图式 1.
氟尿嘧啶的作用机理
Scheme 1.
Mechanism of Fluorouracil

人体中存在着众多生化反应, 而这些生化反应中存在着的各种非共价相互作用, 例如静电相互作用、氢键、范德华力及物质间的疏水亲水作用, 都在很大程度上影响着人体内各种酶的活性, 进而调节着人体的稳态.因此, 长久以来药物的发展都过分注重于以非共价作用与酶结合的药物上.因为非共价相互作用相对强度较小, 使得此类药物的特异性不尽人意, 而药物化学领域一再地企图通过优化药物结构、提高药物用量等方式提高其选择作用性.药物与酶的特异性结合也成为了药物化学中最引人关注的部分.而近来, 人们的目光越来越投向了另一个方向, 便是那些以共价作用与酶结合的药物共价抑制剂.人们认识到, 以共价作用与酶结合的药物, 相较于非共价药物, 由于其特殊的药代动力学性质, 使得共价药物的有效浓度大幅度降低, 有效作用时间大幅度增加.虽然对于共价抑制剂的毒性的恐惧和对其作用机理的探究不足, 阻碍了共价抑制剂的发展, 但更多关于酶化学、药代动力学、结合动力学等方面的了解, 又为我们打开了共价抑制剂发展的大门.
尽管药物研发界在很长一段时间内, 都因为共价抑制剂的高风险性, 尽量避开沿着这条研究方向去开发新药, 相当一部分共价抑制剂也是被偶然发现的, 甚至人们在一些共价抑制剂上市很多年以后才研究清楚它们的作用机制, 但这仍然不能否定共价抑制剂在药物发展史上举足轻重的地位.下面介绍若干个历史上最有名的共价抑制剂.
源自植物的邻羟基苯甲酸, 即水杨酸(salicylic acid), 是人类最早使用的解热镇痛药之一, 但由于水杨酸的酚羟基能与邻位的羧基形成分子内氢键, 这种诱导效应使得水杨酸的酸性较强(pKa=3.0), 对肠胃刺激很大.经过乙酰化改进后的乙酰水杨酸(acetylsalicylic acid), 药品名称阿司匹林(Aspirin, 1), 于1898年由德国拜耳公司研发上市.直到20世纪70年代, 人们才研究清楚以阿司匹林为代表的非甾体抗炎药的作用机制.研究表明, 人体内的花生四烯酸(arachidonic acid, AA)经过代谢, 会生成前列腺素、血栓素和白三烯等生物活性物质.其中, 前列腺素与一系列的炎症、疼痛与发热症状有关, 而血栓素能提升血管张力和血小板的凝聚能力.阿司匹林通过与上述代谢过程中的花生四烯酸环氧合酶(cyclo-oxygen-ase, COX, 2)上的丝氨酸残基(Ser530)反应, 使之乙酰化, 从而不可逆地抑制COX的活性与前列腺素、血栓素的合成(Eq. 1).故阿司匹林等非甾体抗炎药有解热镇痛、预防心肌梗死等功效[1].
|
|
(1) |
1929年, 英国医生Fleming在一次偶然的实验中, 首次发现了青霉素(Penicillin, 4), 从此人们打开了发现β-内酰胺类抗生素(β-lactam antibiotics)的大门.这类抗生素的作用靶标是革兰氏阳性菌的细胞壁, 通过竞争性地与参与细胞壁合成的转肽酶(transpeptidases, 5)的活性中心丝氨酸残基发生不可逆的共价结合, 抑制转肽酶的功能(Eq. 2), 使细菌无法合成细胞壁, 引起细菌溶菌死亡[2].
|
|
(2) |
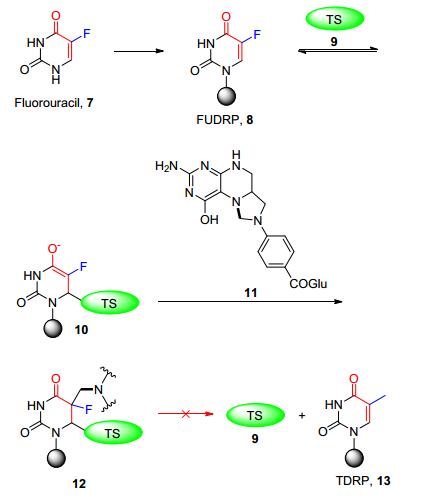
20世纪60年代, 抗癌药物氟尿嘧啶(Fluorouracil, 7)上市.这是一种基于生物电子等排体概念而开发出的抗癌细胞代谢的不可逆共价药物.氟尿嘧啶进入体内被癌细胞摄入后先转化成氟尿嘧啶脱氧核苷酸(FUDRP, 8), 与胸苷酸合成酶(thymidylate synthetase, TS, 9)的亲核基团发生迈克尔加成反应, 紧接着与辅酶5, 10-次甲基四氢叶酸(11)作用.由于F原子电负性高、半径小, C—F键键能很高, 在代谢过程中不易断裂, 导致癌细胞无法合成胸腺嘧啶脱氧核苷酸(TDRP, 13), 且胸苷酸合成酶(9)与反应后的辅酶无法脱落, 从而不可逆地抑制癌细胞的DNA合成(Scheme 1), 最终达到杀死癌细胞的目的[3].
20世纪80年代, 一款直接作用于胃壁细胞的质子泵(proton pump, PP)的抗溃疡药奥美拉唑(Omeprazole)问世. 1998年, 抗血小板药物氯吡格雷(Clopidogrel)率先在美国上市.这两款名药的作用机理均是基于二硫键的形成, 由于其作用机理的特殊性, 在后文的二硫键板块中将做进一步介绍.
天然产物中存在很多结构可以用于共价抑制剂的研发, 这其中绝大多数是迈克尔受体(Michael acceptor).具体的结构特点主要可以划分成以下几种类别: α, β-不饱和内酰胺(α, β-unsaturated lactams)、α, β-不饱和内酯(α, β-unsaturated lactones)和环烯酮(cycloalkenones).以下将分别作简介.
微囊藻毒素(Microcystins)是α, β-不饱和大环内酰胺中最普遍的成员, 它们的大环骨架可以看作一个环七肽.微囊藻毒素有诸多生物活性, 其中有报道称微囊藻毒素-LR (Microcystin-LR, 14)可以与丝氨酸/苏氨酸蛋白磷酸化酶(PP1、PP2A)上的半胱氨酸残基发生迈克尔加成反应形成共价键[4].海藻螺旋酰胺A (Thalassospiramide A, 15)含有一个核心的十二元大环, 据报道, 其能与人calpain 1蛋白酶(HCAN1)上的半胱氨酸残基(Cys115)形成共价键, 以纳摩尔水平抑制其活性[5].海洋小单胞菌酶素A (Rakicidin A, 16)含有对生物活性至关重要的4-酰胺基-2, 4-戊二烯酰胺片段, 其对某些特定酶的半胱氨酸残基的亲和性使其对某些癌细胞系显示出细胞毒性[6].另一些非环肽结构的α, β-不饱和内酰胺天然产物也显示出了出色的抑制活性.例如Pyrrolidine A (17), 据报道其对人早幼粒细胞白血病HL-60细胞具有0.1微摩尔级别的细胞毒性[7].另外还有钩吻素B (Gelegamine B, 18), 尽管这种α-亚甲基内酰胺结构活性没有相应的内酯高[8], 但这仍然不失为一种好的结构研究思路.
|
|
|
|
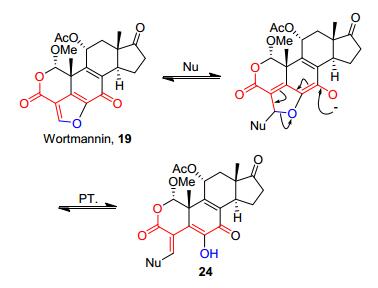
渥曼青霉素(Wortmannin, 19)是胞内磷脂酰肌醇激酶(phosphatidylinositol kinase, PI3K)的共价抑制剂, 其与相应激酶的ATP口袋中的赖氨酸(Lys802)残基以共价键的方式结合, 分子中的呋喃环被打开(Scheme 2)[9].渥曼青霉素及其衍生物与西妥昔单抗或多西他赛组合, 在临床试验中被用于治疗头颈部鳞状细胞癌[10].具有丁烯羟酸内酯环结构的洋地黄毒甙元(Digtitoxigenin, 20)是强心苷(Cardenolides)家族中的一员.研究表明, 其对心脏显示出的毒性与丁烯羟酸内酯环结构有着直接的关系[11].另一类α, β-不饱和内酯天然产物具有环外亚甲基-γ-内酯的结构, 其代表化合物有小白菊内酯(Parthenolide, 21)及亮绿蒿内酯(Arglabin, 22).这类化合物对于宫颈癌、肺癌、黑色素瘤、卵巢癌、结肠癌、甲状腺癌以及乳腺癌等癌细胞系具有微摩尔水平的细胞毒性[12]. 2015年, 雷晓光课题组[13]报道的一种IKK (inhibitor of nuclear factor kappa-B kinase) α/β的天然产物共价抑制剂Ainsliadimer A (23)具有双环外亚甲基-γ-内酯的结构, 其能够与靶标激酶的Cys46共价结合, 抑制激酶的活性.
|
|
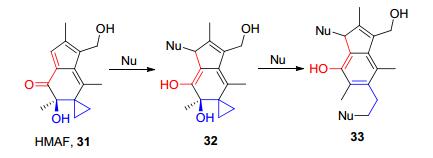
具有大环内酯环烯酮结构的寄端霉素(Hypothemy- cin, 26)能够与丝裂原活化蛋白激酶(mitogen-activated protein kinases, MAPKs)的ATP口袋中的半胱氨酸残基不可逆的共价结合, 具有抗真菌、抗肿瘤、抗细菌和抗病毒等多种生物活性[14].前列腺素A1 (PGA1, 27)是环戊烯酮前列腺素(CyPGs)系列化合物中的一种, 其作为一种内源化合物, 具有丰富的生物学活性, 包括抗炎以及神经保护等作用[15].具有贝壳杉烷骨架的化合物Eriocalyxin B (28)、冬凌草素甲(Oridonin, 29)和延命素(Enmein, 30)均具有环外亚甲基环戊酮的活性结构.据报道, 这类化合物可以与半胱氨酸残基共价结合, 抑制NF-κB信号的传导, 具有广泛的生物活性[16].另一种化合物除了含有常见的烯酮迈克尔受体外, 还含有环丙基芳构化亲电受体, 这类物质就是隐陡头菌素(Illudin)及其衍生物如HMAF (31), 其亲电芳构化机制如Scheme 3所示.研究表明, 其可以不可逆的抑制谷胱甘肽还原酶(Glutathione reductase, GR), 在临床试验中可以作为抗癌药物[17].
|
|
上述的天然产物或存在高毒性、低活性、低选择性或高成本等缺点, 不适合直接用作药物, 但它们新颖多样的结构却能给共价抑制剂的研发带来诸多灵感.
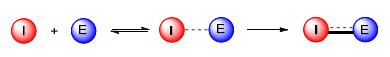
不可逆共价抑制剂分子分为导引头与弹头(warhead)两个部分.进入体内后, 导引头与目标靶蛋白结合位点先形成非共价相互作用, 随后弹头与目标亲核氨基酸残基发生不可逆共价结合(Scheme 4).正因为发生了不可逆共价结合, 使得不可逆共价抑制剂在很大程度上可以增加作用时间, 减少药物用量.本文将主要介绍三种最常见的弹头:迈克尔受体类、磺酰氟类、二硫键类.
肺癌是世界上最常见的恶性肿瘤之一, 而其中非小细胞肺癌占80%, 有生长分裂较慢、扩散转移相对较晚的特点.中国国内的非小细胞肺癌患者, 其表皮生长因子受体(epidermal growth factor receptor, EGFR)基因有较大概率会发生突变, 而针对该突变的抑制剂可以迅速缩小肿瘤, 但对于绝大多数晚期肺癌患者, 服用传统肺癌药物如第一代EGFR抑制剂厄洛替尼(Erlotinib)、吉非替尼(Gefitinib)、阿法替尼(Afatinib, 34)等后1~2年即会出现明显的耐药反应.产生耐药性的原因为其EGFR基因发生了二次突变[EGFR酪氨酸激酶(EGFR TKI) T790M耐药性突变及EGFR酪氨酸激酶的敏化][18].对抗EGFR的二次突变成为了晚期肺癌治疗的关键. AZD9291 (35)为AstraZeneca公司研发的新型口服、高效第三代不可逆EGFR酪氨酸激酶抑制剂, 对于EGFM TKI敏化及T790M二次突变患者有良好的选择性治疗效果, 对于普性的EGFR突变也有较好的活性.第一代EGFR TKI对二次突变的低选择性也在一定程度上表明第一代EGFR TKI对EGFR敏化及T790M二次突变有着诱导作用.而使用AZD9291则不会出现该种获得性耐药[19].
|
|
AZD9291通过与EGFR酪氨酸激酶ATP结合位点的Cys797残基形成共价键结合, 阻断EGFR T790M突变细胞的信号通路与抑制细胞生长来起到抑制作用[20].
AZD9291与其他第三代酪氨酸激酶抑制剂如WZ4002 (36)、CO-1686 (37)的结构不同[21][22], 后二者有许多相同结构, 例如与EGFR上半胱氨酸残基反应的亲电基团的位置、反应基团连接的侧基等.而AZD9291在体系结构上是独特的, 例如其亲电反应基团连接在嘧啶环C-2位、嘧啶五号位无取代等, 该独特结构特征也影响了其强药物活性、高选择性[21].
|
|
FDA于2017年正式批准AstraZeneca公司的Acalabrutinib (ACP-196, 38)上市, 其商品名为Cal- quence, 主要的适应症是套细胞淋巴瘤(Mantle Cell Lymphoma, MCL)与自身免疫疾病[23].作为目前最新的一种第二代BTK (Bruton tyrosine kinase)选择性不可逆共价抑制剂[24], 相比于早些年就被研发出来的其他BTK不可逆共价抑制剂, 如Ibrutinib (39)和Spebrutinib (40), Acalabrutinib在细胞与生化层面上均拥有更高的选择性, 更重要的是, Acalabrutinib并不会受到与不良作用相关的脱靶激酶, 如EGFR和ITK的限制[25], 这些都加强了Acalabrutinib的临床安全性, 改善了其临床的应用效果[26].
|
|
与早些年上市的其他BTK不可逆共价抑制剂如Ibrutinib的丙烯酰胺弹头相比, Acalabrutinib的弹头被改造成了甲基修饰的丙炔酰胺, 然而它们与BTK的作用机理却是非常相似的.与较为常见的丙烯酰胺弹头一样, 这里的甲基修饰的丙炔酰胺弹头在以腺嘌呤衍生类似物为导引头片段的引导下, 与BTK的Cys481残基上的巯基发生迈克尔加成反应, 从而与之形成共价键, 永久性的占用了该BTK分子的活性口袋, 不可逆的抑制了该BTK分子的生化活性[27].
细胞周期蛋白依赖性激酶(Cyclin-dependent kinases, CDKs), 在调节真核细胞周期和转录中发挥了重要作用.化学遗传学的研究证据表明, CDK2活性的异常与癌症的发展直接相关.而发展CDK2抑制剂, 给治疗具有确定遗传特征的肿瘤提供了一种新颖的思路.最近报道的化合物NU6300 (41), 能够选择性的与CDK2中的非保守赖氨酸(Lys89)形成共价键, 而NU6300也是第一个成功应用于生物化学和结构研究的CDK2不可逆共价抑制剂[28].
|
|
对NU6300与CDK2/cyclin A的共价复合物(42)的晶体结构研究表明, 位于靶向蛋白CDK2中口袋铰链区的Glu81、Leu83和Asp86残基中的相应原子与NU6300导引头片段之间形成氢键, 而Lys89上的氨基与NU6300上的乙烯基砜弹头片段发生迈克尔加成反应形成共价键[29], 从而发挥其不可逆共价抑制剂的作用.
|
|
蛋白质激酶的不可逆共价抑制剂大多数是以半胱氨酸的巯基作为亲核基团来开发的, 而基于赖氨酸的氨基来开发的却较为少见. NU6300无疑在这个领域开了一个好头.开发拥有类似弹头基团的药物, 在研究CDKs靶向赖氨酸的药理学上发挥着重要的作用[30].
磺酰氟(Sulfonyl fluoride)亲电子试剂作为蛋白质不可逆共价抑制剂和反应性探针, 在化学生物学、药物化学等领域发挥着越来越重要的作用.早在20世纪20, 30年代, 德国化学家就已经发现了许多磺酰氟的相关反应特性[31], 并最初被用作杀虫剂, 直到20世纪60年代, 其抑制酯酶的作用机制才首次被报道[32].然而磺酰氟在药物方面的相关研究工作却在20世纪50年代几乎中断了.如今, 由于共价药物的市场复兴以及磺酰氟在许多生化反应中表现出的优异性能, 人们又重拾了对于它的研究热情.
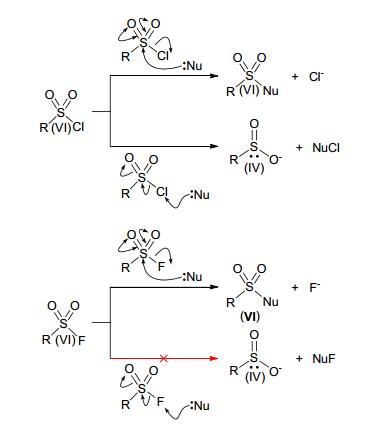
作为蛋白质不可逆共价抑制剂或探针的弹头, 相比于更加常见且反应活性更高的磺酰氯(Sulfonyl chloride), 磺酰氟拥有更长的生物半衰期, 但又在生物相容性(主要包括水稳定性)与蛋白质反应性之间有着适当的平衡[33].另外一方面, 由于氟原子具有极高的电负性, 相比于磺酰氯, 磺酰氟在抗还原性方面有着优异的性能.换句话说, 对于磺酰氟, 亲核试剂只能进攻硫原子, 氟离子离去, 发生加成消除机理的取代反应; 而对于磺酰氯, 则可能发生取代或还原反应两种可能(Scheme 5).除此之外, 相比于丙烯酰胺弹头几乎只能与半胱氨酸残基的巯基发生反应, 磺酰氟弹头除了主要与丝氨酸(Ser)残基的羟基发生反应外, 还能特异性的修饰苏氨酸(Thr)、赖氨酸(Lys)、酪氨酸(Tyr)、半胱氨酸(Cys)和组氨酸(His)的残基[34].上述的这些特点, 使得磺酰氟试剂越来越能够引起药物化学家的研究兴趣, 人们想利用磺酰氟弹头, 开发出一些新颖的不可逆共价抑制剂药物.
作为乙酰胆碱酶(Acetyl Cholinesterase, AChE)选择性的不可逆共价抑制剂, 磺酰氟药物家族中分子量最小的成员:甲基磺酰氟(methyl sulfonyl fluoride, MSF, 43), 已经被成功地用于改善阿尔兹海默症(Alzheimer)患者的记忆力[35].另一个比较有名的例子是NCS 127755 (44), 它被发现能够作为二氢叶酸还原酶(dihydrofolate reductase, DHFR)的不可逆共价抑制剂[36], 特异性的修饰DHFR上的Tyr31[37].除此之外, 人们对于黄嘌呤氧化酶[38](xanthine oxidase, XO)、α-胰凝乳蛋白酶[39] (α-chymotrypsin)、鸟嘌呤脱氨酶[40](guanine deaminase, GAH)、脂肪酸酰胺水解酶[41](fatty acid amide hydrolase, FAAH)以及脂蛋白脂肪酶[42](lipoprotein lipase, LPL)等的磺酰氟类不可逆共价抑制剂(45~48)也已有所研究.
而说到磺酰氟类不可逆共价抑制剂, 就不得不谈到FSBA家族49~52.我们很容易地发现FSBA的结构特点:磺酰氟苯甲酰基取代了原ATP中的三磷酸酰基.开发这种ATP类似物的目的最初是为了探究谷氨酸脱氢酶(glutamate dehydrogenase)结合位点中的亲核残基.进一步的研究表明, FSBA及其衍生物能够与酪氨酸激酶(tyrosine kinases)Src家族的全部9个成员上的赖氨酸残基(Lys295)共价结合[43].而相比于乙烯基砜类共价抑制剂, 磺酰氟类共价抑制剂在对抗脱靶激酶上的效力更高[44].
|
|
二硫键是一种在多肽链中常见的化学键, 连接着不同肽链或同一肽链的不同部分, 且其因稳定性较好, 在蛋白质分子中起着稳定结构的重要作用.
而以形成二硫键为主要功能结构的药物, 也在共价抑制剂药物市场上, 占据着不可或缺的重要作用.常见的如肠道药物奥美拉唑(Omeprazole, 53)、心血管药物氯吡格雷(Clopidogrel, 54)等.该类药物最大的特点为不直接与某一氨基酸发生反应, 而是先在特定的体内环境下发生一定程度的结构变化, 该结构的变化也促进了进一步与半胱氨酸的特异性结合.
|
|
以奥美拉唑(Omeprazole, 53)为例, 该药物的主要功能为治疗胃溃疡及反流性食管炎.胃酸分泌的最后一步为H+/K+-ATP酶催化下的质子转移, 而奥美拉唑即为该质子泵的抑制剂[45].在中性pH条件下, 奥美拉唑分子并不抑制该ATP酶的反应活性, 而在肠胃中胃酸pH条件下却可以特异性的抑制该酶的作用[46].
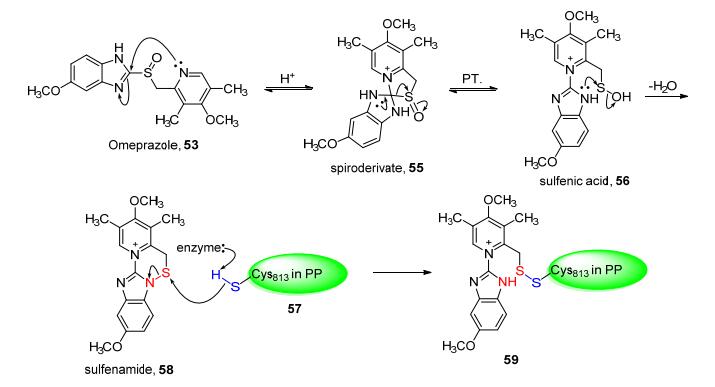
研究表明, 奥美拉唑体外本无活性, 其抑制活性是由它代谢后生成的高反应活性的次磺酰胺结构(58)实现.奥美拉唑在胃酸的酸性条件下, 经由螺环中间体(spiroderivate, 55), 通过Smiles重排形成次磺酸(sulfenic acid, 56), 随后分子内脱去一分子水形成活性代谢物次磺酰胺(sulfonamide, 58).该次磺酰胺结构为奥美拉唑在人体内起抑制作用的最终结构.其抑制活性则体现在, ATP酶中侧链上的Cys813(57)亲核进攻该高活性次磺酰胺结构, 形成稳定的二硫键(59), 不可逆的抑制质子泵的活性(Scheme 6), 从而起到共价抑制的作用[47][48], 发挥其抗溃疡的功效[49].
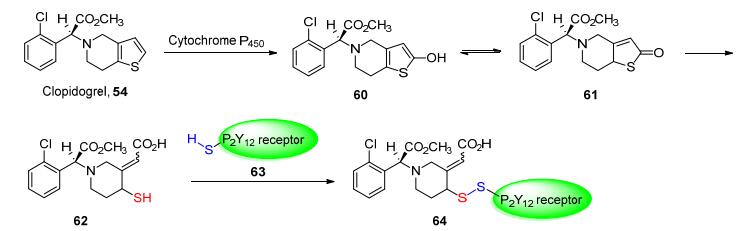
另一个形成二硫键的经典药物为1998年问世的抗血小板高聚集药物氯吡格雷(Clopidogrel, 54).
血小板为人体内重要的凝血物质, 人体血液中的血小板含量会稳定在一定的水平上, 而过高的血小板含量则会造成血小板聚集而阻塞人体动脉血管, 造成各类心血管疾病.
氯吡格雷同奥美拉唑相同, 自身本无活性, 其活性部分为自身的代谢产物.氯吡格雷分子经体内的细胞色素P450氧化后生成羟基噻吩环(60), 羟基噻吩环经过双键的重排后(61)被水解, 生成含有硫醇基团的羧酸开环产物(62), 而其游离出的巯基便是其活性基团.该基团通过选择性的与P2Y12-ADP受体上的半胱氨酸残基(63)发生氧化反应, 构建二硫桥键(64)从而改变受体的构象, 不可逆的抑制该受体的活性(Scheme 7), 从而阻断血小板的聚集[50].
上述提到的不论是奥美拉唑还是氯吡格雷, 距离其问世都过去较长时间了, 而以形成二硫键为模式的药物也被其他不同机理抑制剂的风光所掩盖, 但也不乏依旧有优秀的以形成二硫键为弹头的新药物问世, 普拉格雷(Prasugrel, 65)就是其中之一.
|
|
普拉格雷与氯吡格雷相同, 是一种噻吩吡啶类P2Y12-ADP受体不可逆抑制剂, 于2009年被批准在欧洲使用, 主要针对急性冠动脉综合症(acute coronary syn- drome, ACS)患者的心血管血栓等疾病[51].而其作用机理与氯吡格雷不尽相同, 普拉格雷本身为其活性分子的前驱体, 在进入肠道后, 普拉格雷会迅速代谢水解, 因为其结构中已含有活性结构的前驱结构(乙酰氧基噻吩环), 故不涉及体内酶的氧化, 极大的提高了其药效释放速率.其前驱体水解后发生互变异构, 并再次水解, 游离出其活性的巯基结构.
与氯吡格雷相比, 普拉格雷可以更快、更稳定地抑制腺苷二磷酸(adenosine diphosphate, ADP)诱导的血小板聚集, 所以比氯吡格雷更加高效[52].同时, 普拉格雷不会与质子泵抑制剂(如奥美拉唑)产生拮抗反应, 故应用范围较氯吡格雷更为广泛[53].不足的是, 普拉格雷一定程度上提高了病人大出血的危险性, 也在一定程度上影响了该药物的大规模使用.
可逆共价抑制剂的结构与作用机制与不可逆共价抑制剂均相似, 而不同的是其与目标靶蛋白的共价结合是可逆的(Scheme 8).可逆共价抑制剂的亲电弹头多为氰基、酮羰基等可逆亲核加成反应受体.而其与靶标共价结合的可逆性, 使得可逆共价抑制剂的药代动力学特征介于不可逆共价抑制剂和非共价抑制剂之间.可逆共价抑制剂在一定程度上继承了不可逆共价抑制剂作用时间长、有效浓度低等优点, 同时也减少了脱靶带来的毒性风险.
以特拉匹韦(Telaprevir, 66)和波普瑞韦(Boceprevir, 67)为例, 这两种药物均是抗丙型肝炎病毒(hepatitis C virus, HCV)药物. HCV所导致的丙型肝炎波及全球1.7亿人.而治疗HCV感染的药物靶标是HCV蛋白酶(HCV protease), 抑制这种蛋白的功能就可以抑制HCV的RNA合成与病毒复制.据报道, Telaprevir与Boce- previr能与HCV蛋白酶的丝氨酸残基(Ser139)发生可逆的共价结合, 形成较为稳定半缩酮[54], 从而抑制HCV蛋白酶的功能.
|
|
2012年, Taunton团队[55]报道了经α-氰基修饰的丙烯酰胺类半胱氨酸可逆共价抑制剂.前文中已经提到了具有丙烯酰胺弹头的靶向半胱氨酸不可逆共价抑制剂, 这些不可逆共价抑制剂有些也成功获批上市.然而, 基于对药物脱靶风险的担忧, 考虑如何调节丙烯酰胺弹头的性能也就显得很有必要了.而在丙烯酰胺的α位引入吸电子基团, 例如氰基, 便是一种方法.以化合物68为例[56], 在丙烯酰胺的α位引入氰基不仅可以增加其亲电性, 提高其与半胱氨酸残基的反应性, 同时也大大增强了α-H的酸性, 使得迈克尔加成的逆反应可以在生物体内的条件下发生.另一方面, 通过调节β位基团的位阻大小, 可以调节α-H的脱除速率, 从而调节迈克尔加成逆反应的速率. β位基团的位阻越大, 处于酶口袋中的药物分子上的α-H越难以被碱脱除[57], 迈克尔加成的逆反应的速率也就越低, 从而增加药物的作用时间(表 1).
|
|
 下载:
导出CSV
下载:
导出CSV
| Occupancy/% | 4 h | 20 h |
| a | ≈75 | ≈8 |
| b | ≈80 | ≈30 |
| c | ≈85 | ≈55 |
| d | ≈100 | ≈100 |
不难发现, 对于具有丙烯酰胺弹头的共价抑制剂, 通过调节α位引入的吸电子基团的吸电子能力和β位基团的位阻大小这两种方法, 可以综合调节丙烯酰胺弹头的亲电性能, 这种方法对于开发出更多更安全有效的丙烯酰胺类共价抑制剂具有重要的意义.
从一百二十年前面市的阿司匹林开始, 共价抑制剂对人类健康做出了重要的贡献.而最近几年大量上市的激酶共价抑制剂给癌症化疗领域带来了新的曙光.越来越多的研究发现, 人类目前发现的许多重大疾病如恶性肿瘤等, 都受着激酶的调节, 而这些酶也成为了最引人注目的药物靶点.因此, 共价抑制剂因为其更强的选择性、抑制性、更少的药物用量、更低的耐药性等优势, 越来越成为激酶抑制剂领域的焦点.不仅在激酶治疗领域, 在很多其他治疗领域, 共价抑制剂也有广泛的用途.目前对共价药物的开发也逐渐从以往的偶然发现到精确设计过度, 尽管如此, 共价药物的开发难度依旧较大.而大量存在的非共价抑制剂的现成结构却被加以利用, 作为共价抑制剂的导引头, 通过结构分析, 在合适的部位连接上合适的弹头, 做适当的修改, 即可被改装成共价抑制剂[58].而要想进一步提升共价抑制剂的特异选择性, 开发全新的结构也就变得很有必要了, 这需要组合化学、蛋白质结构化学、计算机科学的共同合作.虽然大多数的共价抑制剂, 尤其是不可逆共价抑制剂, 具有一定的脱靶毒性风险和免疫过敏反应风险, 但是我们仍有理由相信, 只要能降低其副作用, 更好的提高药物的特异性, 那么共价抑制剂将会运用到更广泛的领域之中, 而这些冒险也是值得尝试的.
Zhang, Y. K.; Lei, J. P.; Xie, D. Q. J. Am. Chem. Soc. 2015, 137, 70. doi: 10.1021/ja5112964
Andrieu, J. P.; Guilmi, A. M. D.; Mouz, N.; Hoskins, J.; Jaskunas, S. R.; Gagnon, J.; Dideberg, O.; Vernet, T. J. Bacteriol. 1998, 180, 5652.
Aronson, J. K. Meyler's Side Effects of Drugs, Elsevier Science, Amsterdam, 2016, p. 382.
Drahl, C.; Cravatt, B. F.; Sorensen, E. J. Angew. Chem. Int. Ed. 2005, 44, 5788. doi: 10.1002/(ISSN)1521-3773
Lu, L.; Michael, M.; Gu, Z. L.; Zhang, W. P. Sci. Rep. 2015, 5, 8783. doi: 10.1038/srep08783
Clement, L. L.; Tsakos, M.; Schaffert, E. S.; Scavenius, C.; Enghild, J. J.; Poulsen, T. B. Chem. Commun. 2015, 51, 12427. doi: 10.1039/C5CC04500B
Uesugi, S.; Fujisawa, N.; Yoshida, J.; Watanabe, M.; Dan, S.; Yamori, T.; Shiono, Y.; Kimura, K. J. Antibiot. 2016, 69, 133.
Albrecht, A.; Albrecht, L.; Janecki, T. Eur. J. Org. Chem. 2011, 2011, 2747.
(a) Walker, E. H.; Pacold, M. E.; Perisic, O. Mol. Cell. 2000, 6, 909.
(b) Sorensen, E. J.; Drahl, C.; Cravatt, B. F. Angew. Chem., Int. Ed. 2005, 44, 5788.
Bauman, J. E.; Jimeno, A.; Weissman, C.; Adkins, D.; Schnadig, I.; Beauregard, P.; Bowles, D. W.; Spira, A.; Levy, B.; Seetharamu, N.; Hausman, D.; Walker, L.; Rudin, C. M.; Shirai, K. Oral Oncol. 2015, 51, 383.
Jones, J. B.; Middleton, H. W. Can. J. Chem. 1970, 48, 3819.
Csuk, R.; Schwarz, S.; Siewert, B.; Kluge, R.; Strohl, D. Arch. Pharm. 2012, 345, 215. doi: 10.1002/ardp.v345.3
(a) Lei, X. G.; Yu, X. L.; Li, C. Org. Lett. 2010, 12, 4284.
(b) Lei, X. G.; Dong, T.; Li, C.; Wang, X.; Dian, L. Y.; Zhang, X. G.; Li. L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; He, S. D. Nat. Commum. 2015, 6, 6522.
Wei, L.; Wu, J.; Li, G.; Shi, N. Curr. Pharm. Des. 2012, 18, 1186. doi: 10.2174/138161212799436395
Díez-Dacal, B.; Perez-Sala, D. Cancer Lett. 2012, 320, 150.
Grill, S. P.; Leung, C. H.; Lam, W.; Han, Q. B.; Sun, H. D.; Cheng, Y. C. Mol. Pharmacol. 2006, 70, 1946.
Tanasova, M.; Sturla, S. J. Chem. Rev. 2012, 112, 3578. doi: 10.1021/cr2001367
Cross, D. A. E.; Ashton, S. E.; Ghioghiu, S.; Eberlein, C.; Nebhan, C. A.; Spitzler, P. J.; Orme, J. P.; Finlay, M. R. V.; Ward, R. A.; Mellor, M. J.; Hughes, G.; Rahi, A.; Jacobs, V. N.; Brewer, M. R.; Mireille, E.; Sun, J.; Jin, H.; Ballard, P.; Al-Kadhimi, K.; Rowlinson, R.; Klinowska, T.; Richmond, G. H. P.; Cantarini, M.; Kim, D. W.; Ranson, M. R.; Pao, W. Cancer Discovery 2014, 4, 1046.
Jackson, P. A.; Widen, J. C.; Harki, D. A.; Brummond, K. M. J. Med. Chem. 2017, 60, 839.
Ward, R. A.; Anderton, M. J.; Ashton, S.; Bethel, P. A.; Box, M.; Butterworth, S. J. Med. Chem. 2013, 56, 7025. doi: 10.1021/jm400822z
Zhou, W. J.; Ercan, D.; Chen, L.; Yun, C. H.; Li, D. N.; Capelletti, M.; Cortot, A. B.; Chirieac, L.; Iacob, R. E.; Padera, R.; Engen, J. R.; Wong, K. K.; Eck, M. J.; Gray, N. S.; Janne, P. A. Nature 2009, 462, 1070.
Walter, A. O.; Sjin, R. T.; Haringsma, H. J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z. D.; Wang, Z. G.; Sheets, M.; St Martin, T.; Karp, R.; van Kalken, D.; Chaturvedi, P.; Niu, D. Q.; Nacht, M.; Petter, R. C.; Westlin, W.; Lin, K.; Jaw-Tsai, S.; Raponi, M.; van Dyke, T.; Etter, J.; Weaver, Z.; Pao, W.; Singh, J.; Simmons, A. D.; Harding, T. C.; Allen, A. Cancer Discovery 2013, 3, 1404.
Campo, E.; Rule, S. Blood 2015, 125, 48.
Wu, J. J.; Zhang, M. Z.; Liu, D. L. J. Hematol. Oncol. 2016, 9, 21.
Barf, T.; Covey, T.; Izumi, R.; van der Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; Verkaik, S.; Krantz, F.; Pearson, P. G.; Ulrich, R.; Kaptein, A. J. Pharmacol. Exp. Ther. 2017, 363, 240. doi: 10.1124/jpet.117.242909
Byrd, J. C.; Harrington, B.; O'Brien, S.; Jones, J. A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W. G.; Awan, F. T.; Brown, J. R.; Hillmen, P.; Stephens, D. M.; Ghia, P.; Barrientos, J. C.; Pagel, J. M.; Woyach, J.; Johnson, D.; Huang, J.; Wang, X.; Kaptein, A.; Lannutti, B. J.; Covey, T.; Fardis, M.; McGreivy, J.; Hamdy, A.; Rothbaum, W.; Izumi, R.; Diacovo, T. G.; Jojnson, A. J.; Furman, R. R. New. Engl. J. Med. 2016, 374, 323. doi: 10.1056/NEJMoa1509981
Patel, V.; Balakrishnan, K.; Bibikova, E.; Ayres, M.; Keating, M. J.; Wierda, W. G.; Gandhi, V. Clin. Cancer Res. 2017, 23, 3734.
Owens, T. D.; Yan, L. Compr. Med. Chem. Ⅲ 2017, 76.
Meschini, E.; Mora-Vidal, R.; Martin, M. P.; Anscombe, E.; Staunton, D.; Geitmann, M.; Danielson, U. H.; Stanley, W. A.; Wang, L. Z.; Reuillon, T.; Golding, B. T.; Cano, C.; Newell, D. R.; Nobel, M. E. M.; Wedge, S. R.; Endicott, J. A.; Griffin, R. J. Chem. Biol. 2015, 22, 1159.
Larraufie, M. H.; Yang, W. S.; Jiang, E.; Thomas, A. G.; Slusher, B. S.; Stockwell, B. R. Bioorg. Med. Chem. Lett. 2015, 25, 4787.
Steinkopf, W. J. Prakt. Chem. 1927, 117, 1.
Gold, A. M.; Fahrney, D. J. Am. Chem. Soc. 1963, 85, 997.
Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650.
Dong, J. J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430.
Moss, D. E.; Berlanga, P.; Hagan, M. M.; Sandoval, H. Alzheimer Dis. Assoc. Disord. 1999, 13, 20. doi: 10.1097/00002093-199903000-00003
Corbett, T. H.; Leopold, W. R.; Dykes, D. J.; Roberts, B. J.; Griswold, D. P.; Schabel, F. M. Cancer Res. 1982, 42, 1707.
Kumar, A. A.; Mangum, J. H.; Blankenship, D. T.; Freisheim, J. H. J. Biol. Chem. 1981, 256, 8970.
Baker, B. R.; Wood, W. F. J. Med. Chem. 1969, 12, 214. doi: 10.1021/jm00302a004
Baker, B. R.; Hurlbut, J. A. J. Med. Chem. 1969, 12, 221.
Baker, B. R.; Wood, W. F. J. Med. Chem. 1969, 12, 216. doi: 10.1021/jm00302a005
Karanian, D. A.; Brown, Q. B.; Makriyannis, A.; Kosten, T. A.; Bahr, B. A. J. Neurosci. 2005, 25, 7813.
Kokotos, G.; Kotsovolou, S.; Constantinou-Kokotou, V.; Wu, G. S.; Olivecrona, G. Bioorg. Med. Chem. Lett. 2000, 10, 2803. doi: 10.1016/S0960-894X(00)00566-7
Brummond, K. M.; Jackson, P. A.; Widen, J. C.; Harki, D. A. J. Med. Chem. 2017, 60, 839. doi: 10.1021/acs.jmedchem.6b00788
Gushwa, N. N.; Kang, S. M.; Chen, J.; Taunton, J. J. Am. Chem. Soc. 2012, 134, 20214.
Potashman, M. H.; Duggan, M. E. J. Med. Chem. 2009, 52, 1231. doi: 10.1021/jm8008597
Fellenius, E.; Berglindh, T.; Sachs, G.; Olbe, L.; Elander, B.; Sjçstrand, S. E.; Wallmark, B. Nature 1981, 290, 159.
Gonzμlez-Bello, C. Chem. Med. Chem. 2016, 11, 22. doi: 10.1002/cmdc.v11.1
Andersson, T.; Rohss, K.; Bredberg, E.; Hassan-Alin, M. Aliment. Pharmacol. Ther. 2001, 15, 1563.
Baillie, T. A. Angew. Chem., Int. Ed. 2016, 55, 13408.
Gonzalez-Bello, C. Chem. Med. Chem. 2015, 11, 22.
Baker, W. L.; White, C. M. Am. J. Cardiovasc. Drugs 2009, 9, 213.
Wiviott, S. D.; Braunwald, E.; McCabe, C. H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.; Ardissino, D.; De Servi, S.; Murphy, S. A.; Riesmeyer, J.; Weerakkody, G.; Gibson, C. M.; Antman, E. M. N. Engl. J. Med. 2007, 357, 2001.
John, J.; Koshy, S. J. Am. Board Fam. Med. 2012, 25, 343.
Njoroge, F. G.; Chen, X. X.; Shih, N. Y.; Piwinski, J. J. Acc. Chem. Res. 2008, 41, 50.
Taunton, J.; Serafimova, I. M.; Pufall, M. A.; Krishnan, S.; Duda, K.; Cohen, M. S.; maglathlin, R. L.; McFarlan, J. M.; Miller, R. M.; Frodin, M. Nat. Chem. Biol. 2012, 5, 471.
Moitessier, N.; De Cesco, S.; Kurian, J.; Dufresne, C.; Mit-termaier, A. K. Eur. J. Med. Chem. 2017, 138, 96.
Bradshaw, J. W.; McFarland, J. M.; Paavilainen, V. O.; Bisconte, A.; Tam, D.; Phan, V. T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; Ton, T.; Li, X. Y.; Loughhead, D. G.; Nunn, P. A.; Karr, D. E.; Gerritsen, M. E.; Funk, J. O.; Owen, T. D.; Verner, E.; Brameld, K. A.; Hill, R. J.; Goldstein, D. M.; Taunton, J. Nat. Chem. Biol. 2015, 7, 525.
Liu, Q. S.; Sabnis, Y.; Zhao, Z.; Zhang, T. H.; Buhrlage, S. J.; Jones, L. H.; Gray, N. S. Chem. Biol. 2013, 20, 146. doi: 10.1016/j.chembiol.2012.12.006

图式 5 磺酰氯与磺酰氟的不同反应性
Scheme 5 Different reactivity between sulfonyl chloride and sulfonyl fluoride
表 1 化合物a, b, c, d的时效关系
Table 1. Time-effect relationship of compound a, b, c, d
| Occupancy/% | 4 h | 20 h |
| a | ≈75 | ≈8 |
| b | ≈80 | ≈30 |
| c | ≈85 | ≈55 |
| d | ≈100 | ≈100 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们