
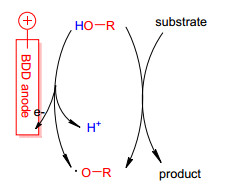
图式 1.
BDD阳极的作用机理
Scheme 1.
Proposed mechanism

有机电化学合成作为有机合成中的一个重要组成部分, 具有绿色可持续、高效率以及高选择性等优点.电化学有机合成一直是药物合成以及精细有机等合成领域中的热点课题, 其主要类型包括:官能团的取代、加成、消除、氧化、还原, 以及利用氧化还原介质进行间接电化学合成[1~3].在一个电化学反应中, 有机化合物分子或氧化还原介质在“电极/溶液”两相的界面上发生电子转移, 从而实现旧键的断裂以及新键的形成.因此, 电极材料的使用、电位和溶剂等条件的控制在电化学转化中至关重要.
通过C—H键的官能化构建C—X (X=C、N、O、S)键一直是有机合成领域中一项热点研究课题[4~12].键能较大以及选择性差是C—H键活化的所要克服的两个难题.工业上, C—H键的转化往往需要繁琐的反应步骤, 能量消耗巨大且副产物生成较多.利用电化学方法可以有效地实现底物在阳极上的氧化, 形成高活性的反应中间体, 是实现C—H键活化的一种绿色合成方法.基于芳香化合物在医药、染料、农药和香料等众多领域具有广阔的应用前景, 本文主要综述了电化学方法促进的此类化合物中C—H键官能化的研究进展.
富电子芳香化合物如酚和芳胺等具有较低的氧化电位, 容易实现电氧化脱氢偶联.早在2000年, Waldvogel等[13, 14]就开始了邻苯二酚衍生物的氧化三聚反应研究, 用于合成相应的三亚苯化合物. 2006年, 他们[15]又报道了硼掺杂金刚石(BDD)作为阳极促进的2, 4-二甲基苯酚的高选择性电氧化脱氢偶联(Eq. 1).在无溶剂条件下, 相比于铂电极, 偶联产物产率有了很大提高. BDD阳极在酸性介质中具有较高的析氧电位, 因此会产生氧自由基促进反应物的转化(Scheme 1).随后, 他们发现使用六氟异丙醇(HFIP)作为电解液可以提高酚邻位偶联的选择性(Eq. 2)[16].使用BDD作阳极, HFIP作为溶剂能起到稳定氧自由基的作用, 此反应在简易的一室型电解池中进行, 氢气是唯一的副产物.基于这些原理, 他们[17]也报道了邻甲氧基苯酚衍生物的阳极偶联反应. 2011年, 他们[18]又将廉价的石墨电极用于酚的偶联反应, 通过加入三氟乙酸(TFA)也可以得到较好的收率和高选择性.
|
|
(1) |
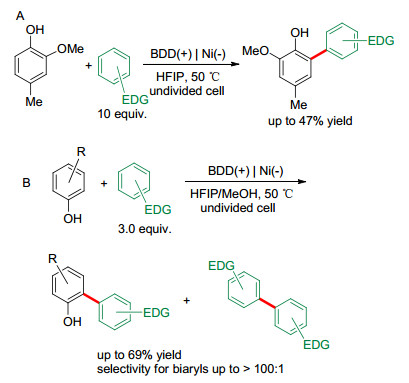
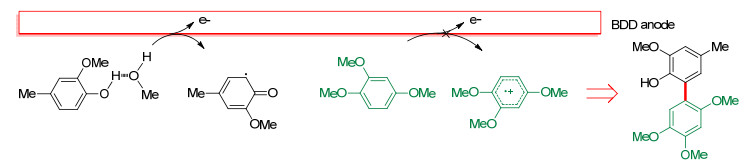
2010年, Waldvogel等[19]报道了酚与芳烃的偶联反应.使用BDD作阳极, 实现了4-甲基-2-甲氧基苯酚和富电子芳烃之间的脱氢交叉偶联(Scheme 2A).尽管HFIP和BDD阳极的使用能促成偶联反应的发生, 但收率较低, 且选择性差.反应中需要使用大量的芳烃底物来避免酚的自偶联反应. 2012年, 该课题组[20]对此类反应进行了改进, 即添加甲醇作为共溶剂.结果表明, 当HFIP/MeOH的体积比为27:6时, 交叉偶联反应的选择性和反应的收率最高(2015年, 他们系统研究了共溶剂对不同底物氧化电位的影响[21]).此外, 使用混合溶剂时, 芳烃底物的用量可减少到3.0 equiv., 并且该反应还适用于更多的酚类底物(Scheme 2B).在HFIP中加入水或甲醇对这一反应至关重要, 甲醇能与酚形成氢键, 破坏溶剂化层, 降低其氧化电位, 从而提供一对匹配的交叉偶联反应前体(Scheme 3).这种溶剂效应的形成和存在依赖于某种特定的反应底物, 它可以改变该底物的氧化电位, 从而提供一对匹配的交叉偶联反应前体.
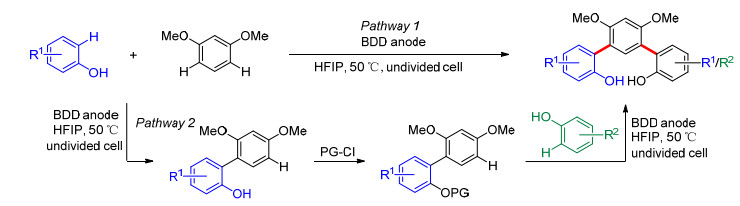
作为酚与富电子芳烃反应的进一步延伸, Waldvogel课题组[22]于2016年还报道了间三联苯-2, 2'-二酚的合成.使用1, 3-二甲氧基苯与2, 4-二取代酚一锅法电解合成对称的间三联苯-2, 2'-二酚(Scheme 4, Pathway 1), 反应产率为28%~41%.而合成非对称间三联苯-2, 2'二酚需要两步电化学偶联: (1)通过电解实现酚和1, 3-二甲氧基苯之间的脱氢交叉偶联; (2)将第一步反应产物中的酚羟基保护, 并与另一种酚进行再次电化学交叉偶联(Scheme 4, Pathway 2).
此外, Waldvogel等[23]也将电化学方法应用于两种酚的直接交叉偶联(Eq. 3).邻甲氧基苯酚衍生物具有较低的氧化电位, 是一种适合偶联的酚类化合物.反应使用BDD阳极和镍网阴极, 并在简易的一室型电解池中进行.将邻甲氧基苯酚衍生物与非富电子酚反应, 可以得到一系列2, 2'-双酚类化合物.此反应与酚-芳烃交叉偶联反应相似, 具有良好的交叉偶联选择性, 且产率较高.为了解决氟化介质可能造成的环境问题, Waldvogel等使用甲酸代替HFIP, 也获得了相似的反应选择性, 但产率较低.
|
|
(3) |
2016年, Waldvogel课题组[24]报道了富电子酚与受保护酚之间的脱氢交叉偶联(Eq. 4), 阳极和阴极均采用BDD电极.硅保护基(如TIPS)对酚的电化学交叉脱氢偶联有显著的促进作用, 反应在相同电解条件下的产率明显提高, 且适用底物范围更广.值得关注的是, 反应在30 mmol的底物用量下依然有较高的反应收率.
芳胺具有与酚类似的低氧化电位, 近年来也有较多关于芳胺的偶联反应的报道. 2007年, Jutand课题组[25]实现了在电化学条件下钯催化N-乙酰芳胺与烯烃之间的Heck偶联反应(Eq. 5).该反应以炭毡为阳极材料, 泡沫镍为阴极材料, 在反应过程中不使用氧化剂; 反应在恒电位条件下进行电解, 以1, 4-苯醌为氧化还原介质, 实现钯的催化循环.此反应在一般情况下只能获得中等的反应产率. 2014年, Kakiuchi课题组[26]将该电化学氧化应用于芳基吡啶的区域选择性偶联反应.
|
|
(4) |
|
|
(5) |
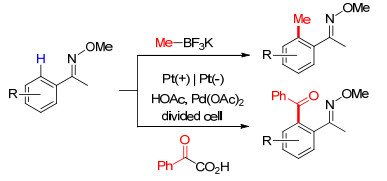
2017年, Mei等[27]将Pd催化剂应用于芳基酮肟与有机硼试剂或α-芳基酮酸的C—C键的偶联反应(Scheme 5).该反应在隔离式电解池中进行, 以醋酸作溶剂, Pt为电极, 可以得到中等至较好收率.
2010年, Jorgensen课题组[28]利用手性胺催化的电化学氧化, 实现了脂肪醛和受保护氨基取代的酚之间的不对称脱氢交叉偶联.在此反应过程中, 酚先被阳极氧化生成亲电性的中间体, 再与原位生成的烯胺进行亲核加成, 实现C—C键的偶联(Scheme 6).当脂肪醛过量时会得到一系列的多取代氨基苯酚.同年, Jang课题组[29]将电化学氧化与有机催化相结合, 实现了脂肪醛与氧杂蒽之间的脱氢交叉偶联.之后, Luo等[30]也报道了同时使用手性伯胺催化, 实现芳基叔胺与简单酮的不对称脱氢交叉偶联.
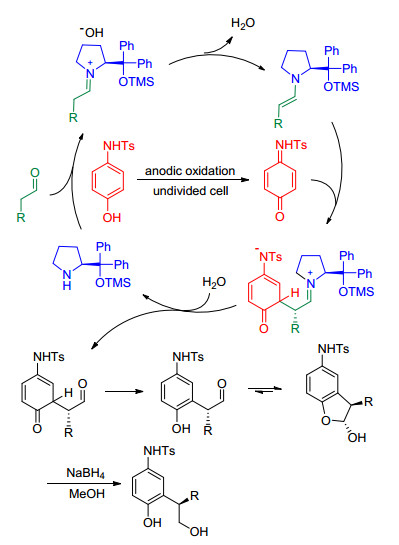
2015年, Zeng和Little课题组[31]以三芳基胺作为氧化还原介质, 实现了烯基酰胺的α-芳基化(Eq. 6).在此反应中, 芳基自由基与烯基酰胺之间发生电子转移, 产生高度亲电的酰亚胺阳离子, 从而实现芳环上的Friedel-Crafts反应.
|
|
(6) |
与酚-酚之间的交叉偶联反应类似, 2017年, Waldvogel课题组[32]报道了两种受保护芳胺化合物的电化学脱氢交叉偶联(Eq. 7).他们选择玻碳材料作为阳极和阴极, 能以较好的产率合成2, 2'-二氨基联苯化合物, 该反应中HFIP仍然是实现交叉偶联高选择性的关键因素.
2017年, Hilt课题组[33]报道了一种由三联苯化合物制备稠环的方法(Scheme 8).该反应在酸性条件下进行, 以2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)作为氧化还原介质.利用间接电氧化, DDQ的用量相较于传统反应可降至催化量, 且产率较高.
|
|
(7) |
|
|
(8) |
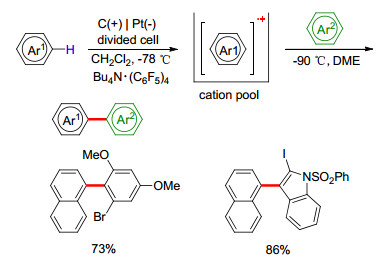
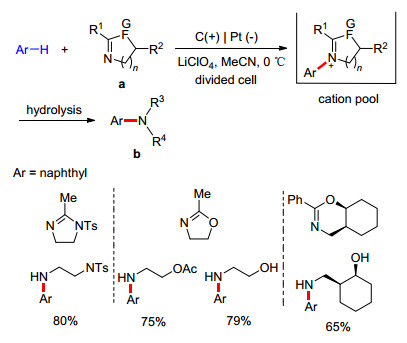
由于氧化电位较高, 非富电子芳香化合物很难利用一般的阳极氧化方法来构建C—C键. 2012年, Yoshida课题组[34]报道了两种未活化芳香化合物之间的脱氢交叉偶联(Scheme 7).反应使用含有超细纤维的碳毡作阳极, 铂作阴极, Bu4N•(C6F5)4作电解液, 在-78 ℃条件下发生芳香化合物(例如萘或蒽)的氧化, 产生自由基阳离子.不稳定的芳基自由基阳离子在“阳离子稳定池”中聚集, 在-90 ℃条件下与另一芳烃底物作用, 生成交叉偶联产物.此反应中C—C键的构建是在非氧化条件下发生的, 避免了底物的非选择性氧化以及偶联产物的过度氧化, 具有良好的官能团适用性、较高的收率和反应速率以及区域选择性. 2015年, Atobe课题组[35]也报道了类似的芳基-芳基偶联反应, 他们使用微型低温反应器, 利用平行层流技术, 也实现了未活化芳香化合物的偶联.
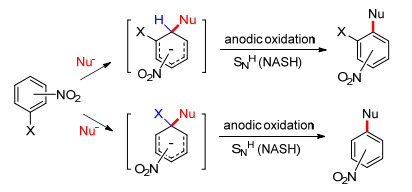
缺电子芳香化合物(如硝基化合物)虽然具有较高的氧化电位, 但这类化合物容易与亲核性物质形成σ-复合物, 利用所形成的Meisenheimer复合物中间体的电化学氧化可以使缺电子芳香化合物间接地实现阳极官能化[36~38].一般而言, 此反应包含两种SNAr反应模式(Scheme 8): (1)通过σ-H-Meisenheimer复合物的氧化来引发质子的消除(Gallardo等[38]将其表示为SNH或NASH过程, 即类似于氢原子被亲核取代的反应); (2)当苯环上连有一个可离去基团(如卤原子)时, 亲核试剂会进攻卤代的碳原子, 从而形成σ-X-复合物, 随后X-离去, 得到最终的亲核取代产物.
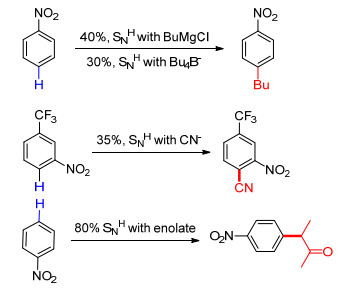
在实际的应用中, 此反应在恒电位条件下进行.以氰根[39, 40]、烷基金属试剂(如格氏试剂、烷基锂)[41, 42]、烯醇[43]等作为亲核试剂, 利用SNH过程实现芳香化合物C—H键的官能化, 用以构建C—C键(Scheme 9).
2017年, Zeng等[44]以α-酮酸为酰化试剂, 在NH4I介导下, 实现了缺电子氮杂芳烃上的C—H键酰基化反应(Eq. 9).反应中, 阳极生成I2首先与α-酮酸反应得到酰基次碘酸盐中间体, 后者随即发生羧酸根阴离子氧化脱羧得到酰基自由基, 最后完成酰基化反应.此过程在简单的一室型电解池中进行, 具有较好的官能团耐受性, 可用于一些药物前体的官能化反应.
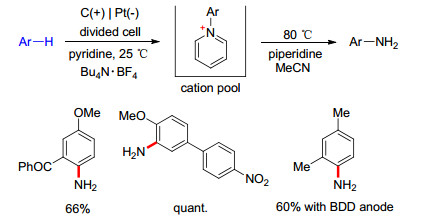
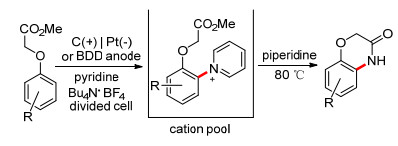
2013年, Yoshida课题组[45]报道了某些芳香化合物C—H键的直接胺化反应(Scheme 10).在吡啶存在下, 芳香化合物的恒流电解形成Zincke型吡啶阳离子, 然后在加热条件下用仲胺(如哌啶)处理, 可以得到芳胺化合物.吡啶在反应中起着关键作用: (1)吡啶的缺电子π键不利于其在阳极条件下的竞争氧化; (2)吡啶氮的强亲核性使其更易与芳香化合物自由基阳离子结合; (3) Zincke型吡啶阳离子带正电荷的吡啶基降低了芳环的电子云密度, 避免了芳香化合物的过度氧化.这种C—H键的胺化反应具有高效、高选择且可预见等优点. Yoshida设计的此反应适用底物主要是富电子芳香化合物(如苯甲醚衍生物).最近, Waldvogel等[46, 47]报道了通过使用BDD电极, 可以将反应的适用范围扩大到电中性的芳香化合物.
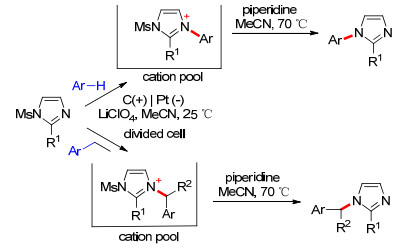
此后, Yoshida课题组[48]报道了甲磺酰基咪唑促进的芳香化合物的胺化(Scheme 11).此反应适用于不含烷烃取代基的底物, 在恒电流条件下, 在阳离子池中先形成咪唑阳离子, 而后脱除甲磺酰基给出偶联产物芳基咪唑.否则咪唑环会进攻芳基的苄位, 最终生成芳甲基咪唑.此反应可用于制备一系列具有生物活性的功能分子.与Zinkle型阳离子类似, 反应中生成的带正电荷的芳基-咪唑中间体也避免了芳香化合物的过度氧化, 从而获得更高的产率.
此外, Yoshida课题组[49]还报道了亚胺作为亲核试剂的芳香化合物的胺化(Scheme 12).这些非芳香杂环化合物(亚胺)在稳定的自由基反应池中与芳基自由基结合, 进而分解为一系列N-含氧(或氮)烷基芳胺.通过这类反应可以实现一些药物分子如茴拉西坦(aniracetam)和非诺贝特(fenofibrate)的修饰.
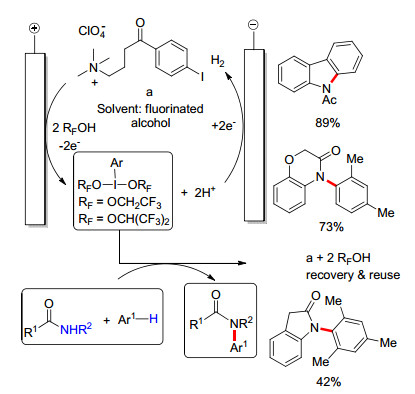
除了形成自由基反应池, 也可以通过氧化还原介质来实现芳香化合物的胺化.碘代芳烃的阳极氧化可以产生高价碘化物, 能作为氧化还原介质参与官能团化[50].例如, 在三氟乙醇存在下, 碘苯的阳极氧化会产生二- (三氟乙氧基)碘苯(PIFE). Nishiyama课题组[51]使用PIFE氧化酰胺, 产生相应的氮鎓离子; 后者与富电子芳香化合物反应形成C—N键.此反应更多的是用于合成含氮芳香杂环化合物. 2016年, Francke课题组[52]使用碘代芳烃制备了一种多功能电化学介质, 即氮鎓离子a.在HFIP中预电解这种介质会产生高价态碘化物, 从而引发酰胺与芳烃的脱氢交叉偶联(Scheme 13).此方法中, 由于氮鎓离子的存在, 反应不需要外加电解质, 并且离子链上存在的羰基可以防止其氧化降解.此外, 这种多功能电化学介质可以在反应结束时回收并循环使用.利用此反应可以得到一系列N-芳基酰胺化合物, 且产率一般较高.
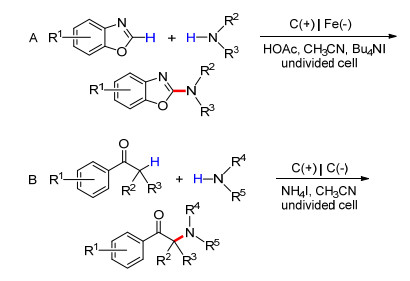
2014年, Zeng和Little等[53]报道了使用四烷基卤化铵作为氧化还原介质的苯并噁唑的胺化(Scheme 14A).该反应采用简单的一室电解池, 以玻璃碳为阳极, 铁为阴极, 于恒电流条件下进行.此方法的局限在于其只适用于仲胺.两年后, 该课题组报道了以NH4I为氧化还原介质的芳基酮的胺化反应, 用以制备α-氨基酮(Scheme 14B)[54].此过程中, 芳基酮与阳极氧化生成的碘单质作用生成α-碘代酮中间体是该反应的关键.
2018年, Lei课题组[55]报道了利用Co催化的N-取代苯甲酰胺邻位C—H键胺化反应, 酰胺基起着导向作用(Eq. 10).在隔离式电解池中, 以吗啉作为底物, 能得到较好的反应收率.此方法底物用量可以拓展至克级且具有较高的反应效率, 但仅限于仲胺.
如第2.2节中所述, 利用Meisenheimer中间体亦可以实现缺电子芳香化合物(如硝基苯)的胺化[40, 56](Eq. 11).
早在1981年, Hussey等[57]就报道了芳香化合物(如蒽、萘、硝基萘和甲苯)在非质子溶剂(如乙腈)中的电化学硝化反应.以四丁基亚硝酸铵或硝基甲烷作硝化试剂, 通过控制电位可以得到芳香化合物的单硝基或多硝基取代产物. 1999年, Sereno等[58]报道了在含有非离子表面活性剂的水溶液中, 亚硝酸盐的存在下, 萘在铂电极上的硝化反应; 主要得到1-和2-硝基萘.
|
|
(10) |
|
|
(11) |
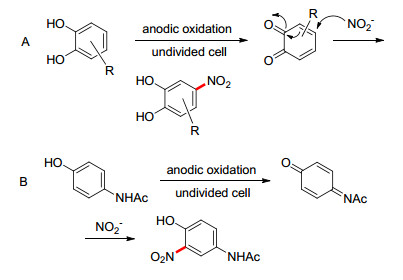
由于邻苯二酚衍生物具有较低的氧化电位, 其在阳极的氧化易形成1, 2-苯醌中间体.加入亲核试剂与中间体发生反应可以用来构建碳-杂键以及一些杂环化合物. 2007年, Nematollahi课题组[59]报道了在水溶液中以亚硝酸根离子为亲核试剂的邻苯二酚类化合物的硝化(Scheme 15A).邻苯二酚的氧化生成苯醌中间体, 后者与亚硝酸根离子发生Michael加成, 得到硝基邻苯二酚产物.硝基的存在也阻止了产物的过度氧化.类似地, 2-氨基酚或4-氨基酚也能在较低电位下被氧化成醌亚胺, 2015年, 同一课题组报道了4-乙酰氨基苯酚的电化学硝化, 且产率较高(Scheme 15B)[60].
2009年, Tajima课题组[61]报道了一种富电子芳香化合物乙酰氧基化的简单方法(Eq. 12).反应使用固体硅胶负载碱, 在酸性条件下原位生成酸根作为亲核试剂.该反应在恒电流条件下发生, 且收率较高.使用三氟乙酸会拓宽底物的氧化电位窗口; 再选择合适的助溶剂(如乙腈或二氯甲烷), 可将降片冰烷或金刚烷等未活化的饱和烷烃C—H键氧化.此反应中, 若对氧化电位进行合理控制, 可以实现底物的选择性氧化.此外, 文中使用的固体硅胶在酸性环境下具有较好的稳定性, 可通过过滤回收进而循环利用.
此后, Budnikova课题组[62, 63]报道了Pd或Ni催化的芳基吡啶的电化学酰氧化(Eq. 13).阳极氧化产生的高氧化态Pd或Ni与芳基吡啶形成配合物, 再与羧酸或全氟羧酸反应, 得到酰氧基化产物.为防止高氧化态金属催化剂的过早阴极还原, 此反应需要在隔离电解池中进行.
|
|
(13) |
2017年, Mei课题组[64]报道了Pd催化下芳基酮肟的电化学酰氧化反应(Eq. 14).以n-Bu4NOAc为电解质, 于40 ℃条件下恒流电解, 产率较好.随后, Sanford等[65]进一步发展了此类反应, 使用四甲基乙酸铵(TMAOAc)作电解质, 并加入苯醌作为氧化还原介质实现Pd的催化循环, 在隔离式电解池中完成了一系列含导向基芳环上的乙酰氧基化(Eq. 15).
|
|
(14) |
|
|
(15) |
2017年, Ackermann课题组[66]报道了室温下Co催化的C—H键氧化, 用于芳基醚的合成(Eq. 16).以网状玻璃碳(RVC)为阳极, N-取代苯甲酰胺与由阳极氧化生成的高氧化态Co形成配合物, 再与醇发生反应得到芳基醚产物.此反应底物使用范围广, 具有较好的区域选择性.除合成芳基醚外, 该方法也可用于合成烯醇醚.
|
|
(16) |
除了第2.1节中提到的芳基C—H/C—H之间的直接偶联外, 酚类化合物也能够与卤代物反应形成芳基醚. Nishiyama课题组[67]系统报道了邻卤代酚的阳极氧化.文中指出, 一卤代酚的阳极氧化会完全生成C—H/C—H偶联产物; 而2, 5-二卤代苯酚则会通过阳极氧化以及阴极处的C—X键断裂, 形成二芳基醚(Scheme 16)[68, 69].在反应过程中, 经电化学卤化合成的二卤代酚与在甲醇中电解产生的苯氧基自由基偶联生成二聚醚中间体, 后者在阴极处发生还原得到二芳基醚.此反应中, 二卤代酚底物上的一个卤原子促就了反应的区域选择性. Nishiyama课题组利用此法成功合成了邻-甲基罗氏唐松草碱[69, 70]和黄酮化合物[71]等.
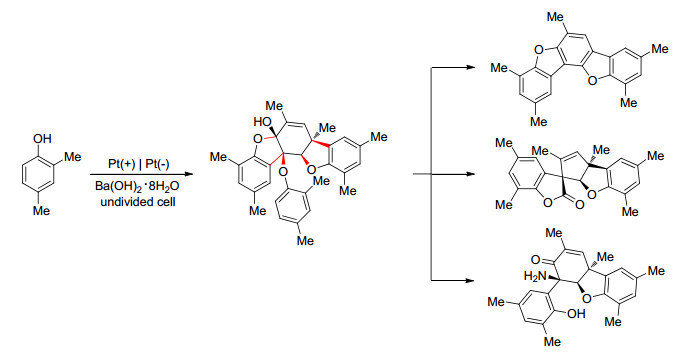
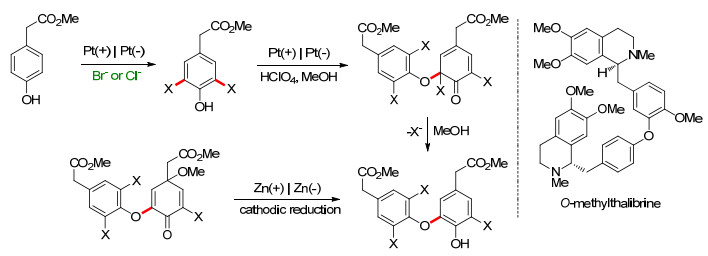
若不经过卤代酚步骤, 直接进行酚的阳极氧化, 通常会得到一系列具有复杂碳骨架的芳基醚产物, 这些产物在有机合成领域通常是很有价值的.例如, Waldvogel课题组[72~75]报道了在一室型电解池中以铂为电极, Ba(OH)8•8H2O的甲醇溶液作电解质, 2, 4-二甲基苯酚的阳极氧化.反应会生成脱氢四聚体, 产率最高可达52%.此反应单次的底物用量可达23 g, 可实现大量制备; 由于其四聚体的溶解性差, 得到的产品沉淀于电解槽中, 易于分离.此外, 产物可以进一步被修饰进而合成各种具有复杂结构的化合物[72], 部分修饰产物如Scheme 17所示.
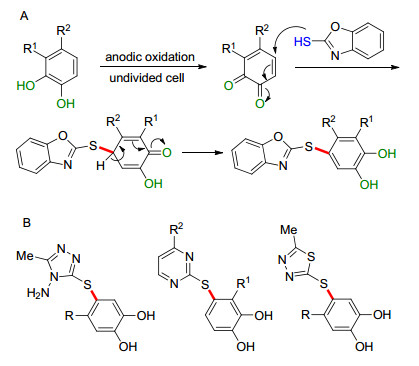
邻苯二酚和对氨基酚等化合物具有较低的氧化电位, 可通过在阳极的氧化形成邻二苯醌或醌亚胺, 进而顺利实现芳基C—H键的活化生成芳基硫醚. 2005年, Tammari课题组[76]将2-巯基苯并噁唑与邻苯二酚衍生物置于一室型电解池中, 通过一锅法高效地合成了二芳基硫醚, 产率中等(Scheme 18A). Zeng等[77-79]系统研究了醋酸缓冲溶液中噻二唑硫醇、1, 3, 4-三唑硫醇及嘧啶硫醇参与的硫醚的电化学合成.通过筛选电极材料、电位和电解池等条件, 高效地合成了相应具有生理活性的硫醚产物(Scheme 18B). 2008年, Khodaei课题组[80]报道了四唑硫醇和邻(对)二苯酚参与的硫醚的合成.除杂芳基硫醇作亲核试剂外, 2009年, Fotouhi等[81]报道了以2-二甲氨基乙硫醇为亲核试剂, 4-叔丁基邻苯二酚的硫醚化反应, 反应过程中同样经历了苯醌中间体的形成.
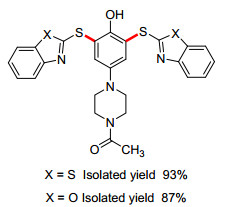
对氨基苯酚化合物也是合成二芳基硫醚较为理想的底物. 2012年, Nematollahi课题组[82]报道了巯基苯并噻(噁)唑作为亲核试剂, 1-(4-(4-羟基苯基)-哌嗪-1-基)乙酮经醌亚胺中间体的C—H键活化(图 1).反应所得的硫醚产物继续电解, 通常给出二聚硫醚产物, 且产率较高.此反应提供了一种高效环保的芳基硫代苯并唑的合成方法.
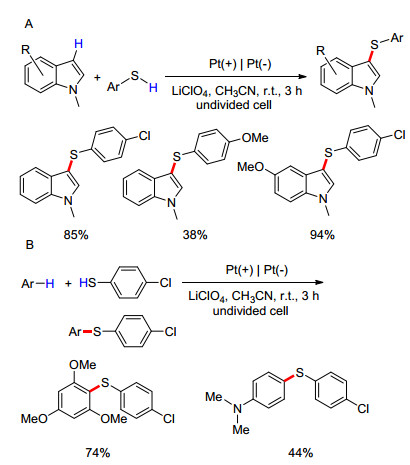
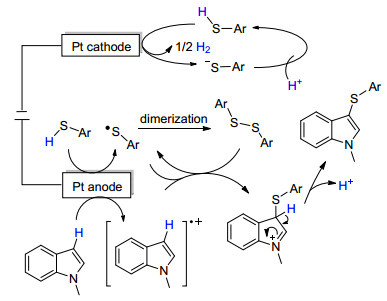
2017年, Lei课题组[83]报道了硫酚与吲哚化合物的电化学脱氢偶联用于构建C—S键, 形成芳基硫醚.反应以LiClO4作电介质, 乙腈为溶剂, 在恒电流条件下使用Pt作为电极, 并在一室型电解池中进行, 具有高效、环保等优点.在普适性方面, 除少数硫酚底物(如4-甲氧基硫酚)外, 该反应均能获得较高的收率; 使用吲哚作为底物, 也都能得到较高产率的硫醚(Scheme 19A).此外, Lei课题组也考察了使用其他富电子芳香化合物(如酚、胺、芳香醚、吡咯、噻吩等)作底物的反应情况(Scheme 19B), 得到了较好的反应结果, 证明了此反应具有广泛的应用前景.文中提出了此类反应的可能机理为:硫酚经阳极氧化生成的自由基二聚形成二硫醚, 后者再与吲哚的阳极氧化产物(自由基阳离子)发生偶联反应, 给出最终产物(Scheme 20).
2003年, Nematollahi课题组[84]报道了在芳基亚磺酸存在下4-叔丁基邻苯二酚的磺酰化反应(Scheme 21A).此反应与邻苯二酚的硫醚化和芳基的硝化等反应类似, 是经中间体醌与芳磺酸发生Michael加成反应得到产物.反应使用(4-甲基)苯亚磺酸作为磺酰化试剂, 得到了较高的收率. 2013年, 该课题组[85]系统研究了邻苯二酚及其衍生物电化学磺酰化反应的动力学过程. 2007年, Zeng课题组[86]报道了在水溶液中咖啡酸的磺酰化反应, 用于合成一系列具有潜在抑制HIV整合酶生物活性的芳基磺酰基咖啡酸衍生物.此反应具有简便、产率良好和产品纯度高等优点. 2013年, Zeng等[87]报道了氨基苯酚化合物的磺酰化反应.与邻苯二酚类似, 反应过程也经历了Michael加成, 并且磺酰基化会区域选择性地发生在氨基的间位(Scheme 21B).值得注意的是, 未保护氨基取代的苯酚并不能经电氧化得到预期产物.
2017年, Chen和Yu等[88]报道了亚磺酸盐存在下吲哚及其衍生物的α-磺酰化反应(Eq. 17).该反应使用Pt作电极, CH3CN/H2O为溶剂, TBAI(四丁基碘化铵)作催化剂, 在室温下反应能够得到较高产率的吲哚基芳基砜.此反应也具有较好的官能团耐受性, 可用于合成具有生物活性的5-HT6调节剂.
|
|
(17) |
稠环和杂环化合物在药物、染料和农药等的合成中具有广泛的应用.利用分子内偶联构建C—C/N/O/S键, 或通过两种分子之间的电化学双官能化, 可以构筑各类不同的稠环和杂环化合物.
2016年, Xu课题组[89]报道了含有炔基的芳胺化合物分子内的C—H/N—H脱氢[3+2]环化反应, 用于合成吲哚和氮杂吲哚(Eq. 18).使用二茂铁(Cp2Fe)作为氧化还原介质, 利用阳极氧化间接生成氮中心自由基.这些自由基可以与苯环或芳杂环进行分子内加成, 得到环化产物.此反应在一室型电解池中进行, 具有较好的反应速率及官能团耐受性. 2017年, Xu等[90]将此反应进一步发展, 用于多环N-杂芳香化合物的电化学合成.
|
|
(18) |
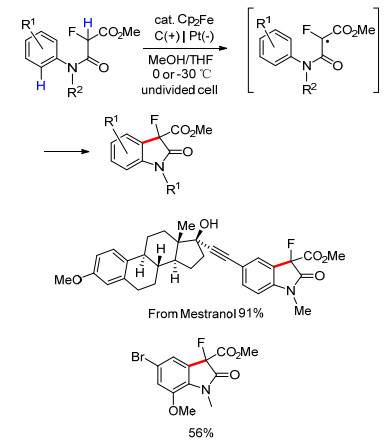
利用二茂铁介导产生碳中心自由基, Xu等[91]也实现了N-芳基酰胺的分子内脱氢偶联, 用于合成3-氟-2-吲哚酮衍生物(Scheme 22).此反应有很好的官能团耐受性, 可以用于修饰各种复杂底物, 如炔雌醇甲醚(Mestranol).
2018年, 该课题组开发了一种新型的二氟甲基化试剂CF2HSO2NHNHBoc, 其通过二茂铁介导下产生二氟甲基自由基, 再与炔基发生加成反应, 得到相应氟化的二苯并氮杂七元环产物[92](Eq.19).此方法具有简易高效等优点, 可用于多种含氮杂七元环结构商业药物的合成, 如米安色林(Mianserin)、米氮平(Mirtazapine)等.
2017年, Lei课题组[93]报道了以碘化物为氧化还原介质的N-芳基烯胺的电化学分子内脱氢环化, 用于合成吲哚衍生物(Eq. 20).此反应在一室型电解池中进行, 采用铂板作电极, 可以得到良好至极高的产率.通过高价碘间接氧化生成的氮中心自由基是该反应的关键.此外, 当使用N-吡啶基烯胺作为底物时, 可以制备咪唑并[1, 2-a]吡啶.同年, 该课题组[94]报道了芳基炔酮与亚磺酸的电化学脱氢串联环化反应, 用于合成磺化茚酮(Eq. 21).该反应以Pt作电极, Bu4NI作氧化还原催化剂, 在一室型电解池中进行.此反应的机理为: Bu4NI经阳极氧化生成的I+将亚磺酸氧化生成相应的磺酰基, 后者对炔酮进行自由基加成; 生成的中间体经分子内环化和氧化后得到茚酮产物.这种串联环化反应具有良好的官能团耐受性和高反应效率.
|
|
(20) |
Xu课题组[95]于2017年报道了一种通过阳极N—H键裂解, 实现C—H/N—H脱氢交叉偶联的电化学方法(Eq. 22).反应具有适用底物范围广、选择性高和产率较高等优点, 用以合成官能化的四环苯(吡啶)并咪唑衍生物. 2017年, 该课题组[96]报道了由联芳醛和NH3合成菲啶的C—H/N—H交叉偶联反应(Eq. 23).该方法使用廉价易得的氨作为氮供体, 具有较高原子经济性, 且产率较好.
|
|
(21) |
|
|
(22) |
|
|
(23) |
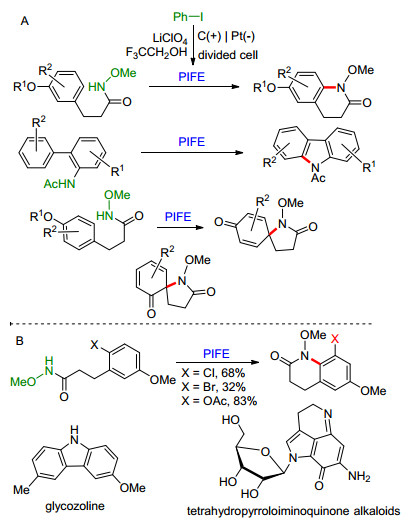
在三氟乙醇存在下, 碘苯的氧化可以产生二(三氟乙氧基)碘苯(PIFE). Nishiyama课题组[51]使用PIFE将酰胺或异羟肟酸进行氧化生成相应的氮自由基.这些自由基与富电子芳香化合物进行环化, 可以得到喹啉骨架[97]、咔唑[98]或螺环[99](取决于苯环上的取代形式) (Scheme 23A).此反应可用于吡咯类生物碱(tetrahy- dropyrroloiminoquinone alkaloids)和山小桔灵(glycol- zoline)的合成.当苯环上邻位被卤素或类卤素取代时, 环化反应进行的同时会发生取代基迁移(Scheme 23B)[99].
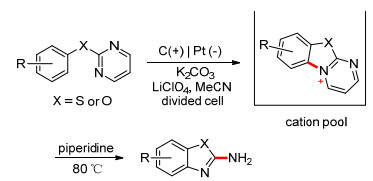
如C—H键的胺化一节中所提到的, 利用Zincke型吡啶阳离子池等也可以高效地合成某些杂环化合物. 2015年, Yoshida课题组[100]利用分子内的C—H胺化, 合成出苯并噁唑(噻唑)化合物(Scheme 24). 2017年, Waldvogel等[101]成功地合成了1, 4-苯并噁嗪-3-酮(Scheme 25).作为一些天然产物和生物活性物质中的重要结构单元, 1, 4-苯并噁嗪-3-酮此前广泛采用过渡金属催化反应来合成; 此方法提供了一种绿色可持续的合成途径, 且具有一定的官能团耐受性及较广的底物适用范围.
2017年, Xu课题组[102]报道了由N-苄基酰胺合成4H-1, 3-苯并噁嗪的电化学C—H键氧化反应(Eq. 24).该过程中, 首先发生富电子苯环上的电氧化, 生成阳离子自由基, 随后活化的C—H键与酰胺环合, 形成噁嗪环.
同年, Waldvogel等[103, 104]利用电化学方法由N-芳基酰胺合成苯并噁唑(Eq. 25).反应中酰胺基氮正离子的阳极生成使环化反应和C—O键的构建得以发生.此过程使用简单的一室型电解池, 具有较好的产率及官能团耐受性.
|
|
(24) |
|
|
(25) |
2017年Zeng等[105]报道了芳香羧酸的电化学分子内脱氢酯化反应(Eq. 26).该方法使用简单的一室型电解池, 底物用量可拓展至40 g以上, 能用于多种内酯的合成, 且具有较高区域选择性.
|
|
(26) |
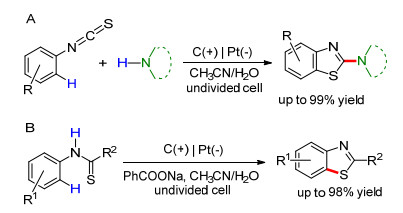
2017年, Xu课题组[106]研究了基于硫自由基的C—S键构建, 报道了在一室型电解池中, 以四甲基哌啶氮氧化物(TEMPO)作氧化还原介质N-芳基硫代酰胺的分子内脱氢偶联, 用于高效合成苯并噻唑及其衍生物(Eq. 27).同年, Lei课题组[107]报道了芳基异硫氰酸酯与仲胺的串联反应, 用来合成苯并噻唑(Scheme 26A).与Xu等的方法不同, 此反应的电解体系中无需添加氧化还原介质.此外, 通过在电解体系中加入2 equiv. PhCOONa, Lei等也实现了无氧化还原介质下的N-芳基硫代酰胺的分子内脱氢偶联(Scheme 26B). 2018年, Xu课题组[108]进一步改进了此类反应, 即应用连续流动化学的方法, 无需添加支持电解质即可完成N-芳基(吡啶基)硫代酰胺的偶联反应, 合成苯(吡啶)并噻唑(Eq. 28).利用流动化学的方法, 使用较小反应器即可完成大量底物的转化.
|
|
(27) |
芳香化合物可以发生分子间的电化学双官能化, 从而制备相应的苯并杂环或稠环化合物. 2000年, Chiba课题组[109]报道了酚与烯烃之间的电化学偶联[3+2]环加成反应, 用于合成二氢苯并呋喃(Scheme 27).该反应在一室型电解池中进行, 适用于三取代和四取代的脂肪族烯烃以及苯乙烯, 且产率较高. 2002年, Nishiyama课题组[110]报道了萘酚与富电子烯烃的[3+2]或[5+2]环加成反应, 萘酚阳离子中间体的形成是反应的关键.随后, Chiba等[111, 112]采用温控多相溶剂系统进一步发展了酚与烯烃之间的电化学[3+2]反应.此反应过程中, 酚存在于强极性的MeNO2一层中, 而环加成产物聚集在含MeNO2与环己烷的弱极性中间层中.弱极性中间层的形成增强了苯氧离子中间体与非极性烯烃之间的相互作用, 同时阻止了环加成产物的过度氧化.

|
|
(28) |
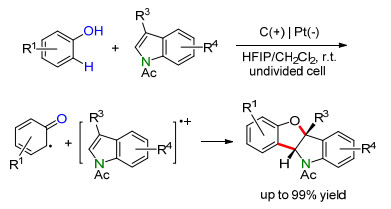
2017年, Lei课题组[113]报道了酚与N-乙酰基吲哚衍生物之间的[3+2]环化反应, 得到一系列苯并呋喃并吲哚化合物(Scheme 28).此反应适用于带有不同C-3和C-2取代基的N-乙酰基吲哚, 且产率很高.然而, 只有使用富电子的苯酚才能取得较高的反应效率.从机理上看, 此反应中的酚和N-乙酰基吲哚均发生了阳极氧化, 通过其生成的自由基中间体实现环化反应.
2018年, Ackermann等[114]利用Co催化, 在室温下实现了水相中N-取代苯甲酰胺上C—H/N—H键与炔烃的环化反应(Eq. 29).反应过程中, 阳极氧化生成的高氧化态Co先与苯甲酰胺底物络合, 随后发生炔烃的插入, 完成环化.该方法使用RVC阳极, 甲醇水溶液为溶剂, 在室温下电解, 有很好的区域和化学选择性, 且底物适用范围广.
|
|
(29) |
随后, Xu课题组[115]报道了Ru催化下N-取代芳胺上C—H/N—H键与炔烃的环化反应, 用于合成吲哚类化合物(Eq. 30).此反应在水溶液体系中进行, 采用简单的一室型电解池, 具有较高的产率和底物适用范围.
|
|
(30) |
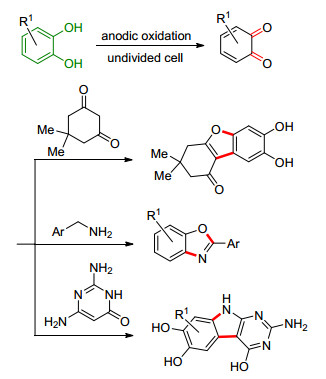
邻(对)苯二酚易发生阳极的氧化反应生成苯醌中间体, 利用此中间体也可以进行某些稠杂环化合物的合成(Scheme 29).例如, Nematollahi课题组[116, 117]报道了邻(对)苯二酚在1, 3-二酮衍生物或苯甲胺存在下发生阳极氧化, 分别得到苯并呋喃衍生物及2-芳基-苯并噁唑[118].此外, Zeng课题组[119, 120]报道了N, O-烯酮缩醛存在下邻苯二酚阳极氧化, 可以制备出相应的吲哚衍生物.
芳香化合物的电化学官能化已用于构建某些碳-碳键、碳-杂键以及苯并噁(噻)唑、吲哚等稠环化合物, 具有绿色可持续、高选择性以及高效率等优点.然而, 前期的工作中对于非富电子芳香化合物的C—H键官能化主要采用构建自由基反应池或Meisenheimer中间体的方法, 适用范围有限.因此开发更多基于非富电子化合物的电化学C—H键活化方法和技术是未来研究的一个目标.在电极材料方面, 未来的研究可以对电极表面进行修饰(如负载催化剂等), 以提高电极材料的性能和化学反应的效率.此外, 开发新型的电催化剂、廉价的电极以及优化电解槽的设计等可以将更多的电化学有机合成应用于工业化生产.最后, 电化学有机合成领域缺乏标准化的仪器设备, 尽管利用实验室中自制的简易设备也可以顺利完成一些电有机反应, 但这可能会导致实验结果难以重复.
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230. doi: 10.1021/acs.chemrev.7b00397
Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302. doi: 10.1021/acscentsci.6b00091
Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485. doi: 10.1021/acs.chemrev.7b00271
Li, Z.; Li, C. J.; Herrerías, C. I.; Yao, X. Chem. Rev. 2007, 107, 6.
Bergman, R. G. Nature 2007, 466, 391.
Sun, C. L.; Li, B. J.; Shi, Z. J. Chem. Rev. 2011, 111, 1293. doi: 10.1021/cr100198w
Liao, K.; Pickel, T. C.; Boyarskikh, V.; Bacsa, J.; Musaev, D. G.; Davies, H. Nature 2016, 47, 230.
Evan, J. H.; Brandon, R. R.; Yong, C.; Tang, J. Z.; Ke, C.; Eastgate, M. D.; Baran, P. S. Nature 2016, 533, 77. doi: 10.1038/nature17431
刘薇, 郑昕宇, 曾建国, 程辟, 有机化学, 2017, 37, 1. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345749.shtmlLiu, W.; Zheng, X. Y.; Zeng, J. G.; Cheng, P. Chin. J. Org. Chem. 2017, 37, 1(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345749.shtml
Liu, Y. R.; Hu, B. L.; Zhang, X. G. Chin. J. Chem. 2017, 35, 307 doi: 10.1002/cjoc.v35.3
裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15. doi: 10.3969/j.issn.0253-2409.2017.01.003Pei, P. K.; Zhang, F.; Yi, H.; Lei, A. W. Acta Chimi. Sinica 2017, 75, 15(in Chinese). doi: 10.3969/j.issn.0253-2409.2017.01.003
Li, T. T.; Jin, P. Y.; Lin, S.; Zhi, Y. G.; Liu, X. Y. Chin. J. Chem. 2016, 34, 490. doi: 10.1002/cjoc.201500891
Schopohl, M. C.; Faust, A.; Mirk, D.; Fr hlich, R.; Kataeva, O. Eur. J. Org. Chem. 2010, 2987.
Waldvogel, S. R.; Mirk, D. Tetrahedron Lett. 2000, 31, 4769.
Malkowsky, I. M.; Griesbach, U.; Pütter, H.; Waldvogel, S. R. Eur. J. Org. Chem. 2006, 4569.
Kirste, A.; Martin, N.; Malkowsky, I. M.; Florian, S.; Andreas, F.; Waldvogel, S. R. Chem. Eur. J. 2009, 15, 2273 doi: 10.1002/chem.v15:10
Kirste, A.; Gregor, S.; Waldvogel, S. R. Org. Lett. 2011, 13, 3126. doi: 10.1021/ol201030g
Kirste, A.; Shotaro, H.; Gregor, S.; Malkowsky, I. M.; Florian, S.; Andreas, F.; Toshio, F.; Waldvogel, S. R. Chem.-Eur. J. 2011, 17, 14164. doi: 10.1002/chem.v17.50
Kirste, A.; Gregor, S.; Florian, S.; Andreas, F.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2010, 49, 971. doi: 10.1002/anie.200904763
Kirste, A.; Elsler, B.; Schnakenburg, G.; Waldvogel, S. R. J. Am. Chem. Soc. 2012, 43, 3571.
Bernd, E.; Anton, W.; Dieter, S.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. Chem.-Eur. J. 2015, 21, 12321. doi: 10.1002/chem.201501604
Sebastian, L.; Anton, W.; Bernd, E.; Dieter, S.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2016, 55, 10872. doi: 10.1002/anie.201605865
Bernd, E.; Dieter, S.; Marie, D. K.; Franke, R.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2014, 53, 5210.
Anton, W.; Dieter, S.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2016, 55, 11180.
Amatore, C.; Cammoun, C.; Jutand, A. Adv. Synth. Catal. 2007, 349, 292. doi: 10.1002/(ISSN)1615-4169
Saito, F.; Aiso, H.; Kochi, T.; Kakiuchi, F. Organometallics 2014, 33, 6704. doi: 10.1021/om500957a
Ma, C.; Zhao, C. Q.; Li, Y. Q.; Zhang, L. P.; Xu, X. T.; Zhang, K.; Mei, T. S. Chem. Commun. 2017, 53, 12189. doi: 10.1039/C7CC07429H
Jensen, K. L.; Franke, P. T.; Nielsen, L. T.; Daasbjerg, K.; Joergensen, K. A. Angew. Chem., Int. Ed. 2010, 122, 133. doi: 10.1002/ange.200904754
Ho, X. H.; Mho, S. I.; Kang, H.; Jang, H. Y. Eur. J. Org. Chem. 2010, 4436.
Fu, N.; Li, L.; Yang, Q.; Luo, S. Z. Org. Lett. 2017, 19, 2122. doi: 10.1021/acs.orglett.7b00746
Li, L. J.; Jiang, Y. Y.; Lam, C. M.; Zeng, C. C.; Hu, L. M.; Little, R. D. J. Org. Chem. 2015, 80, 11021. doi: 10.1021/acs.joc.5b02222
Lara, S.; Enders, M.; Bernd, E.; Dieter, S.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. Angew. Chem. Int. Ed. 2017, 56, 4877. doi: 10.1002/anie.201612613
R se, P.; Emge, S.; K nig, C. A.; Hilt, G. Adv. Synth. Catal. 2017, 359, 1359. doi: 10.1002/adsc.v359.8
Morofuji, T.; Shimizu, A.; Yoshida, J. Angew. Chem., Int. Ed. 2012, 51, 7259. doi: 10.1002/anie.201202788
Arai, T.; Hiroyuki, T.; Nakabayashi, K.; Kashiwagi, T.; Mahito, A. Chem. Commun. 2015, 51, 4891. doi: 10.1039/C5CC01253H
Gallardo, I.; Gonzalo, G.. Eur. J. Org. Chem. 2008, 2463.
Moutiers, G.; Pinson, J.; Terrier, F.; Goumont, R. Chem.-Eur. J. 2001, 7, 1712. doi: 10.1002/(ISSN)1521-3765
Charushin, V. N. ; Chupakhin, O. N. In Topics in Heterocyxlic Chemistry, Ed. : Selig, P., Springer International Publishing, Cham. 2013, Vol. 37, pp. 1~50.
Gallardo, I.; Guirado, G.; Marquet, J. Chem.-Eur. J. 2001, 7, 1759. doi: 10.1002/(ISSN)1521-3765
Gallardo, I.; Gonzalo, G.; Marquet, J. J. Org. Chem. 2002, 67, 2548. doi: 10.1021/jo010847t
Gallardo, I.; Gonzalo, G.; Marquet, J. J. Org. Chem. 2003, 68, 631. doi: 10.1021/jo025898k
Gallardo, I.; Gonzalo, G.; Marquet, J. J. Org. Chem. 2003, 68, 7334. doi: 10.1021/jo030158c
Gallardo, I.; Guirado, G.; Marquet, J. Eur. J. Org. Chem. 2002, 261.
Wang, Q. Q.; Xu, K.; Jiang, Y. Y.; Liu, Y. G.; Sun, B. G.; Zeng, C. C. Org. Lett. 2017, 19, 5517. doi: 10.1021/acs.orglett.7b02589
Morofuji, T.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 5000. doi: 10.1021/ja402083e
M hle, S.; Herold, S.; Richter, F.; Nefzger, H.; Waldvogel, S. R. ChemElectroChem 2017, 4, 2196. doi: 10.1002/celc.201700476
Herold, S.; M hle, S.; Zirbes, M.; Richter, F.; Nefzger, H.; Waldvogel, S. R. Eur. J. Org. Chem. 2016, 1274.
Morofuji, T.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2014, 136, 4496. doi: 10.1021/ja501093m
Morofuji, T.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2015, 137, 9816. doi: 10.1021/jacs.5b06526
Watts, K.; Gattrell, W.; Beilstein, T. J. Org. Chem, 2011, 7, 1108.
Nishiyama, S.; Amano, Y. Tetrahedron Lett. 2006, 37, 6505.
Broese, T.; Francke, R. Org. Lett. 2016, 18, 5896. doi: 10.1021/acs.orglett.6b02979
Gao, W. J.; Li, W. C.; Zeng, C. C.; Tian, H. Y.; Hu, L. M.; Little, R. D. J. Org. Chem. 2014, 79, 9613. doi: 10.1021/jo501736w
Liang, S.; Zeng, C. C.; Tian, H. Y.; Sun, B. G.; Luo X. G.; Ren F. Z. J. Org. Chem. 2016, 2016, 11157.
Gao, X. L.; Wang, P.; Zeng, L.; Tang, S.; Lei, A. W. J. Am. Chem. Soc. 2018, 140, 4195 doi: 10.1021/jacs.7b13049
Gallardo, I.; Guirado, G.; Marquet, J. Eur. J. Org. Chem. 2002, 2002, 251. doi: 10.1002/1099-0690(20021)2002:2<251::AID-EJOC251>3.0.CO;2-A
Hussey, C. L.; Achord, J. M. Annal. Dogmatic Theology 1981, 128, 2556.
Cortona, M. N.; Vettorazzi, N. R.; Silber, J. J.; Sereno, L. E. J. Electroanal. Chem. 1999, 470, 157. doi: 10.1016/S0022-0728(99)00232-6
Nematollahi, D.; Ariapad, A.; Rafiee, M. J. Electrochem. Soc. 2007, 602, 37.
Salahifar, E.; Nematollahi, D.; Bayat, M.; Mahyari, A.; Rudbari, H. A. Org. Lett. 2015, 17, 4666. doi: 10.1021/acs.orglett.5b01837
Tajima, T.; Kishi, Y.; Atsushi, N. Electrochim. Acta 2009, 54, 5959. doi: 10.1016/j.electacta.2009.05.067
Dudkina, Y. B.; Mikhaylov, D. Y.; Gryaznova, T. V.; Tufatullin, A. I.; Kataeva, O. N.; David, A. V.; Budnikova, Y. H. Organometallics 2013, 32, 4785. doi: 10.1021/om400492g
Dudkina, Y. B.; Gryaznova, T. V.; Sinyashin, O. G.; Budnikova, Y. H. Russ. Chem. Bull. 2015, 64, 1713. doi: 10.1007/s11172-015-1067-3
Li, Yi. Q.; Yang, Q. L.; Fang, P.; Mei, T. S.; Zhang, D. Y. Org. Lett. 2017, 19, 2905. doi: 10.1021/acs.orglett.7b01138
Shrestha, A.; Lee, M.; Dunn, A. L.; Sanford, M. S. Org. Lett. 2018, 20, 204. doi: 10.1021/acs.orglett.7b03559
Sauermann, N.; Meyer, T. H.; Tian, C.; Ackermann, L. J. Am. Chem. Soc. 2017, 139, 18452. doi: 10.1021/jacs.7b11025
Nishiyama, S.; Yamamura, S. Synlett 2002, 533.
Kazuki, M.; Takahashi, M.; Yamamura, S.; Nishiyama, S. Tetra-hedron 2001, 57. 5527. doi: 10.1016/S0040-4020(01)00478-1
Kawabata, Y.; Naito, Y.; Saitoh, T.; Kawa, K.; Fuchigami, T.; Nishiyama, S. Eur. J. Org. Chem. 2013, 99.
Naito, Y.; Tanabe, T.; Yuki, K.; Ishikawa, Y.; Nishiyama, S. Tetrahedron Lett. 2010, 51, 4776. doi: 10.1016/j.tetlet.2010.07.037
Tanabe, T.; Doi, F.; Ogamino, T.; Nishiyama, S. Tetrahedron Lett. 2004, 45, 3477. doi: 10.1016/j.tetlet.2004.02.154
Barjau, J.; Schnakenburg, G.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2011, 50, 1415. doi: 10.1002/anie.v50.6
Michael, M.; Lars, A.; Caroline, S.; Joaquin, B.; Dieter, S.; Till, O.; Arne, L.; Siegfried, R. W. Eur. J. Org. Chem. 2015, 4876.
Barjau, J.; Schnakenburg, G.; Waldvogel, S. R. Synthesis 2011, 2054.
Barjau, J.; K nigs, P.; Kataeva, O.; Waldvogel, S. R. Synlett 2008, 2309.
Nematollahi, D.; Tammari, E. J. Org. Chem. 2005, 70, 7769. doi: 10.1021/jo0508301
Zeng, C. C.; Liu, F. J.; Ping. D. W.; Cai, Y. L.; Zhong, R. G.; Becker, J. Y. J. Electroanal. Chem. 2009, 625, 131. doi: 10.1016/j.jelechem.2008.10.019
Zeng, C. C.; Liu, F. J.; Ping, D. W.; Hu, L. M.; Cai, Y. L.; Zhong, R. G.. Tetrahedron 2009, 65, 4505. doi: 10.1016/j.tet.2009.03.101
Zeng, C. C.; Ping, D. W.; Zhang, S. C.; Zhong, R. G.; Becker, J. Y. J. Electroanal. Chem. 2008, 622, 90. doi: 10.1016/j.jelechem.2008.05.009
Khodaei, M. M.; Alizadeh, A.; Pakravan, N. J. Org. Chem. 2008, 73, 2527. doi: 10.1021/jo702327m
Fotouhi, L.; Asadi, S.; Tammari, E.; Heravi, M. M.; Nematollahi, D. Anal. Bioanal. Electrochem. 2009, 1, 216.
Amani, A.; Nematollahi, D. J. Org. Chem. 2012, 77, 11130.
Wang, P.; Tang, S.; Huang, P. F.; Lei, A. W. Angew. Chem., Int. Ed. 2017, 56, 3009. doi: 10.1002/anie.201700012
Nematollahi, D. N.; Ramazanali, R.; Maryam, M. Synth. Commun. 2003, 34, 2269.
Nematollahi, D.; Beiginejad, H.; Varmaghani, F.; Bayat, M.; Salehzadeh, H. J. Electrochem. Soc. 2013, 160, 142.
Zeng, C. C.; Liu, C. F.; Zeng, J.; Zhong, R. G.. J. Electroanal. Chem. 2007, 608, 85. doi: 10.1016/j.jelechem.2007.02.005
Xiao, H. L.; Yang, C. W.; Zhang, N. T.; Zeng, C. C.; Hu, L. M.; Tian, H. Y.; Little, R. D. Tetrahedron 2013, 69, 658. doi: 10.1016/j.tet.2012.11.005
Feng, M. L.; Xi, L. Y.; Chen, S. Y.; Yu, X. Q. Eur. J. Org. Chem. 2017, 2017, 2746. doi: 10.1002/ejoc.201700269
Hou, Z. W.; Mao, Z. Y.; Zhao, H. B.; Melcamu, Y. Y.; Lu, X.; Song, J.; Xu, H. C. Angew. Chem., Int. Ed. 2016, 55, 9168. doi: 10.1002/anie.201602616
Hou, Z. W.; Mao, Z. Y.; Song, J.; Xu, H. C. ACS Catal. 2017, 7, 5810. doi: 10.1021/acscatal.7b02105
Wu, Z. J.; Xu, H. C. Angew. Chem., Int. Ed. 2017, 56, 4631. doi: 10.1002/anie.201702378
Xiong, P.; Xu, H.; Song, J. S.; Xu, H. C. J. Am. Chem. Soc. 2018, 140, 2460. doi: 10.1021/jacs.8b00391
Tang, S.; Gao, X. L.; Lei, A. W. Chem. Commun. 2017, 53, 3354. doi: 10.1039/C7CC00410A
Wen, J. W.; Shi, W. Y.; Zhang, F.; Liu, D.; Tang, S.; Wang, H. M.; Lin, X. M.; Lei, A. W. Org. Lett. 2017, 19, 3131. doi: 10.1021/acs.orglett.7b01256
Zhao, H. B.; Hou, Z. W.; Liu, Z. J.; Zhou, Z. F.; Song, J. S.; Xu, H. C. Angew. Chem. Int. Ed. 2017, 56, 587. doi: 10.1002/anie.201610715
Zhao, H. B.; Liu, Z. J.; Song, J.; Xu, H. C. Angew. Chem., Int. Ed. 2017, 56, 12732. doi: 10.1002/anie.201707192
Inoue, K.; Ishikawa, Y.; Nishiyama, S. Org. Lett. 2010, 12. 436.
Kajiyama, D.; Inoue, K.; Ishikawa, Y.; Nishiyama, S. Tetrahedron 2010, 66, 9779. doi: 10.1016/j.tet.2010.11.015
Amano, Y.; Inoue, K.; Nishiyama, S. Synlett. 2008, 134.
Morofuji, T.; Shimizu, A.; Yoshida, J. Chem.-Eur. J. 2015, 21, 3211. doi: 10.1002/chem.201406398
Wesenberg, L. J.; Herold, S.; Shimizu, A.; Yoshida, J.; Waldvogel, S. R. Chem.-Eur. J. 2017, 23, 12096. doi: 10.1002/chem.201701979
Xu, F.; Qian, X. Yang.; Li, Y. J.; Xu, H. C. Org. Lett. 2017, 19, 6332. doi: 10.1021/acs.orglett.7b03152
Tile, G.; Anton, K.; Dieter, S.; Moeller, K. D.; Waldvogel, S. R. Chem. Commun. 2017, 53, 2974. doi: 10.1039/C7CC00927E
Tile, G.; Anton, K.; Dieter, S.; Moeller, K. D.; Waldvogel, S. R. J. Am. Chem. Soc. 2017, 139, 12317 doi: 10.1021/jacs.7b07488
Zhang, S.; Li, L. J.; Wang, H. Q.; Li, Q.; Liu, W. M.; Xu. K, Zeng, C. C. Org. Lett. 2018, 20, 252. doi: 10.1021/acs.orglett.7b03617
Qian, X. Y.; Li, S. Q.; Song, J. S.; Xu, H. C. ACS Catal. 2017, 7, 2730. doi: 10.1021/acscatal.7b00426
Wang, P.; Tang, S.; Lei, A. W. Green Chem. 2017, 19, 2092. doi: 10.1039/C7GC00468K
Folgueiras-Amador, A. A.; Qian, X. Y.; Xu, H. C.; Wirth, T. Chem.-Eur. J. 2018, 24, 487. doi: 10.1002/chem.v24.2
Chiba, K.; Fukuda, M.; Kim, S.; Kitano, Y.; Tada, M. J. Org. Chem. 2000, 31, 7654.
Elseedi, H. R.; Yamamura, S.; Nishiyama, S. Tetrahedron 2002, 58, 7485. doi: 10.1016/S0040-4020(02)00829-3
Kim, S.; Noda, S.; Hayashi, K.; Chiba, K. Org. Lett. 2008, 10, 1827. doi: 10.1021/ol8004408
Kim, S.; Hirose, K.; Uematsu, J.; Mikami, Y.; Chiba, K. Chem.-Eur. J. 2012, 18, 6284. doi: 10.1002/chem.201103630
Liu, K.; Tang, S.; Huang, P. F.; Lei, A. W. Nat. Commun. 2017, 8, 775. doi: 10.1038/s41467-017-00873-1
Tian, C.; Massignan, L.; Meyer, Tjark. H.; Ackermann, L. Angew. Chem., Int. Ed. 2018, 57, 2383. doi: 10.1002/anie.201712647
Xu, F.; Li, Y. Jie.; Huang, C.; Xu, H. C. ACS Catal. 2018, 8, 3820.. doi: 10.1021/acscatal.8b00373
Nematollahi, D.; Habibi, D.; Rahmati, M.; Rafiee, M. J. Org. Chem. 2004, 69, 2637. doi: 10.1021/jo035304t
Hosseiny Davarani, S. S.; Nematollahi, D.; Shamsipur, M.; Najafi, N. M.; Masoumi, L.; Ramyar, S. J. Org. Chem. 2006, 71, 2139. doi: 10.1021/jo0523767
Salehzadeh, H.; Nematollah, D.; Hesari, H. Green Chem. 2014, 45, 2441.
Zeng, C. C.; Liu, F. J.; Ping, D. W.; Hu, L. M.; Cai, Y. L.; Zhong, R. G.. J. Org. Chem. 2010, 41, 6386.
Bai, Y. X.; Ping, D. W.; Little, R. D.; Tian, H. Y.; Hu, L. M.; Zeng, C. C. Tetrahedron 2011, 67, 9334. doi: 10.1016/j.tet.2011.09.126

图式 2 酚与芳烃的电化学脱氢交叉偶联
Scheme 2 Electrochemical dehydrogenative cross-coupling between phenols and arenes

图式 3 酚与富电子芳香化合物的交叉偶联反应机理
Scheme 3 Proposed mechanism of cross-coupling between phenols and electron-rich aromatic compounds

图式 5 钯催化酮肟与有机硼试剂或α-酮酸的偶联反应
Scheme 5 Pd-catalyzed coupling of ketoximes with organoboron reagents or α-keto acids

图式 6 电化学氧化与手性胺催化联合实现脱氢交叉偶联
Scheme 6 Dehydrogenation cross-coupling by the combination of electrochemical oxidation with chiral amine catalysis

图式 7 基于自由基阳离子池的芳基-芳基交叉偶联反应
Scheme 7 Aryl-aryl cross-coupling reaction based on radical-cation pools

图式 8 缺电子芳香化合物官能化的两种形式
Scheme 8 Two forms in the functionalization of electron-deficient aromatic compounds

图式 9 基于Meisenheimer中间体的缺电子芳香化合物的官能化
Scheme 9 Functionalization of electron-deficient aromatic compounds based on Meisenheimer intermediates

图式 10 基于Zincke中间体的芳香化合物的直接胺化
Scheme 10 Direct amination of aromatic compounds based on Zincke intermediates

图式 11 咪唑与芳香化合物间的C—N偶联反应
Scheme 11 C—N coupling reaction between imidazole and aromatic compounds

图式 13 可循环碘代芳香化合物促进的酰胺芳基化反应
Scheme 13 Arylation of amide with a recyclable iodoarene as mediator

图式 17 2, 4-二甲基苯酚阳极产物的修饰
Scheme 17 Modification of an anodic product derived from 2, 4-dimethylphenol

图式 18 由邻苯二酚衍生物电氧化合成芳基硫醚
Scheme 18 Synthesis of aryl sulfides by electrochemical oxidation of catechol

图式 19 电催化C—H/S—H脱氢交叉偶联
Scheme 19 Electrocatalytic dehydrogenative C—H/S—H cross-coupling

图式 20 电催化C—H/S—H交叉偶联反应机理
Scheme 20 Proposed mechanism of C—H/S—H cross-cou- pling by electrocatalytic dehydrogenation

图式 21 由邻苯二酚及氨基苯酚衍生物电氧化合成芳基砜
Scheme 21 Synthesis of aryl sulfones by electrochemical oxidation of catechol with aminophenols

图式 22 Cp2Fe介导的3-氟-2-吲哚酮衍生物的电化学合成
Scheme 22 Electrochemical synthesis of 3-fluoro-oxindoles mediated by Cp2Fe

图式 24 电化学C—H胺化合成苯并噁唑(噻唑)
Scheme 24 Synthesis of benzoxazolesor (benzothiazoles) by electrochemical C—H amination

图式 25 电化学C—H胺化合成1, 4-苯并噁嗪-3-酮
Scheme 25 Synthesis of 1, 4-benzoxazin-3-ones by electrochemical C—H amination

图式 26 电化学分子内脱氢构建C—S键
Scheme 26 Construction of C—S bond by electrochemical intramolecular dehydrogenation

图式 27 酚与烯烃之间的电化学[3+2]成环反应
Scheme 27 Electrochemical [3+2] annulation between phenols and olefins

图式 28 酚与N-乙酰基吲哚之间的电化学[3+2]成环反应
Scheme 28 Electrochemical [3+2] annulation between phenols and N- acetylindoles

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


























 下载:
下载:







