图 1.
类二芳基甲基哌嗪类δ阿片受体激动剂
Figure 1.
Benzhydrylpiperazine-like δ OR agonists

吗啡、芬太尼等阿片类镇痛药是用于中重度疼痛和癌痛治疗的临床常用药物.然而, 此类药物伴有一系列严重副反应[1], 例如欣快感引起强烈的成瘾和滥用倾向, 而伴随的呼吸抑制则可能危及生命.由于监管不力, 近年来美国出现大量阿片成瘾和过量致死案例, 成为关注焦点. 2017年6月, Endo公司的羟吗啡酮缓释制剂Opana ER因滥用风险被撤市; 同年10月, 美国政府宣布进入全国公共卫生紧急状态以应对阿片类药物滥用危机.此外, 阿片类药物还存在尿潴留、恶心、便秘、呕吐、头痛、眩晕和嗜睡等其它副作用.因此, 寻找更加安全有效的镇痛药物是阿片类药物研究领域的重大课题.
阿片类药物的靶标是分布于中枢和外周神经系统的阿片受体.阿片受体有四种亚型, 即μ阿片受体、δ阿片受体、κ阿片受体和阿片受体样受体1 (ORL-1受体)[1].不同的阿片类药物对四种阿片受体有着不同的活性, 产生的药理效应也不同.根据与阿片受体的关系, 一般将阿片类药物分为相应的激动剂、拮抗剂、部分激动剂、混合激动-拮抗剂等[2].
为克服阿片类药物的副作用, 并拓展临床用途, 新型阿片类药物的研发一直受到广泛关注. 1980~2000年陆续发现了全部阿片受体的选择性激动剂和拮抗剂.在此基础上, 同时靶向两种以上阿片受体的小分子药物成为近期研究热点.同时, 由于阿片受体晶体结构和信号传导[3~6]等相关领域的进展, G蛋白偏向性阿片受体激动剂研究也备受研究者关注.在此, 我们将对小分子阿片类药物研究中的一些热点进行归纳总结.
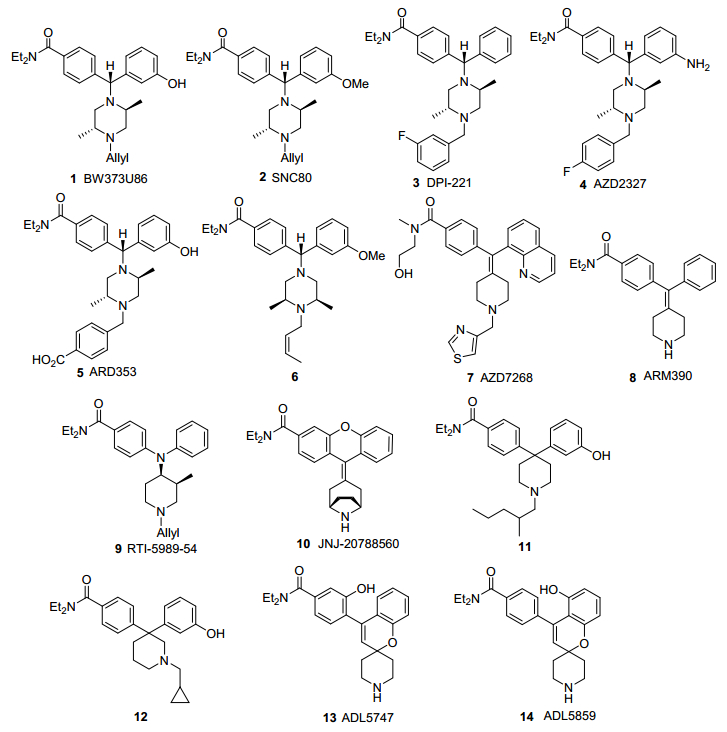
δ阿片受体药物研究始于20世纪80年代, Portoghese课题[7]组基于脑啡肽与δ受体相互作用的“信号-地址”模型, 发现了吗啡喃类δ受体拮抗剂纳曲吲哚.然而, 纳曲吲哚并无明显的药理作用.另一方面, 20世纪90年代BW373U86 (1, 图 1)等选择性激动剂的发现使得δ受体成为受关注的药物靶点[8].现已明确δ受体激动剂具有多方面的药理作用, 如镇痛、抗抑郁、抗焦虑、心脏保护和神经保护作用等.目前已经报道了大量δ受体激动剂, 其中类二芳基甲基哌嗪类化合物是最重要的类别[9~11].结构上, 此类化合物又可细分为6种结构类型:二芳基甲基哌嗪类、二芳基亚甲基哌啶类、二芳基氨基哌啶类、莨菪烷类、偕二芳基哌啶类和螺环哌啶类.
二芳基甲基哌嗪类包括BW373U86 (1)、SNC80 (2)、DPI-221 (3)、AZD2327 (4)和ARD-353 (5)等化合物, 是最早发现的δ受体激动剂结构类型.化合物1[12]是Bur- roughs Wellcome(现GSK)研发的首个非肽类小分子δ阿片受体激动剂.在动物研究中, 化合物1镇痛作用弱, 仅对慢性疼痛具有镇痛作用, 对急性疼痛无效, 有惊厥副作用.化合物2[13]是Rice小组报道的化合物1的O-甲基化衍生物, 活性有所下降, 但δ受体选择性显著提高.含有羧基的化合物如5亲脂性弱[14], 是作用于外周的药物, 无惊厥等中枢副作用.此类骨架中哌啶环取代基的手性和氮取代基对激动活性也有显著影响, 例如, 取代基构型改变并带有N-顺丁烯基的化合物6为δ受体拮抗剂[15].
二芳基亚甲基哌啶类包括AZD7268 (7)和ARM390 (8)等, 是由阿斯利康研发的化合物, 以带有环外碳碳双键的哌啶环替代化合物1中的哌嗪环.化合物8为δ受体完全激动剂, 不引起δ受体磷酸化或内吞, 能有效激活G蛋白信号通路, 产生镇痛作用[16].
二芳基氨基哌啶类代表性化合物有Carroll课题组[17]报道的RTI-5989-54 (9), 带有4-位二芳基氨基取代的哌啶环取代化合物1中的哌嗪环, 也具有良好的δ受体激动活性.
莨菪烷类代表性化合物有JNJ-20788560 (10), 莨菪烷环3-位二芳基氨基或二芳基亚甲基取代时具有良好的δ受体激动活性[18].
偕二芳基哌啶类包括4, 4-偕二芳基哌啶和3, 3-偕二芳基哌啶两类相近的结构, δ受体激动活性较弱, 例如最近辉瑞报道的化合物11和12属于δ受体拮抗剂[19].
螺环哌啶类包括ADL5859 (13)和ADL5747 (14)等由Adolor公司(现默克)研发的一类δ受体药物[20, 21].化合物13的δ受体亲和力达亚纳摩尔级, 在炎症疼痛敏化大鼠模型中能逆转δ受体介导的疼痛敏化, 起效快速, 无明显副作用, 不引起惊厥或增加自发活动, 安全性良好.该药物也具有抗抑郁作用.化合物14也能够有效镇痛, 同时不影响自主活动.与化合物13相比, 14在大鼠模型中口服吸收更好, 抗疼痛敏化作用更强, 并且代谢半衰期更长.
上述化合物中有多种相继进入临床试验[22], 但尚未获准上市. Adolor和辉瑞合作进行了化合物13的临床研究, 在临床一期研究中口服吸收和耐受性方面性质良好, 但在多项临床二期研究中, 化合物13并没有显著的镇痛作用, 因此其研发已于2010年终止.化合物14进入了临床一期研究, 但研发也已终止.阿斯利康对化合物4和7进行了抗抑郁治疗临床试验, 但未见后续报道.由于δ受体选择性激动剂的临床价值尚不明确, 相关研究逐渐转向混合型阿片受体药物.
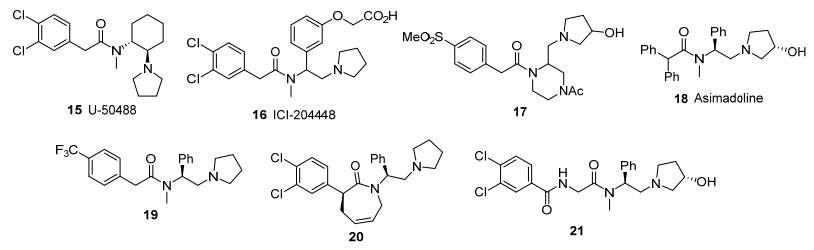
κ阿片受体药物研发可追溯到20世纪80年代初期, Upjohn公司(现辉瑞)在靶向非µ受体药物的研究中发现了首个κ受体激动剂U-50488 (15, 图 2)[23].化合物15及其类似物Spiradoline属于芳基乙酰胺类化合物, 镇痛活性良好, 无阿片样副作用, 并能对抗µ激动剂依赖.然而, 这些中枢选择性激动剂具有心境恶劣、幻觉、嗜睡、多尿等严重副作用, 应用前景不尽人意.因此, 外周选择性κ激动剂是近年来的研发重点, 已有多种化合物进入临床试验, 有望获准上市[24].
ICI制药公司研发的ICI-204448 (16, 图 2)是最早的外周选择性κ激动剂[25].由于在分子结构中引入羧基, 化合物的亲脂性下降, 难以进入中枢神经系统.化合物16的受体亲核性强, 对炎症疼痛具有良好的镇痛活性, 但口服活性较弱.同期由Galaxo(现GSK)研发的化合物17(图 2)分子结构中具有亲水性的羟基和砜基, 也具有良好的外周选择性[26].
Asimadoline (18, 图 2)是德国默克和Tioga公司研发的首个进入临床试验的外周选择性芳基乙酰胺类κ受体激动剂[27], 通过皮下或口服给药, 对炎症痛觉过敏模型表现出剂量依赖性的镇痛效果, 但对非炎症疼痛模型没有活性, 能减轻结肠压迫性膨胀、肠易激综合征、发热以及其它内脏损伤引起的疼痛, 并在镇痛剂量内产生利尿作用.化合物18几乎不通过血脑屏障, 因此中枢副作用轻微.化合物18用于治疗腹泻型肠易激综合征处于临床三期, 已获美国食品药品管理局(FDA)批准进入快速通道.
Adolor也研发了多种外周选择性芳基乙酰胺类κ受体激动剂.在构效关系研究中, 发现酰基片段的芳基取代基对外周选择性有显著影响, 含4-三氟甲基苯基的化合物19(图 2)的外周选择性较ICI-199441提高4倍以上[28]. Adolor的ADL-10-0101、ADL-10-0116和ADL-10-0398相继进入临床试验[24], 但具体结构未被报道. ADL-10-0101在炎性疼痛模型中经皮下或口服给药均有镇痛作用; 在搔痒模型中皮下给药可起止痒作用. ADL-10-0101治疗内脏疼痛和皮肤搔痒的临床二期试验已经完成, 用于术后疼痛和其它炎性疼痛的研究进入临床二期. ADL-10-0116和ADL-10-0398的口服活性和外周选择性更好, 作用时间更长.在大鼠直肠膨胀性疼痛模型中, ADL-10-0116经皮下或口服给药可产生剂量依赖的镇痛作用, 作用时间均超过60 min, 与吗啡相当. ADL-10-0116用于治疗炎性疼痛的研究进入一期临床阶段. ADL-10-0398处于临床前研究阶段, 在炎性疼痛模型中的口服镇痛活性分别为ADL-10-0116的1/40和1/9.
Adolor还以芳基乙酰胺为先导物, 设计合成了氮杂环庚-2-酮类κ受体激动剂, 其中活性最高的化合物20(图 2)在啮齿动物模型中有强效镇痛作用, 具有良好的外周选择性[29].芳基乙酰胺的芳基部分也可以肽链替代, 轭合物21对κ受体具有高亲和力和选择性, 并且亲脂性较低, 不易进入中枢系统[30].
ORL1受体广泛分布于大脑、脊髓和外周组织, 其内源性配体为孤啡肽/痛敏肽(Orphanin FQ/nociception, NOP), 与疼痛、抑郁、焦虑、依赖和耐受、学习和记忆、帕金森症等机体功能或疾病有关, 是有潜力的药物靶点[31].近期研究表明, ORL1拮抗剂可用于抑郁、疼痛、认知障碍、肥胖症、帕金森症的治疗, 因此受到广泛关注, 已有一些化合物进入临床试验[31].
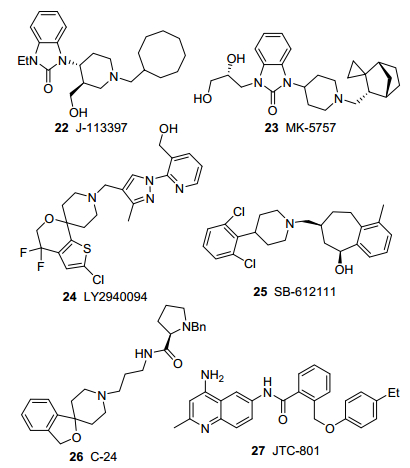
苯并咪唑酮类化合物J-113397[32](22, 图 3)是由Banyu公司1999年报道的首个ORL1受体小分子拮抗剂, 也是首个ORL1受体药物, 亲和性达到纳摩尔级, 受体选择性比其它阿片受体高600倍以上, 是目前药理研究常用的工具药物.化合物22在体内和体外均可有效拮抗NOP和其它ORL1受体激动剂的作用, 如能拮抗NOP引起的小鼠痛敏, 也可逆转NOP抑制EFS刺激所引起的GPI和MVD收缩.在细胞水平上, 可抑制佛司可林引起的cAMP生成.由于化合物22的药代动力学性质和口服利用度不佳, Banyu开展了进一步优化, 通过以其它环烷基替代环辛基、增强哌啶环疏水性、在苯并咪唑酮环上引入N-亲水取代基等方法, 得到了性质更为理想的衍生物MK-5757[33](23, 图 3).目前化合物23正处于临床二期, 用于治疗精神分裂症的认知障碍.
礼来公司研发的4-芳基哌啶类ORL1拮抗剂LY2940094[34](24, 图 3)用于治疗重度抑郁症[35]和酒精依赖, 在临床一期研究中完成了安全性和药代动力学评价, 已进入临床二期.化合物24在2~800 mg剂量单次给药以及40~200 mg多次给药14 d后耐受性良好, 未发现明显副作用.在健康志愿者中, 单次40 mg剂量的ORL1受体占有率为480% (2.5 h)和470% (26.5 h), 表明可对ORL1受体发挥持续高度拮抗作用.
SB-612111 (25, 图 3)是GSK报道的4-芳基哌啶类ORL1拮抗剂[36], 在大鼠炎性疼痛模型中无镇痛作用, 但可逆转吗啡耐受性.化合物25在大鼠帕金森症模型中可改善帕金森病症状, 并能与左旋多巴(L-DOPA)发挥协同作用[37].
Calò课题组[38]报道了4-芳基哌啶类ORL1拮抗剂C-24 (26, 图 3), 在大鼠镇痛实验中化合物26可拮抗NOP颅内注射的致痛作用和鞘内注射的镇痛作用.此外, Stevens课题组[6]以化合物26为配体完成了ORL1受体晶体结构的解析.
JTC-801 (27, 图 3)是JT公司发现的4-氨基喹啉类ORL1受体拮抗剂[39], 受体亲和性较好(Ki 44.6 nmol/L), 能完全拮抗孤啡肽所引起的对cAMP积累的抑制, 这种作用不能被纳诺酮抑制.
联合用药研究发现各阿片受体介导的生理效应之间存在一定的相互制衡和增强的关系.例如, δ受体激动剂镇痛作用弱, 但可加强μ受体激动剂的镇痛作用, 减弱μ受体激动剂引起的呼吸抑制和肌肉僵直, 同时μ受体激动剂可消除δ受体激动剂的惊厥副作用[15, 40]; δ受体激动剂无依赖性和成瘾性等阿片样副作用[41], 与μ受体激动剂一起使用能减弱其成瘾性[42].再如, κ受体选择性激动剂可强效镇痛[43], 其厌恶感和幻觉副作用可对抗µ受体激动剂的成瘾性[44].此外, δ受体拮抗剂与μ受体激动剂联用可显著减弱镇痛耐受.因此, 寻找阿片受体混合活性化合物是近期的研发热点之一, 多种混合型阿片激动剂和阿片受体激动剂/拮抗剂相继进入临床前和临床研究, 部分已获准上市, 表现出良好的研发前景.
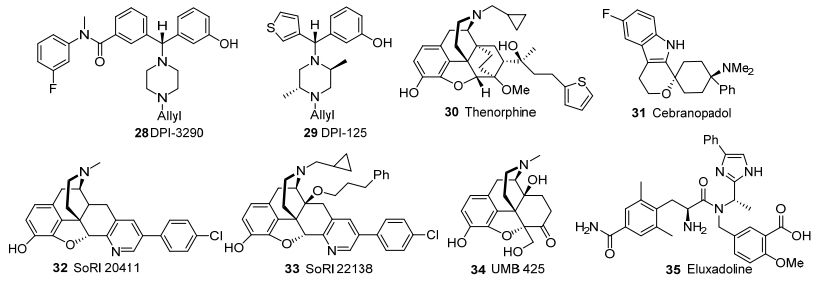
由Ardent公司研发的DPI-3290 (28)和DPI-125 (29)(图 4)属于二芳基甲基哌嗪δ、μ和κ受体混合激动剂, 结构上与δ激动剂BW373U86接近.在二芳基甲基哌嗪类δ激动剂的构效关系研究中, Chang等[45, 46]发现二芳基甲基哌嗪骨架中左上苯基的酰胺取代位置从对位改为间位时, δ受体激动活性基本保持, 但受体选择性下降, µ和κ受体活性增强, 所得化合物具备强效镇痛活性, 而且呼吸抑制和成瘾性副作用均明显减轻.在二苯基甲基哌嗪化合物中筛选得到的化合物28作为镇痛药进入临床二期试验.二芳基甲基哌嗪骨架中的左上苯基以噻吩基替代, 得到的噻吩苯基甲基哌嗪化合物也具有类似的药理性质[46], 其中化合物29进入临床一期试验.
Chang和Shen课题组[47, 48]近期对化合物28、29及其类似物的药理研究表明, 此类混合激动剂具有较为均衡的δ、μ和κ受体活性.例如, 化合物29激活δ、μ和κ阿片受体的活性差距较小(EC50=4, 11, 17 nmol/L).值得注意的是, 受体活性顺序为δ>μ~κ时, 能同时有效减弱μ受体激动导致的呼吸抑制和成瘾性, 因此是一类高效安全的镇痛药物[47].在呼吸抑制大鼠模型中, 化合物29表现出高呼吸安全特性, 而且μ和δ受体的激动活性EC50比与呼吸安全指数正相关, 表明该化合物的呼吸安全性来源于其高δ受体活性.在猕猴自身给药模型中, 化合物29表现出低成瘾性, 这与其对μ和κ受体具有相近活性有关.
由军事医学科学研究院研制的吗啡喃类化合物噻诺菲(30, 图 4)属于混合型μ和κ受体激动剂[49], 目前处于临床二期, 用于治疗阿片药物成瘾.与丁丙喏啡相比, 化合物30有镇痛能力强、作用时间更长、依赖性低和安全系数大等优点, 有望成为高效低毒的镇痛药和安全有效的戒毒药物.
由格兰泰研发的吲哚类阿片镇痛药Cebranopadol (31, 图 4)为阿片受体混合激动剂[50], 对μ和δ受体是完全激动剂, 对ORL1和κ受体是部分激动剂, 目前处于临床三期.在大鼠甩尾试验中, 化合物31静脉注射和口服均有效, 与芬太尼(30 min)和吗啡(3 h)相比, 等效剂量镇痛作用可持续7 h.在关节炎痛、骨癌痛、糖尿病性多发性神经痛和脊神经结扎慢性疼痛模型中, 静脉注射化合物31可有效镇痛.
Ananthan课题组[51]报道的吡啶并吗啡喃衍生物SoRI20411 (32, 图 4)是μ受体激动剂/δ受体拮抗剂, 颅内给药的镇痛效果比吗啡约低10倍, 但耐受性降低.与化合物32相比, 14-烷氧基类似物SoRI22138 (33)是更加有效的μ受体激动剂/δ受体拮抗剂[52], 在μ受体和δ受体共表达细胞中不产生依赖性和耐受性.在大鼠甩尾实验中, 化合物33颅内注射的镇痛效果与吗啡相当, 并且耐受性降低.
Matsumoto课题组[53]报道的UMB 425 (34, 图 4)结构上与纳曲酮接近, 具有μ受体激动剂/δ受体拮抗剂活性, 对μ受体的亲和力为纳摩尔级, 对δ受体有拮抗活性.大鼠实验显示, 化合物34的镇痛效果类似吗啡, 长时间使用不产生耐受, 无明显毒性.
由Janssen公司研发的外周选择性苯基咪唑类阿片药物艾沙度林(35, 图 4)表现出μ和κ受体激动活性及δ阿片受体拮抗活性[54, 55], 对μ受体亲和力较高(Ki=1.7 nmol/L), 对κ受体亲和力中等(Ki=55 nmol/L), 对δ受体亲和力较低(Ki=367 nmol/L), 用于治疗肠易激综合症的腹泻和腹部疼痛症状, 也可用于对抗单纯激活μ受体产生的耐受和滥用.艾沙度林已于2015年在美国获准上市.
阿片受体的信号传导主要有G蛋白和β-arrestin两种途径, 传统的µ受体激动剂镇痛药同时作用于μ受体的G蛋白和β-arrestin通路.目前认为, 在μ受体的信号传导中, G蛋白通路介导镇痛作用, β-arrestin通路则与呼吸抑制、便秘等副作用有关, 如能在选择性激活G蛋白通路的同时避免激活β-arrestin通路, 将有可能获得副作用轻微的新型镇痛药[56, 57].因此, μ受体偏向性激动剂成为近期备受关注的研发热点.
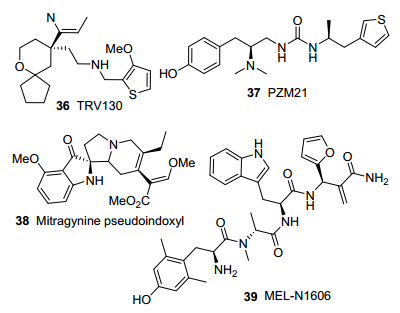
基于μ受体晶体结构, Trevena公司研发了选择性激活μ受体G蛋白通路的偏向性激动剂TRV130 (36, 图 5)[58, 59].化合物36是以μ受体β-arrestin通路活性较低的4-苯基-4-(2-苄胺乙基)四氢吡喃类似物为先导化合物, 通过在四氢吡喃环上引入手性中心, 减弱β-arrestin通路活性而优化得到. cAMP累积法测得化合物36的G蛋白通路活性为吗啡的84%, 而PathHunter酶互补法测得的β-arrestin通路活性仅约15%, 呈现出显著的G蛋白激活偏向性.临床二期试验结果表明, 53%的吗啡组病人发生呼吸抑制、恶心、呕吐等不良反应, 而化合物36组只有15%~31%病人出现这些反应.化合物36镇痛强效快速, 耐受性、成瘾性、便秘和呼吸抑制作用比吗啡明显减弱, 已经进入临床三期.
Shoichet等课题组[60]通过合作, 对小分子结构与μ受体晶体结构进行分子对接和虚拟筛选, 继而进行实验验证和优化, 筛选得到G蛋白偏向性μ受体选择性激动剂PZM21 (37, 图 5).化合物37左侧苯环的酚羟基能与μ受体的His297残基形成氢键网络而增强活性.化合物37的G蛋白通路活性与吗啡相当, 但无β-arrestin通路活性.在大鼠模型实验中, 化合物37表现强镇痛活性, 而且便秘和呼吸抑制等不良反应显著减轻.
Majumdar课题组[61]近期发现吲哚类天然产物衍生物mitragynine pseudoindoxyl (38, 图 5)为G蛋白偏向的μ受体激动剂/δ受体拮抗剂.化合物38在功能实验中对μ受体有低纳摩尔级激动活性, 但对于β-arrestin-2通路没有激活作用.在大鼠甩尾和热板镇痛试验中, 化合物38表现出较强的镇痛作用, 耐受性、呼吸抑制、便秘、成瘾性等副作用比吗啡显著降低.
王锐课题组[62]近期报道了对G蛋白信号通路具有偏向激动性的内吗啡肽合成类似物MEL-N1606 (39, 图 5), 通过残基结构修饰, 显著提高了μ受体亲和性(EC50=0.00177 nmol/L, 内吗啡肽-1的EC50=14.4 nmol/L). MEL-N1606的β-arrestin-2募集活性较弱, 具有显著的G蛋白通路选择性.化合物39在动物模型中表现出强镇痛作用, 比内吗啡肽更易于通过血脑屏障, 外周注射后能够发挥中枢镇痛作用, 副作用明显减弱.
阿片类药物主要用于镇痛, 但伴有多种严重的副作用, 限制了临床应用.因此, 新型阿片类药物的研发具有重要的科学及社会意义, 一直受到广泛关注.现已获得各阿片受体的高选择性激动剂和拮抗剂, 但值得注意的是, 研究结果表明提高受体选择性无法克服副作用.得益于阿片类药物药理和阿片受体三维结构、信号通路等方面的研究进展, 同时靶向多种阿片受体的混合型药物和选择性激活G蛋白信号通路的偏向性药物是目前普遍认为较有前景的发展方向.相信随着研究的不断深入, 将有望获得药效强、副作用轻微、易于应用的新型阿片类药物.
Feng, Y.; He, X. Z.; Yang, Y. L.; Chao, D. M.; Lazarus, L. H.; Xia, Y. Curr. Drug Targets 2012, 13, 230. doi: 10.2174/138945012799201612
Smith, N. J.; Bennett, K. A.; Milligan, G. Mol. Cell. Endocrinol. 2011, 331, 241. doi: 10.1016/j.mce.2010.07.009
Huang, W. J.; Manglik, A.; Venkatakrishnan, A. J.; Laeremans, T.; Feinberg, E. N.; Sanborn, A. L.; Kato, H. E.; Livingston, K. E.; Thorsen, T. S.; Kling, R.; Granier, S.; Gmeiner, P.; Husbands, S. M.; Trayner, J. R.; Weis, W. I.; Steyaert, J.; Dror, R. O.; Kobilka, B. K. Nature 2015, 524, 315. doi: 10.1038/nature14886
Fenalti, G.; Giguere, P. M.; Katritch, V.; Huang, X. P.; Thompson, A. A.; Cherezov, V.; Roth, B. L.; Stevens, R. C. Nature 2014, 506, 191. doi: 10.1038/nature12944
Wu, H. X.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G. W.; Vardy, E.; Liu, W.; Thompson, A. A.; Huang, X. P.; Carroll, F. I.; Mascarella, S. W.; Westkaemper, R. B.; Mosier, P. D.; Roth, B. L.; Cherezov, V.; Stevens, R. C. Nature 2012, 485, 327. doi: 10.1038/nature10939
Thompson, A. A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H. X.; Vardy, E.; Huang, X. P.; Trapella, C.; Guerrini, R.; Calo, G.; Roth, B. L.; Cherezov, V.; Stevens, R. Nature 2012, 485, 395. doi: 10.1038/nature11085
Portoghese, P. S.; Sultana, M.; Nagase, H. J. Med. Chem. 1988, 21, 281. http://europepmc.org/abstract/MED/2828619
Chang, K. J.; Yi, S. P.; Shen, Y. H. Neural Functions of the Delta-Opioid Receptor, Ed.: Xia, Y., Springer, Switzerland, 2015, pp. 45~65.
Calderon, S. N.; Coop, A. Curr. Pharm. Des. 2004, 10, 733. doi: 10.2174/1381612043453054
Calderon, S. N. Top. Curr. Chem. 2011, 299, 121.
Bishop, M. J.; McNutt, R. W. The Dela Receptor, Marcel Dekker, New York, Basel, 2004, pp. 113~138.
Chang, K. J.; Rigdon, G. C.; Howard, J. L.; McNutt, R. W. J. Pharmacol. Exp. Ther. 1993, 267, 852. http://www.ncbi.nlm.nih.gov/pubmed/8246159
Calderon, S. N.; Rothman, R. B.; Porreca, F.; Flippen-Anderson, J. L.; McNutt, R. W.; Xu, H.; Smith, L. E.; Bilsky, E. J.; Davis, P.; Rice, K. C. J. Med. Chem. 1994, 37, 2125. doi: 10.1021/jm00040a002
Watson, M. J.; Holt, J. D. S.; O'Neill, S. J.; Wei, K.; Pendergast, W.; Gross, G. J.; Gengo, P. J.; Chang, K. J. J. Pharmacol. Exp. Ther. 2006, 316, 423.
Su, Y. F.; Mcnutt, R. W.; Chang, K. J. J. Pharmacol. Exp. Ther. 1998, 287, 815. http://europepmc.org/abstract/MED/9864259
Marie, N.; Landemore, G.; Debout, C.; Jauzac, P.; Allouche, S. Life Sci. 2003, 73, 1691. doi: 10.1016/S0024-3205(03)00489-2
Thomas, J. B.; Herault, X. M.; Rothman, R. B.; Atkinson, R. N.; Burgess, J. P.; Mascarella, S. W.; Dersch, C. M.; Xu, H.; Flippen-Anderson, J. L.; George, C. F.; Carroll, F. I. J. Med. Chem. 2001, 44, 972. doi: 10.1021/jm000427g
Carson, J. R.; Coats, S. J.; Codd, E. E.; Dax, S. L.; Lee, J.; Martinez, R. P.; Neilson, L. A.; Pitis, P. M.; Zhang, S. P. Bioorg. Med. Chem. Lett. 2004, 14, 2109. doi: 10.1016/j.bmcl.2004.02.051
Liras, S.; McHardy, S. F.; Allen, M. P.; Segelstein, B. E.; Heck, S. D.; Bryce, D. K.; Schmidt, A. W.; Vanase-Frawley, M.; Callegari, E.; McLean, S. Bioorg. Med. Chem. Lett. 2010, 20, 503. doi: 10.1016/j.bmcl.2009.11.113
Le Bourdonnec, B.; Windh, R. T.; Ajello, C. W.; Leister, L. K.; Gu, M. H.; Chu, G. H.; Tuthill, P. A.; Barker, W. M.; Koblish, M.; Wiant, D. D.; Graczyk, T. M.; Belanger, S.; Cassel, J. A.; Feschenko, M. S.; Brogdon, B. L.; Smith, S. A.; Christ, D. D.; Derelanko, M. J.; Kutz, S.; Little, P. J.; DeHaven, R. N.; DeHaven-Hudkins, D. L.; Dolle, R. E. J. Med. Chem. 2008, 51, 5893. doi: 10.1021/jm8008986
Le Bourdonnec, B.; Windh, R. T.; Leister, L. K.; Zhou, Q. J.; Ajello, C. W.; Gu, M. H.; Chu, G. H.; Tuthill, P. A.; Barker, W. M.; Koblish, M.; Wiant, D. D.; Graczyk, T. M.; Belanger, S.; Cassel, J. A.; Feschenko, M. S.; Brogdon, B. L.; Smith, S. A.; Derelanko, M. J.; Kutz, S.; Little, P. J.; DeHaven, R. N.; DeHaven-Hudkins, D. L.; Dolle, R. E. J. Med. Chem. 2009, 52, 5685. doi: 10.1021/jm900773n
Spahn, V.; Stein, C. Expert Opin. Invest. Drugs 2017, 26, 155. doi: 10.1080/13543784.2017.1275562
Szmuszkovicz, J.; Von Voigtlander, P. F. J. Med. Chem. 1982, 25, 1125. doi: 10.1021/jm00352a005
DeHaven-Hudkins, D. L.; Dolle, R. E. Curr. Pharm. Des. 2004, 10, 743. doi: 10.2174/1381612043453036
Shaw, J. S.; Carroll, J. A., Alcock, P.; Main, B. G. Br. J. Pharmacol. 1989, 96, 986. doi: 10.1111/bph.1989.96.issue-4
Birch, P. J.; Hayes, A. G.; Johnson, M. R.; Lea, T. A.; Murray, P. J.; Rogers, H.; Scopes, D. I. C. Bioorg. Med. Chem. Lett. 1992, 2, 1275. doi: 10.1016/S0960-894X(00)80229-2
Kramer, H. J.; Uhl, W.; Ladstetter, B.; B cker, A. Br. J. Clin. Pharmacol. 2000, 50, 227. http://www.ncbi.nlm.nih.gov/pubmed/10971307
Kumar, V.; Marella, M. A.; Cortes-Burgos, L.; Chang, A. C.; Cassel, J. A.; Daubert, J. D.; DeHaven, R. N.; DeHaven-Hudkins, D. L.; Gottshall, S. L.; Mansson, E.; Maycock, A. L. Bioorg. Med. Chem. Lett. 2000, 10, 2567. doi: 10.1016/S0960-894X(00)00519-9
Tuthill, P. A.; Seida, P. R.; Barker, W.; Cassel, J. A.; Belanger, S.; DeHaven, R. N.; Koblish, M.; Gottshall, S. L.; Little, P. J.; DeHaven-Hudkins, D. L.; Dolle, R. E. Bioorg. Med. Chem. Lett. 2004, 14, 5693. doi: 10.1016/j.bmcl.2004.08.041
Kumar, V.; Guo, D. Q.; Daubert, J. D.; Cassel, J. A.; DeHaven, R. N.; Mansson, E.; DeHaven-Hudkins, D. L.; Maycock, A. L. Bioorg. Med. Chem. Lett. 2005, 15, 1279. doi: 10.1016/j.bmcl.2005.01.038
Zaveri, N. T. J. Med. Chem. 2016, 59, 7011. doi: 10.1021/acs.jmedchem.5b01499
Kawamoto, H.; Ozaki, S.; Itoh, Y.; Miyaji, M.; Arai, S.; Nakashima, H.; Kato, T.; Ohta, H.; Iwasawa, Y. J. Med. Chem. 1999, 42, 5061. doi: 10.1021/jm990517p
Satoh, A.; Sagara, T.; Sakoh, H.; Hashimato, M.; Nakashima, H.; Kato, T.; Goto, Y.; Mizutani, S.; Azuma-Kanoh, T.; Tani, T.; Okuda, S.; Okamoto, O.; Ozaki, S.; Iwasawa, Y.; Ohta, H.; Kawamoto, H. J. Med. Chem. 2009, 52, 4091. doi: 10.1021/jm900581g
Toledo, M. A.; Pedregal, C.; Lafuente, C.; Diaz, N.; Martinez-Grau, M. A.; Jiménez, A.; Benito, A.; Torrado, A.; Mateos, C.; Joshi, E. M.; Kahl, S. D.; Rash, K. S.; Mudra, D. R.; Barth, V. N.; Shaw, D. B.; McKinzie, D.; Witkin, J. M.; Statnick, M. A. J. Med. Chem. 2014, 57, 3418. doi: 10.1021/jm500117r
Post, A.; Smart, T. S.; Krikke-Workel, J.; Dawson, G. R.; Harmer, C. J.; Browning, M.; Jackson, K.; Kakar, R.; Mohs, R.; Statnick, M.; Wafford, K.; McCarthy, A.; Barth, V.; Witkin, J. M. Neuropsychopharmacology 2016, 41, 1803. doi: 10.1038/npp.2015.348
Zaratin, P. F.; Petrone, G.; Sbacchi, M.; Garnier, M.; Fossati, C.; Petrillo, P.; Ronzoni, S.; Giardina, G. A. M.; Scheideler, M. A. J. Pharmacol. Exp. Ther. 2004, 308, 454.
Marti, M.; Mela, F.; Budri, M.; Volta, M.; Malfacini, D.; Molinari, S.; Zaveri, N. T.; Ronzoni, S.; Petrillo, P.; Calò, G.; Morari, M. Br. J. Pharmacol. 2013, 168, 863. doi: 10.1111/j.1476-5381.2012.02219.x
Fischetti, C.; Camarda, V.; Rizzi, A.; Pelà, M.; Trapella, C.; Guerrini, R.; McDonald, J.; Lambert, D. G.; Salvadori, S.; Regoli, D.; Calò, G. Eur. J. Pharmacol. 2009, 614, 50. doi: 10.1016/j.ejphar.2009.04.054
Shinkai, H.; Ito, T.; Iida, T.; Yamada, H.; Uchida, I. J. Med. Chem. 2000, 43, 4667. doi: 10.1021/jm0002073
O'Neill, S. J.; Collins, M. A.; Pettit, H. O.; McNutt, R. W.; Chang, K. J. J. Pharmacol. Exp. Ther. 1997, 282, 271. http://europepmc.org/abstract/MED/9223564
Negus, S. S.; Gatch, M. B.; Mello, N. K.; Zhang, X. Y.; Rice, K. J. Pharmacol. Exp. Ther. 1998, 286, 362. http://www.ncbi.nlm.nih.gov/pubmed/9655881
Lee, P. H. K.; Mcnutt, R. W.; Chang, K. J. J. Pharmacol. Exp. Ther. 1993, 267, 883. http://europepmc.org/abstract/MED/8246163
Lahti, R. A.; Vonvoigtlander, P. F.; Barsuhn, C. Life Sci. 1982, 31, 2257. doi: 10.1016/0024-3205(82)90132-1
Schenk, S.; Partridge, B. Pharmacol. Biochem. Behav. 2001, 68, 629. doi: 10.1016/S0091-3057(00)00478-0
Bishop, M. J.; Carrido, D. M.; Boswell, G. E.; Collins, M. A.; Harris, P. A.; McNutt, R. W.; O'Neill, S. J.; Chang, K. J. J. Med. Chem. 2003, 46, 623. doi: 10.1021/jm020395s
Gengo, P. J.; Chang, K. J. The Dela Receptor, Marcel Dekker, New York, Basel, 2004, pp. 231~244.
Yi, S. P.; Kong, Q. H.; Li, Y. L.; Pan, C. L.; Cui, B. Q.; Wang, Y. F.; Wang, G. L.; Zhou, P. L.; Wang, L. L.; Gong, Z. H.; Su, R. B.; Shen, Y. H.; Yu, G.; Chang, K. J. Acta Pharmacol. Sin. 2017, 38, 977. doi: 10.1038/aps.2017.14
李玖玲, 孔庆宏, 郁洁, 伊首璞, 李亚民, 王冠林, 沈悦海, 张宽仁, 中国药理学通报, 2016, 32, 652. doi: 10.3969/j.issn.1001-1978.2016.05.012Li, J. L.; Kong, Q. H.; Yu, J.; Yi, S. P.; Li, Y. M.; Wang, G. L.; Shen, Y. H.; Chang, K. J. Chin. Pharmacol. Bull. 2016, 32, 652(in Chinese). doi: 10.3969/j.issn.1001-1978.2016.05.012
Yu, G.; Li, S. H.; Cui, M. X.; Yan, L. D.; Yong, Z.; Zhou, P. L.; Su, R. B.; Gong, Z. H. CNS Neurosci. Ther. 2014, 20, 282. doi: 10.1111/cns.2014.20.issue-3
Linz, K.; Christoph, T.; Tzschentke, T. M.; Koch, T.; Schiene, K.; Gautrois, M.; Schröder, W.; Kögel, B. Y.; Beier, H.; Englberger, W.; Schunk, S.; De Vry, J.; Jahnel, U.; Frosch, S. J. Pharmacol. Exp. Ther. 2014, 349, 535. doi: 10.1124/jpet.114.213694
Ananthan, S.; Khare, N. K.; Saini, S. K.; Seitz, L. E.; Bartlett, J. L.; Davis, P.; Dersch, C. M.; Porreca, F.; Rothman, R. B.; Bilsky, E. J. J. Med. Chem. 2004, 47, 1400. doi: 10.1021/jm030311v
Ananthan, S.; Saini, S. K.; Dersch, C. M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E. J.; Rothman, R. B. J. Med. Chem. 2012, 55, 8350. doi: 10.1021/jm300686p
Healy, J. R.; Bezawada, P.; Shim, J.; Jones, J. W.; Kane, M. A.; MacKerell, A. D. Jr.; Coop, A.; Matsumoto, R. R. ACS Chem. Neurosci. 2013, 4, 1256. doi: 10.1021/cn4000428
Breslin, H. J.; Diamond, C. J.; Kavash, R. W.; Cai, C. Z.; Dyatkin, A. B.; Miskowski, T. A.; Zhang, S. P.; Wade, P. R.; Hornby, P. J.; He, W. Bioorg. Med. Chem. Lett. 2012, 22, 4869. doi: 10.1016/j.bmcl.2012.05.042
Levy-Cooperman, N.; McIntyre, G.; Bonifacio, L.; McDonnell, M.; Davenport, J. M.; Covington, P. S.; Dove, L. S.; Sellers, E. M. J. Pharmacol. Exp. Ther. 2016, 359, 471. doi: 10.1124/jpet.116.236547
Link, A.; Müller, C. E. Angew. Chem., Int. Ed. 2016, 55, 15962. doi: 10.1002/anie.201609015
Pradhan, A. A.; Smith, M. L.; Kieffer, B. L.; Evans, C. J. Br. J. Pharmacol. 2012, 167, 960. doi: 10.1111/j.1476-5381.2012.02075.x
Chen, X. T.; Pitis, P.; Liu, G. D.; Yuan, C.; Gotchev, D.; Cowan, C. L.; Rominger, D. H.; Koblish, M.; DeWire, S. M.; Crombie, A. L.; Violin, J. D.; Yamashita, D. S. J. Med. Chem. 2013, 56, 8019. doi: 10.1021/jm4010829
DeWire, S. M.; Yamashita, D. S.; Rominger, D. H.; Liu, G. D.; Cowan, C. L.; Graczyk, T. M.; Chen, X. T.; Pitis, P. M.; Gotchev, D.; Yuan, C.; Koblish, M.; Lark, M. W.; Violin, J. D. J. Pharmacol. Exp. Ther. 2013, 344, 708. doi: 10.1124/jpet.112.201616
Manglik, A.; Lin, H.; Aryal, D. K.; McCorvy, J. D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R. C.; Bernat, V.; Hübner, H.; Huang, X. P.; Fassano, M. F.; Giguère, P. M.; Löber, S.; Duan, D.; Scherrer, G.; Kobilka, B.; Gmeiner, P.; Roth, B. L.; Shoichet, B. K. Nature 2016, 537, 185. doi: 10.1038/nature19112
Váradi, A.; Marrone, G. F.; Palmer, T. C.; Narayan, A.; Szabó, M. R.; Le Rouzic, V.; Grinnell, S. G.; Subrath, J. J.; Warner, E.; Kalra, S.; Hunkele, A.; Pagirsky, J.; Eans, S. O.; Medina, J. M.; Xu, J.; Pan, Y. X.; Borics, A.; Pasternak, G. W.; McLaughlin, J. P.; Majumdar, S. J. Med. Chem. 2016, 59, 8381. doi: 10.1021/acs.jmedchem.6b00748
Liu, X.; Zhao, L.; Wang, Y.; Zhou, J. J.; Wang, D.; Zhang, Y. X.; Zhang, X. H.; Wang, Z. J.; Yang, D. X.; Mou, L. Y.; Wang, R. ACS Chem. Neurosci. 2017, 8, 2180. doi: 10.1021/acschemneuro.7b00097

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



