好的底物兼容性, 各类芳基酮亚胺和杂环酮亚胺(噻吩、呋喃和吡啶)都能够以中等以上的收率转化为[1, 2-吡啶]嘧啶-4-酮衍生物.
此外, 考虑到重氮化合物特别是具有羰基功能基的受体/受体或者受体/供体卡宾前体化合物易于与金属催化剂形成金属卡宾而参与不同的有机反应[19]; 同时N-吡啶酮亚胺在Pd(Ⅱ)催化作用下可形成六元环钯中间体, 该中间体极有可能被重氮化合物所捕获而形成金属卡宾.基于此想法, 我们继续探索了N-吡啶酮亚胺与重氮化合物进行偶联环化反应的可能性.经过不断努力, 我们[20]发现Rh(Ⅲ)催化剂在吡啶功能基的螯合协助下, 可实现亚胺基α-位甲基C—H键的卡宾体插入/环化反应, 该反应可通过N-吡啶酮亚胺与重氮化合物的[3+2]环化偶联有效制备多功能基取代的吡咯化合物(Scheme 9).
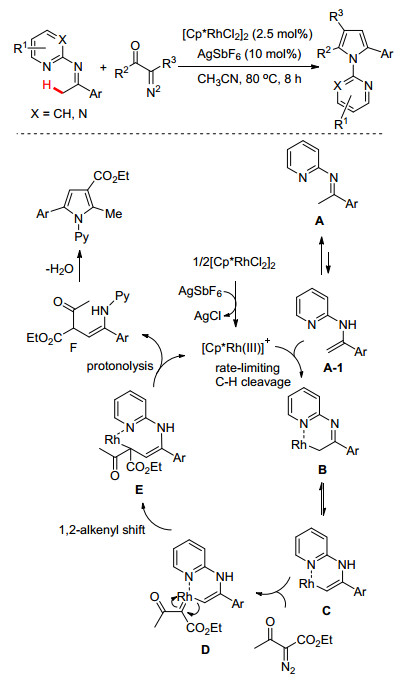
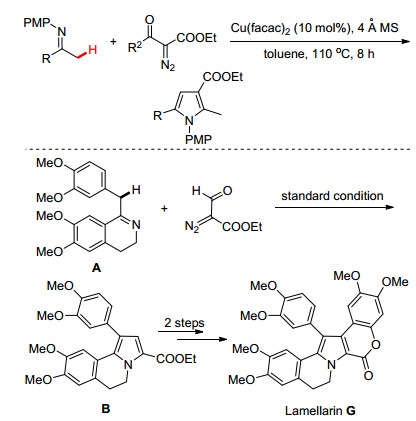
几乎在同一时间里, Yoshikai小组[21]也报道了类似的研究工作(Scheme 10).不同之处是Yoshikai使用的是Cu(Ⅱ)催化体系, 该体系通过铜卡宾和亚胺分子间的脱氢偶联反应同样构建了多取代吡咯化合物.作者还利用此方法设计合成了生物活性分子片螺素(Lamellarin G).其关键的有机反应是直接利用亚胺α-位的C(sp3)—H键与重氮化合物的偶联环化反应合成了二氢吡咯[2, 1-a]异喹啉中间体B, 因此该策略为片螺素衍生物的高效制备提供了绿色简便的合成路线.
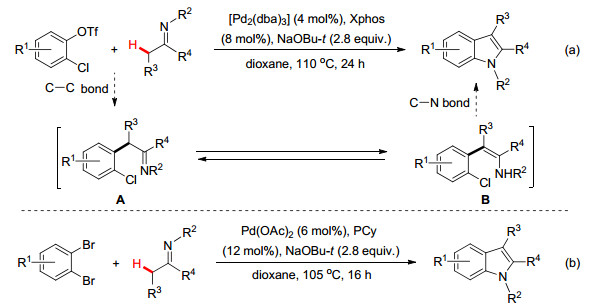
除了炔烃、CO和重氮化合物可与亚胺类化合物在过渡金属催化作用下通过环化反应构建不同的氮杂环分子外, Valdes小组[22]及Langer小组[23]还考查过将邻氯苯磺酸酯和邻二溴苯常作为C2单元与亚胺化合物通过环化反应构建特定杂环骨架分子的可能性.他们利用钯催化剂, 先后通过Pd(0)对C(sp2)—O键或C(sp2)—Br键氧化加成和烯亚胺的亲电钯化等反应历程, 为高效制备了吲哚化合物提供了不同的制备途径(Scheme 11).
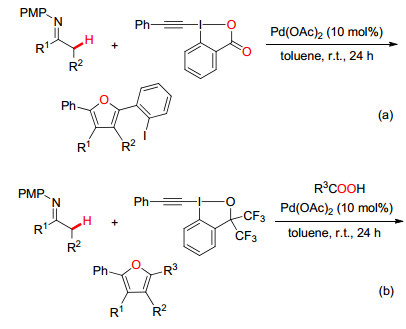
炔基高价碘试剂[24]属于一类活泼的亲电试剂, 常被广泛应用于合成化学中. 2014年, Yoshikai等[25]报道了Pd(OAc)2-催化N-芳基酮亚胺与炔基高价碘试剂的环化反应, 该催化体系导致了意料之外的四取代呋喃衍生物的形成(Scheme 12a).由于其中的取代基含有碘苯功能基, 可以通过偶联反应进一步衍生成含有呋喃骨架的有用的药物分子.作者也通过一系列的同位素标记实验证明了呋喃中的氧原子来自于高价碘试剂.该小组[26]于2015年又继续发展了酮亚胺/高价炔基碘/羧酸三组分的催化体系同样高效地构建了多取代呋喃化合物(Scheme 12b), 并通过同位素标记实验验证了呋喃的氧原子来自于羧酸底物, 并非来自之前报道的高价碘试剂.
除了利用亚胺基C(sp3)—H键的亲电金属化/螯合导向活化C(sp3)—H策略来实现烷基C(sp3)—H键官能化外, 自由基引发亚胺基α-位的C(sp3)—H键的功能化/环化反应在构建氮杂环骨架结构方面也受到了化学者们的青睐. 2015年, 李金恒小组[27]报道了无金属催化的N-芳基亚胺与亚硝酸叔丁基酯的硝化/环化反应, 该反应可高效制备3-硝基吲哚类化合物(Scheme 13a), 其反应体系中亚硝酸叔丁基酯即作为自由基引发剂又充当了硝基源.与此同时, 焦宁小组[28]也报道了相同反应底物在四丁基溴化铵条件下的无金属催化体系, 但是反应得到的产物并非3-硝基吲哚, 而是喹啉氮氧化合物(Scheme 13b), 其中氮氧杂烯丙基自由基中间体B是形成喹啉氮氧化合物的关键中间体.
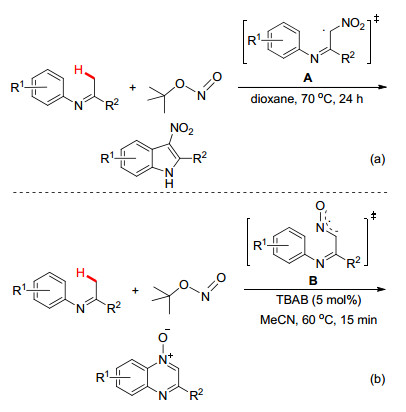
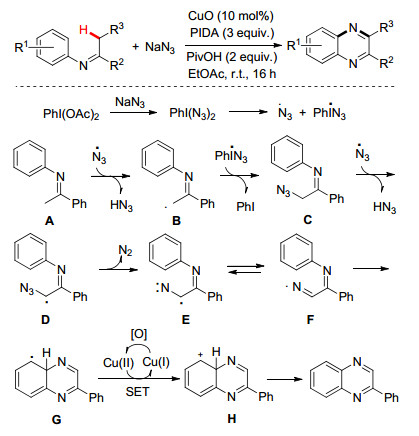
此外, 我们[29]最近利用廉价易得的酮亚胺和叠氮化钠为起始原料, 通过Cu催化[5+1]环化反应可简捷而高效制备喹喔啉化合物(Scheme 14).机理研究表明N-芳基酮亚胺在叠氮自由基的诱导下产生α-亚胺甲基碳自由基B, 随后与叠氮自由基偶联形成α-叠氮基取代的酮亚胺C, 并再次通过自由基攫氢生成亚胺基氮自由基F, 接着通过自由基环化/芳构化产生喹喔啉目标产物.
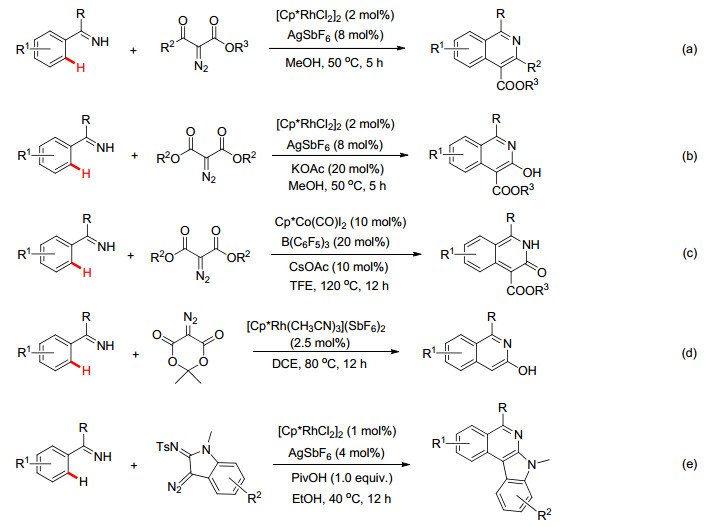
近十年来, 合成化学家同行利用各种类型的导向基团(如吡啶、酰胺基、羧基及羟基等)为构建结构复杂的杂环化合物提供最具有潜力和最方便的途径.其中, 亚胺功能基作为一种新型的导向基团, 其在过渡金属催化高选择性C—H键官能化反应中也逐步引起广泛关注.
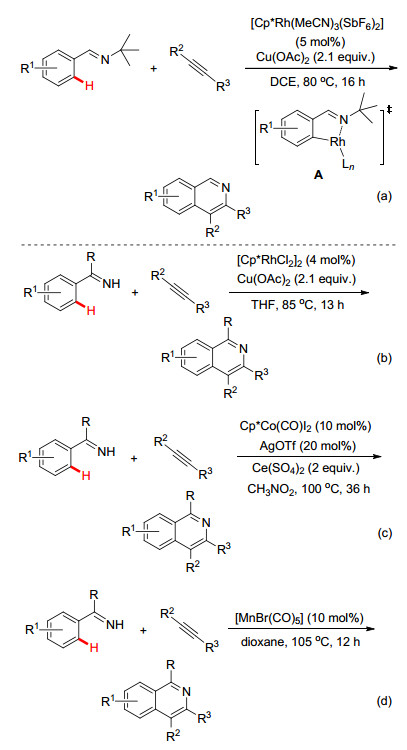
如早在2009年, Fagnou小组[30]利用N-叔丁基亚胺作为导向基, 通过亚胺N原子与金属Rh配位, 经由五元环铑中间体A完成芳环邻位C—H键的活化, 最后通过与炔烃的迁移插入和还原消除形成C—N键的同时脱除叔丁基并生成异喹啉目标产物(Scheme 15a).随后, 李兴伟小组[31]、王华东小组[32]和王重洋小组[33]及其他课题组[34~37]也先后利用[RhCp*Cl2]2, Cp*Co(CO)I2和[MnBr(CO)5]作为催化剂, 同样实现了亚胺基导向的C—H键与不同炔烃的偶联/环化反应(Scheme 15, b~d), 为构建多样性异喹啉和异喹啉季胺盐开辟了新的合成途径.
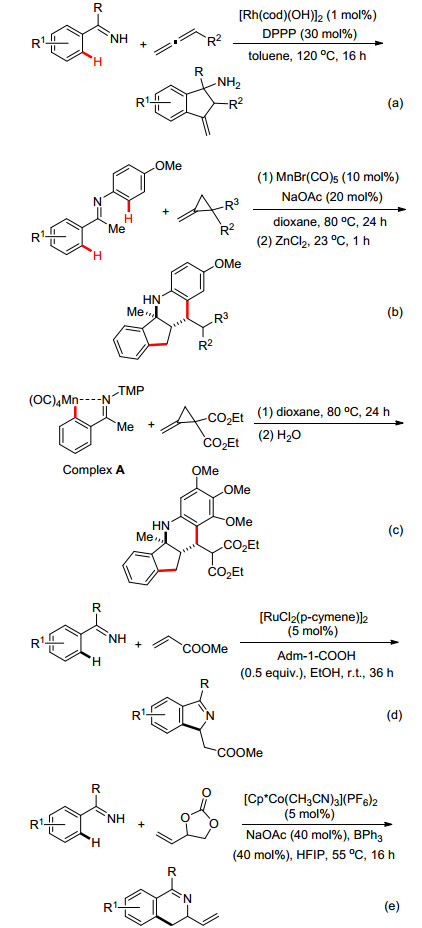
除了炔烃之外, Cramer小组[38]则报道了利用联烯作为偶联试剂, 通过类似的C—H活化机理实现了亚胺导向的[3+2]环化反应, 该反应可高选择性地在二氢茚类衍生物的1, 3-位引入氨基和烯基功能基(Scheme 16a).最近, Ackermann小组[39]则以烯烃为偶联试剂, 发展了廉价Mn催化N-芳基苯乙酮亚胺和亚甲基环丙烷串联环化反应, 该反应通过一锅法合成了多环核心骨架结构(Scheme 16b).随后作者还分离得到五元环锰络合物A, 并且通过X射线单晶衍射分析予以确认, 同时利用该络合物与亚甲基环丙烷在没有Mn(Ⅰ)催化剂的条件下, 也能够有效地生成目标产物(Scheme 16c), 这无疑地证明了该五元环锰络合物A是整个反应历程的关键中间体.与此同时, Jeganmohan小组[40]和Ackermann小组[41]分别报道了Ru(Ⅱ)和Co(Ⅲ)催化亚胺与烯烃, 经由C—H键活化/环化反应快速构建二氢异吲哚(Scheme 16d)和二氢异喹啉化合物(Scheme 16e).
羰化反应是有机合成化学研究的主要基元反应之一, 直接的C—H键羰化反应可高原子和高步骤经济性地构建羰基化合物[42]. 2016年, 黄汉民小组[43]报道了利用CO作为羰基源, 以亚胺功能基作为导向基, 利用[RhCp*Cl2]2催化活化芳基C(sp2)—H键并形成环铑中间体, 从而通过插羰/还原消除过程快速地制备了异吲哚酮衍生物(Scheme 17).为证实反应机理, 作者同样分离并通过晶体衍射和后续的化学转换证实了五元环铑金属络合物(Ⅱ)的存在.
近几年, 过渡金属催化的螯合导向C—H键的卡宾功能化反应, 特别是涉及卡宾插入的串联环化反应研究, 因可快速合成多样性杂环化合物而受到关注.在这方面, 朱进等[44]早期报道了利用亚胺功能基与Rh螯合实现芳环C—H键的活化, 并与α-羰基重氮酯经由卡宾迁移插入及脱水环化反应合成了多取代异喹啉化合物(Scheme 18a), 但当作者把α-羰基重氮酯换成丙二酸酯重氮化合物作为偶联试剂时, 则生成了脱醇环化的3-羟基取代的异喹啉衍生物(Scheme 18b).
随后, Glorius小组[45]则利用廉价Co催化剂代替Rh催化剂, 同样实现亚胺导向的C—H键活化与丙二酸酯重氮化合物的[4+2]环化反应, 但产物为异喹啉酮化合物(Scheme 18c).在此体系中, Co盐扮演了双重角色:一是作为金属催化剂活化C—H键; 二是作为路易斯酸活化酯羰基, 促进了分子内亲核环化反应.此外, 鉴于重氮化梅尔德伦酸酯是一类环状的高活性的重氮化合物, 容易脱去一分子丙酮和CO2进行开环, 刘培念小组[46]利用其特点还发展了Rh催化N-芳基酰亚胺与重氮化梅尔德伦酸酯的偶联环化反应, 该反应可导致3-羟基异喹啉化合物的形成(Scheme 18d).紧接着, 李兴伟小组[47]报道了Rh(Ⅲ)催化亚胺与α-亚胺基重氮化合物的[4+2]环化反应简便合成了喹啉并吲哚类衍生物(Scheme 18e).
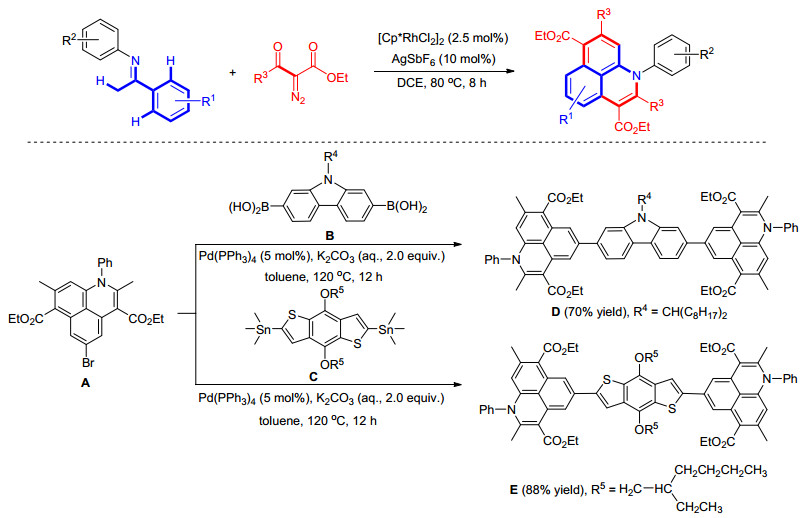
尽管重氮化合物参与的C—H键烷基化/环化反应取得了重大进展, 但大部分都局限于芳环单个C(sp2)—H键的卡宾功能化反应.而对于重氮参与芳环多个C—H键的接力卡宾功能反应的研究比较少.在这方面, 我们[48]最近发展了Rh接力催化酮亚胺与α-羰基重氮酯通过两次C—H键活化/烷基化/环化反应高效地合成1-氮杂非那烯衍生物(Scheme 19).考虑到1-氮杂非那烯属于一类比较强的受体结构, 我们进一步将1-氮杂非那烯分别与给体结构的咔唑或苯并二噻吩, 通过Suzuki和Still偶联反应构建为超π-共轭的小分子, 研究了这些小分子的光电性能.通过循环伏安法(CV)研究发现小分子D和E都具有比较低的HOMO能级, 分别为-5.76和-5.57 eV, 而较低的HOMO能级是高性能光电材料的必备条件之一, 该策略为今后合成高效率光电材料分子提供了独特的合成方法.
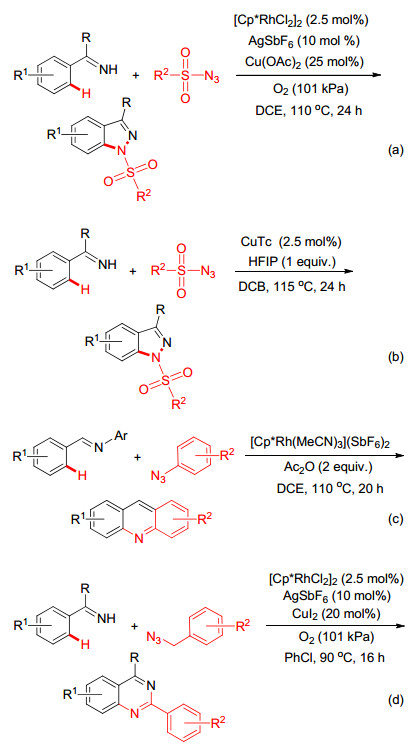
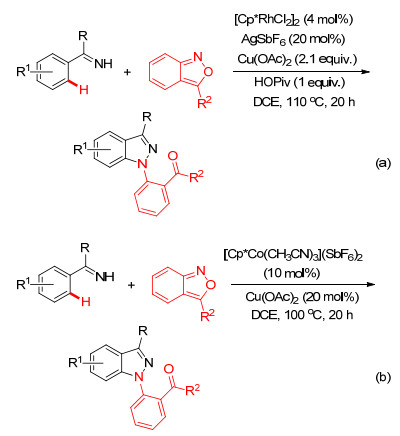
有机叠氮化合物作为一种高活性的胺化试剂, 常应用于过渡金属催化的胺化反应之中[49].如Glorius小组[50]和朱强小组[51]相继报道了Rh(Ⅲ)-和Cu(Ⅰ)-催化芳基酰亚胺与磺酰叠氮胺环化反应性能(Scheme 20a, 20b), 此反应体系的底物适用性广, 且氮气作为唯一的副产物, 为高效合成多取代的吲唑化合物提供了绿色的合成方法.除了磺酰基叠氮可作为氮源实现C—H键的胺环化反应外, Ellman小组[52]和焦宁小组[53]发现芳基叠氮和烷基叠氮也可作为氮源, 在Rh(Ⅲ)的催化作用下通过与N-芳基醛亚胺的偶联环化反应来构建吖啶(Scheme 20c)和喹唑啉衍生物(Scheme 20d).不过从绿色化学理念来看, Ellman的方法需要脱去一分子芳胺, 原子经济性较差.
除了叠氮化合物之外, 二氧氮茂酮[54]作为一种新型的胺化试剂, 也可应用于亚胺基导向的C—H键胺化/环化反应.在这方面, 朱进小组[55]于2016年曾报道了通过Rh催化亚胺基协助的C—H键活化策略, 实现了芳烷基酮亚胺与二氧氮茂酮经由分子间的[4+2]环化反应, 合成了喹唑啉衍生物(Scheme 21a).考虑到廉价Co(Ⅲ)催化剂在C—H键活化方面具有独特的优势, 同年李兴伟小组[56]和Glorius小组[57]几乎在同一时间报道了利用二氧氮茂酮作为氮源, 以Cp*Co(CO)I2为催化剂, 实现了二氧氮茂酮与芳烷基酮亚胺的[4+2]环化反应, 产物为喹唑啉骨架化合物(Scheme 21b), 其中Glorius小组还进一步比较了Co、Rh、Ir三种催化剂的催化活性并发现Co催化体系因于其本身在活化C—H键的同时还可作为路易斯酸活化酰羰基, 有利于分子内的脱水环化.
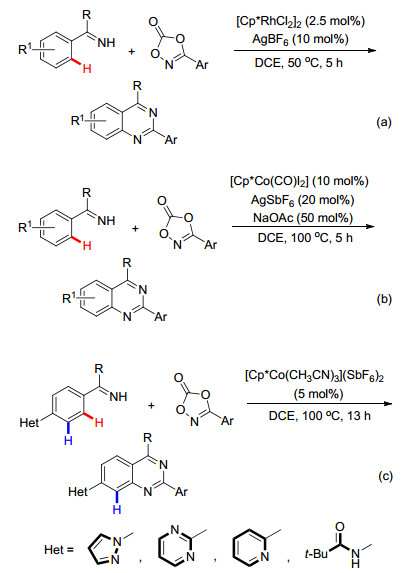
另一方面, Ackermann小组[58]还通过在亚胺基苯的对位同时引入了吡啶、吡唑、嘧啶和酰胺等功能基(Scheme 21c), 深入研究了酮亚胺与这些功能基在钴催化体系下导向活化芳基C—H键的选择性, 发现酮亚胺能选择性地与Co螯合配位并活化邻位C—H键, 所形成的环钴中间体可进一步被二氧氮茂酮捕获, 从而实现高区域选择性的环胺化反应(Scheme 21c), 而吡唑等其他导向基则很难以通过导向螯合作用活化芳基C—H键.
受到过渡金属催化芳基C—H键与叠氮化合物和二氧氮茂酮胺环化反应的鼓舞, 最近李兴伟小组则继续利用氨茴内酐作为胺化试剂, 以酮亚胺为螯合功能基, 相继实现了Rh(Ⅲ)[59]或Co(Ⅲ)[60]催化芳基C—H键的胺化反应, 并通过C—N键和N—N键的形成构建了N-芳基取代的吲唑类衍生物(Scheme 22).
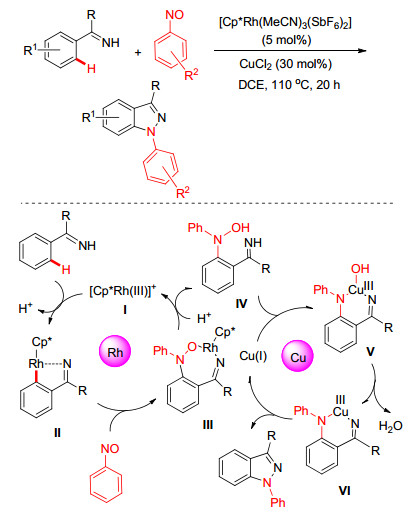
令人兴奋的是, 该小组[61]后来发现若利用亚硝基苯作为氮源, 在Rh(Ⅲ)/Cu(Ⅱ)的协同催化下可实现亚胺基导向的[4+1]胺环化反应(Scheme 23).反应涉及两个催化循环过程:首先是由五元环铑中间体(Ⅱ)对亚硝基苯的N=O双键迁移插入及随后的金属质解实现C—H键的胺化反应, 生成中间体(Ⅳ); 随后通过Cu(Ⅰ)催化分子内羟胺与酮亚胺的偶联环化完成目标化合物的构建.
在过去十几年里, 过渡金属催化的导向基诱导的C(sp3)—H键和C(sp2)—H键选择性活化都取得了巨大的进步, 但是导向基团当量的使用及除去, 不仅使得合成步骤增多, 而且部分导向基团与底物官能团不相容, 也大大限制了反应的使用效率和适用范围.为了解决这一问题, 通过原位生成瞬态导向基并与金属中心结合, 实现位点选择性官能团化后自行脱除则更具有高原子步骤经济性.近几年来, 利用醛酮底物与催化量的胺类化合物通过形成瞬态的亚胺功能基, 实现导向的官能团化反应已经取得重大进展[62].然而大部分工作都是局限于C—H键直接的官能团化, 而经由瞬态导向基诱导C—H键官能团化引发后续串联环化反应的相关报道甚少.
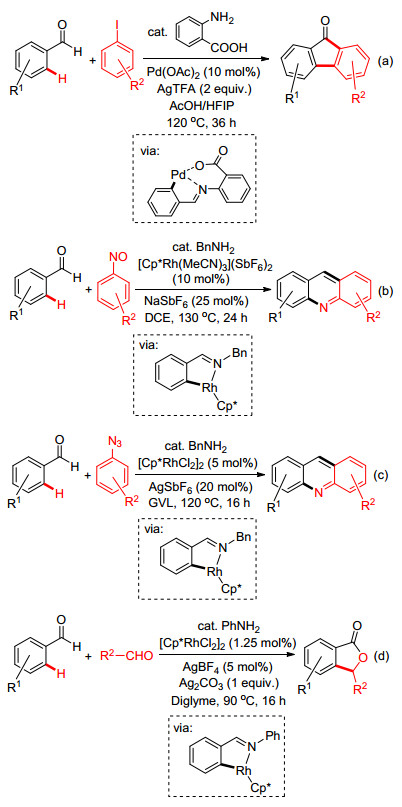
在这个方面, Sorensen小组[63]最近报道了利用Pd(OAc)2和邻氨基苯甲酸作为共催化剂, 通过邻氨基苯甲酸和底物芳香醛原位生成催化量的亚胺无痕导向基, 并与Pd经由双齿螯合作用活化芳环C—H键, 实现与碘苯的偶联/环化反应快速生成了芴酮衍生物(Scheme 24a).与此同时, 成江小组[64]、何明阳小组[65]和Seayad小组[66]也分别报道了利用亚硝基苯、芳基叠氮及醛类化合物作为偶联试剂, 通过瞬态导向策略实现了Rh催化的C—H键活化/环化反应构建了多样性的吖啶(Scheme 24, b~c)和苯酞类化合物(Scheme 24d).
主要论述了亚胺参与的C(sp3)—H键和C(sp2)—H键的直接官能化/环化的各种类型反应, 并就其相关机理予以阐述.亚胺/烯胺异构或者亚胺直接参与过渡金属的螯合作用是实现C—H键官能化反应的关键过程, 并为结构多样性的氮杂环化合物的高效构建提供了绿色低碳的合成方法.遗憾的是, 目前的大部分工作集中于亚胺导向的芳基C(sp2)—H键或者活泼烷基C(sp3)—H键官能化反应.通过底物分子的巧妙设计, 进一步研究亚胺螯合协助的远程非活泼烷基C(sp3)—H键的官能化, 特别是不对称C(sp3)—H键的官能团化将是今后颇具有挑战性的研究方向.
(a) Dolzhenko, A. V. ; Chui, W. K. Heterocycles 2008, 75, 1575.
(b) Gundla, R. ; Kazemi, R. ; Sanam, R. ; Muttineni, R. ; Sarma, J. A. R. P. ; Dayam, R. ; Neamati, N. J. Med. Chem. 2008, 51, 3367.
(c) Van Horn, K. S. ; Burda, W. N. ; Fleeman, R. ; Shaw, L. N. ; Manetsch, R. J. Med. Chem. 2014, 57, 3075.
(a) Zhang, J. ; Liu, J. ; Ma, Y. ; Cheng, P. Chin. J. Org. Chem. 2017, 37, 555(in Chinese).
(张金, 刘佳, 马养民, 程佩, 有机化学, 2017, 37, 555. )
(b) Zhang, Z. ; Zheng, X. ; Guo, C. Chin. J. Org. Chem. 2016, 36, 1241(in Chinese).
(张钊瑞, 郑晓霖, 郭长彬, 有机化学, 2016, 36, 1241. )
(a) Ramirez T. A. ; Zhao B. ; Shi Y. Chem. Soc. Rev. 2012, 41, 931.
(b) Wang, S. ; Yan, F. ; Wang, L. ; Zhu, L. Chin. J. Org. Chem. 2018, 38, 291(in Chinese).
(汪珊, 严沣, 汪连生, 朱磊, 有机化学, 2018, 38, 291. )
(c) Chen, J. ; Hu, X. ; Lu, L. ; Xiao, W. Chem. Rev. 2015, 115, 5301.
(a) Lamas, M. C. ; Vaillard, S. E. ; Wibbeling, B. ; Studer, A. Org. Lett. 2010, 12, 2072.
(b) Ricardo, G. P. ; Kiyoi, T. ; Whiting, A. Org. Biomol. Chem. 2011, 9, 3105.
(c) Rishikesh, N. ; Potowski, M. ; Jia, Z. J. ; Antonchick, A. P. ; Waldmann, H. Acc. Chem. Res. 2014, 47, 1296.
(a) Xiao, X. ; Zhang, W. ; Lu, X. ; Deng, Y. ; Jiang, H. ; Zeng, W. Adv. Synth. Catal. 2016, 358, 2497.
(b) Dang, L. ; Liang, L. ; Qian, C. ; Fu, M. ; Ma, T. ; Xu, D. ; Jiang, H. ; Zeng, W. J. Org. Chem. 2014, 79, 769.
(c) Fu, S. ; Jiang, H. ; Deng, Y. ; Zeng, W. Adv. Synth. Catal. 2011, 353, 2795.
(d) Zhu, S. ; Lu, X. ; Luo, Y. ; Zhang, W. ; Jiang, H. ; Yan, M. ; Zeng, W. Org. Lett. 2013, 15, 1440.
(e) Zhu, S. ; Dong, J. ; Fu, S. ; Jiang, H. ; Zeng, W. Org. Lett. 2011, 13, 4914.
(f) Chen, J. ; Lu, X. ; Lou, W. ; Ye, Y. ; Jiang, H. ; Zeng, W. J. Org. Chem. 2012, 77, 8541.
(g) Luo, Y. ; Lu, X. ; Ye, Y. ; Guo, Y. ; Jiang, H. ; Zeng, W. Org. Lett. 2012, 14, 5640.
黎秋, 汪雨, 胡孟金, 陈鹏, 游文玮, 赵培亮, 有机化学, 2017, 37, 967. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345857.shtmlLi, Q.; Wang, Y.; Hu, M.; Chen, P.; You, W.; Zhao, P. Chin. J. Org. Chem. 2017, 37, 967(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345857.shtml
Wei, Y.; Deb, I.; Yoshikai, N. J. Am. Chem. Soc. 2012, 134, 9098. doi: 10.1021/ja3030824
Chen, Z.; Lu, B., Ding, Z.; Gao, K.; Yoshikai, N. Org. Lett. 2013, 15, 1966. doi: 10.1021/ol400638q
Meng, L.; Wu, K.; Liu, C.; Lei, A. Chem. Commun. 2013, 52, 5853.
Shi, Z.; Suri, M.; Glorius, F. Angew. Chem., Int. Ed. 2013, 52, 4892. doi: 10.1002/anie.v52.18
Wu, K.; Meng, L.; Huai, M.; Huang, Z.; Liu, C.; Qi, X.; Lei, A. Chem. Commun. 2017, 53, 2294. doi: 10.1039/C6CC09332A
Cappelli, A.; Giuliani, G.; Anzini, M.; Riitano, D.; Giorgi, G.; Vomero, S. Bioorg. Med. Chem. 2008, 16, 6850. doi: 10.1016/j.bmc.2008.05.067
Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232. doi: 10.1021/ol503165b
Xiao, X.; Xie, Y.; Bai, S.; Deng, Y.; Jiang, H.; Zeng, W. Org. Lett. 2015, 17, 3998. doi: 10.1021/acs.orglett.5b01868
Bagdi, A. K.; Rahman, M.; Santra, S.; Majee, A.; Hajra, A. Adv. Synth. Catal. 2015, 355, 1741.
Zhang, Y.; Chen, Z.; Wu, W.; Zhang, Y.; Su, W. J. Org. Chem. 2013, 78, 12494. doi: 10.1021/jo402134x
Xie, Y.; Chen, T.; Fu, S.; Li, X.; Deng, Y.; Jiang, H.; Zeng, W. Chem. Commun. 2014, 50, 10699. doi: 10.1039/C4CC04676E
Xie, Y.; Chen, T.; Fu, S.; Jiang, H.; Zeng, W. Chem. Commun. 2015, 51, 9377. doi: 10.1039/C5CC02631H
王亮, 李站, 万康, 瞿星, 胡思前, 王锋, 有机化学, 2016, 36, 889. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345417.shtmlWang, L.; Li, Z.; Wang, K.; Xu, X.; Hu, S.; Wang, F. Chin. J. Org. Chem. 2016, 36, 889(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345417.shtml
Chen, X.; Xie, Y.; Xiao, X.; Li, G.; Deng, Y.; Jiang, H.; Zeng, W. Chem. Commun. 2015, 51, 15328. doi: 10.1039/C5CC06428G
Tan, W. W.; Yoshikai, N. Chem. Sci. 2015, 6, 6448. doi: 10.1039/C5SC02322J
Barluenga, J.; Jimenez-Aquino, A.; Aznar, F.; Valdes, C. Chem. Eur. J. 2010, 16, 11707. doi: 10.1002/chem.v16:38
Pham, N. N.; Dang, T. T.; Ngo, N. T. Villinger, A.; Ehlers, P.; Langer, P. Org. Biomol. Chem. 2015, 13, 6047. doi: 10.1039/C5OB00720H
陈静, 曲红梅, 彭静, 陈超, 有机化学, 2015, 35, 937. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345052.shtmlChen, J.; Qu, H.; Peng, J.; Chen, C. Chin. J. Org. Chem. 2015, 35, 937(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345052.shtml
Lu, B.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2014, 136, 11598. doi: 10.1021/ja5059795
Wu, J.; Yoshikai, N. Angew. Chem. Int. Ed. 2015, 54, 11107. doi: 10.1002/anie.201504687
Deng, G. B.; Zhang, J. L.; Liu, Y. Y.; Liu, B.; Yang, X. H.; Li, J. H. Chem. Commun. 2015, 51, 1886. doi: 10.1039/C4CC08498E
Chen, F.; Huang, X.; Li, X.; Shen, T.; Zou, M.; Jiao, N. Angew. Chem. Int. Ed. 2014, 53, 10495. doi: 10.1002/anie.201406479
Chen, T.; Chen, X.; Wei, J.; Lin, D.; Xie, Y.; Zeng, W. Org. Lett. 2016, 18, 2078. doi: 10.1021/acs.orglett.6b00709
Guimond, N.; Fagnou, K. J. Am. Chem. Soc. 2009, 131, 12050. doi: 10.1021/ja904380q
Wei, X.; Zhao, M.; Du, Z.; Li, X. Org. Lett. 2011, 13, 4636. doi: 10.1021/ol2018505
Zhang, S.; Liu, X.; Chen, S.; Tan, D.; Jiang, C.; Wu, J.; Li, Q.; Wang, H. Adv. Synth. Catal. 2016, 358, 1705. doi: 10.1002/adsc.v358.10
He, R.; Huang, Z.; Zheng, Q.; Wang, C. Angew. Chem., Int. Ed. 2014, 53, 4950. doi: 10.1002/anie.201402575
Badra, K.; Kumar, G. S. Med. Res. Rev. 2011, 31, 821. doi: 10.1002/med.v31.6
Jayakumar, J.; Parthasarathy, K.; Cheng, C. H. Angew. Chem., Int. Ed. 2012, 124, 201. doi: 10.1002/ange.201105755
Lao, Y.; Zhang, S.; Liu, X.; Jiang, C.; Wu, J.; Li, Q.; Huang, Z.; Wang, H. Adv. Synth. Catal. 2016, 358, 2186. doi: 10.1002/adsc.v358.13
Zhang, W.; Wang, C.; Lin, H.; Dong, L.; Xu, Y. Chem.-Eur. J. 2016, 22, 907. doi: 10.1002/chem.201504245
Tran, D. N.; Cramer, N. Angew. Chem., Int. Ed. 2012, 122, 8357.
Liang, Yu.; Müller, V.; Liu, W.; Münch, A.; Stalke, D.; Ackermann, L. Angew. Chem., Int. Ed. 2017, 56, 9415. doi: 10.1002/anie.v56.32
Manikandan, R.; Tamizmani, M.; Jeganmohan, M. Org. Lett. 2017, 19, 6678. doi: 10.1021/acs.orglett.7b03405
Wang, H.; Lorion, M. M.; Ackermann, L. ACS Catal. 2017, 7, 3430. doi: 10.1021/acscatal.7b00756
Li, X.; Li, X.; Jiao, N. J. Am. Chem. Soc. 2015, 137, 9246. doi: 10.1021/jacs.5b05843
Gao, B.; Liu, S.; Lan, Y.; Huang, H. Organometallics 2016, 35, 1480. doi: 10.1021/acs.organomet.6b00072
Wang, J.; Zha, S.; Chen, K.; Zhang, F.; Zhu, J. Org. Biomol. Chem. 2016, 14, 4848. doi: 10.1039/C6OB00901H
Kim, J. H.; Greßies, S.; Glorius, F. Angew. Chem., Int. Ed. 2016, 55, 5577. doi: 10.1002/anie.201601003
Li, X.; Sun, M.; Jin, Q.; Liu, K.; Liu, P. N. J. Org. Chem. 2016, 81, 3901. doi: 10.1021/acs.joc.6b00264
Wang, H.; Li, L.; Yu, S.; Li, Y.; Li, X. Org. Lett. 2016, 18, 2914. doi: 10.1021/acs.orglett.6b01284
Chen, X.; Liu, X.; Su, S. J.; Li, J.; Zeng, W. Chem. Commun. 2016, 52, 5856. doi: 10.1039/C6CC00254D
Cenini, S.; Gallo, E.; Caselli, A.; Ragaini, F.; Fantauzzi, S.; Piangiolino, C. Coord. Chem. Rev. 2006, 250, 1234. doi: 10.1016/j.ccr.2005.10.002
Yu, D. G.; Suri, M.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 8802. doi: 10.1021/ja4033555
Peng, J.; Xie, Z.; Chen, M.; Wang, J.; Zhu, Q. Org. Lett. 2014, 16, 4702. doi: 10.1021/ol502010g
Lian, Y.; Hummel, J. R.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2013, 135, 12548. doi: 10.1021/ja406131a
Wang, X.; Jiao, N. Org. Lett. 2016, 18, 2150. doi: 10.1021/acs.orglett.6b00774
Park, Y.; Park, K. T.; Kim, J. G.; Chang, S. J. Am. Chem. Soc. 2015, 137, 4534. doi: 10.1021/jacs.5b01324
Wang, J.; Zha, S.; Chen, K.; Zhang, F.; Song, C.; Zhu, J. Org. Lett. 2016, 18, 2062. doi: 10.1021/acs.orglett.6b00691
Wang, F.; Wang, H.; Wang, Q.; Yu, S.; Li, X. Org. Lett. 2016, 18, 1306. doi: 10.1021/acs.orglett.6b00227
Wang, X.; Lerchen, A.; Glorius, F. Org. Lett. 2016, 18, 2029.
Wang, H.; Lorion, M. M.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 10386. doi: 10.1002/anie.201603260
Yu, S.; Tang, G.; Li, Y.; Zhou, X.; Lan, Y.; Li, X. Angew. Chem., Int. Ed. 2016, 55, 8696. doi: 10.1002/anie.201602224
Li, L.; Wang, H.; Yu, S.; Yang, X.; Li, X. Org. Lett. 2016, 18, 3662. doi: 10.1021/acs.orglett.6b01716
Wang, Q.; Li, X. Org. Lett. 2016, 18, 2102. doi: 10.1021/acs.orglett.6b00727
(a) Zhu, R. -Y. ; Liu, L. -Y. ; Yu, J. -Q. J. Am. Chem. Soc. 2017, 139, 12394.
(b) Xu, J. ; Liu, Y. ; Wang, Y. ; Li, Y. ; Xu, X. ; Jin, Z. Org. Lett. 2017, 19, 1562.
(c) Yao, Q. -J. ; Zhang, S. ; Zhan, B. -B. ; Shi, B. -F. Angew. Chem., Int. Ed. 2017, 56, 6617.
(d) Wu, Y. ; Chen, Y. -Q. ; Liu, T. ; Eastgate, M. D. ; Yu, J. -Q. J. Am. Chem. Soc. 2016, 138, 14554.
(e) Liu, Y. ; Ge, H. Nat. Chem. 2016, 9, 26.
(f) Xu, Y. ; Young, M. C. ; Dong, G. J. Am. Chem. Soc. 2017, 139, 5716.
(g) Yada, A. ; Liao, W. ; Sato, Y. ; Murakami, M. Angew. Chem., Int. Ed. 2017, 56, 1073.
Chen, X. Y.; Ozturk, S.; Sorensen, E. J. Org. Lett. 2017, 19, 1140. doi: 10.1021/acs.orglett.7b00161
Hu, W.; Zheng, Q.; Sun, S.; Cheng, J. Chem. Commun. 2017, 53, 6263. doi: 10.1039/C7CC03006A
Shen, J.; Liu, X.; Wang, L.; Chen, Q.; He, M. Synth. Commun. 2018, 9, 1.
Tan, P. W.; Juwaini, N. A. B.; Seayad, J. Org. Lett. 2013, 15, 5166. doi: 10.1021/ol402145m

图式 1 亚胺参与的C—H键官能团化/环化反应
Scheme 1 C—H bond functionalization/cyclization involving imines

图式 2 Pd-催化N-芳基亚胺的分子内环化反应
Scheme 2 Pd-catalyzed intramolecular cyclization of N-aryl imines

图式 3 Pd-催化N-烯丙基亚胺的分子内环化反应
Scheme 3 Pd-catalyzed intramolecular cyclization reaction of N-allyl imines

图式 4 Pd-催化N-环烯基亚胺的分子内环化反应
Scheme 4 Pd-catalyzed intramolecular cyclization reaction of N-cycloalkenyl imines

图式 5 Pd-催化N-(2-甲基)-烯丙基亚胺的分子内环化反应
Scheme 5 Pd-catalyzed intramolecular cyclization reaction of N-(2-methyl)-allyl imines

图式 6 Cu-或无金属催化N-吡啶亚胺的分子内环化反应
Scheme 6 Cu or metal free-catalyzed intramolecular cyclization of N-pyridyl imines

图式 7 Cu-催化N-吡啶亚胺的分子内环化反应
Scheme 7 Cu-catalyzed intramolecular cyclization of N-pyridyl imines

图式 8 Pd-催化N-吡啶亚胺与炔烃的[3+2]环化反应
Scheme 8 Pd-catalyzed [3+2] cyclization of N-pyridyl imines with alkynes

图式 9 Rh-催化N-吡啶亚胺与重氮化合物的[3+2]环化反应
Scheme 9 Rh-catalyzed [3+2] cyclization of N-pyridyl imines with diazo compounds

图式 10 Cu-催化N-芳基亚胺与重氮化合物的[3+2]环化反应
Scheme 10 Cu-catalyzed [3+2] cyclization of N-aryl imines with diazo compounds

图式 11 Pd-催化亚胺与二功能基取代芳烃的[3+2]环化反应
Scheme 11 Pd-catalyzed [3+2] cyclization of imines with o-difunctionalized arenes

图式 12 Pd-催化亚胺与高价碘试剂的[3+2]环化反应
Scheme 12 Pd-catalyzed [3+2] cyclization of imines with hypervalent iodine

图式 13 无金属催化亚胺与亚硝酸叔丁基酯的环化反应
Scheme 13 Metal-free catalyzed cyclization of imines with hypervalent iodines

图式 14 Cu-催化N-芳基亚胺与叠氮化钠的[5+1]环化反应
Scheme 14 Cu-catalyzed [5+1] cyclization of N-aryl imines with sodium azides

图式 15 过渡金属催化亚胺与炔烃的[4+2]环化反应
Scheme 15 Transition metal-catalyzed [4+2] cyclization of imines with alkynes

图式 16 过渡金属催化亚胺与烯烃或联烯的环化反应
Scheme 16 Transition metal-catalyzed cyclization of imines with allenes or alkenes

图式 17 Rh-催化酮亚胺与CO的[4+1]环化反应
Scheme 17 Rh-catalyzed [4+1] cyclization of ketimines with CO

图式 18 Rh-催化酮亚胺与重氮化合物的[4+2]环化反应
Scheme 18 Rh-catalyzed [4+2] cyclization of ketimines with diazo compounds

图式 19 Rh-催化芳基酮亚胺C—H键的接力卡宾功能化反应
Scheme 19 Rh-catalyzed relay carbenoid functionalization of aryl ketimine C—H bonds

图式 20 过渡金属催化亚胺与叠氮化合物的环化反应
Scheme 20 Transition metal-catalyzed cyclization of imines with azides

图式 21 过渡金属催化亚胺与二氧氮茂酮的[4+2]环化反应
Scheme 21 Transition metal-catalyzed [4+2] cyclization of imines with dioxazolones

图式 22 过渡金属催化亚胺与氨茴内酐的[4+1]环化反应
Scheme 22 Transition metal-catalyzed [4+1] cyclization of imines with anthranils

图式 23 Rh-催化苯甲酰亚胺与亚硝基苯的[4+1]环化反应
Scheme 23 Rh-catalyzed [4+1] cyclization of benzimides with nitrosobenzenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
