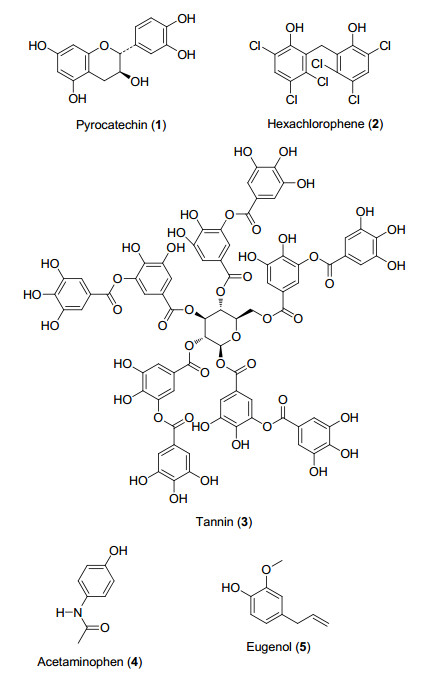
图 1.
含酚羟基的重要分子
Figure 1.
Important molecules containing phenol

苯酚及其衍生物广泛地存在于自然界中, 在医药、农药和材料科学中具有重要的意义[1].很多天然化合物具有酚羟基的结构, 比如人们喜爱饮用的饮料——茶中含有的茶多酚(GTP, 又名茶单宁), 是茶叶中黄酮类、儿茶素类、酚酸类和花色素类化合物的总称.其中主要成分儿茶素(Pyrocatechin)属于酚类化合物, 它不仅具有抗氧化性, 还具有清除人体内的自由基、抑制肿瘤的形成、减缓人体衰老等功效[2].此外, 酚及其衍生物还广泛应用于医药和材料科学.比如鞣酸(Tannins)可以做收敛剂, 不仅能沉淀蛋白质, 还可以与生物碱、甙及重金属等形成不溶性复合物, 也常用于印染行业中; 对乙酰氨基酚(Acetaminophen)具有解热镇痛的作用, 主要用于治疗感冒发烧、关节痛、神经痛等; 丁香油酚(Eugenol)不仅可以作为香皂的香料, 还可以用来制作抗菌、麻醉和降血压的药物.上述的化合物都具有酚羟基的结构(图 1).除此之外, 酚及其衍生物还可以用于过渡金属催化合成反应方法中的配体, 制备酚酯聚合物等, 其活泼的反应活性使它成为非常重要的有机合成中间体.
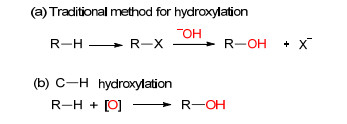
传统的酚类化合物的合成方法主要是基于卤素化合物水解(Scheme 1a)[3], 这种方法存在着程序较为繁琐, 反应产生卤素原子导致浪费等缺点.因此, 有机化学家一直在探索新的更为绿色高效的酚类化合物的合成方法.近几年来, 随着过渡金属催化C—H键构建新的C—C键方法的成熟发展, 通过C—H键活化构建C—O键的方法也越来越受到有机化学家的关注.其中C—H羟基化为苯酚类化合物的制备提供了更为便捷的途径(Scheme 1b).此类方法步骤更为简单, 并具有较高的原子经济性, 符合绿色化学的基本原则.本篇对C—H羟基化的方法进行了综述, 相信对酚类化合物高效、绿色的新合成方法探索研究具有一定的帮助.
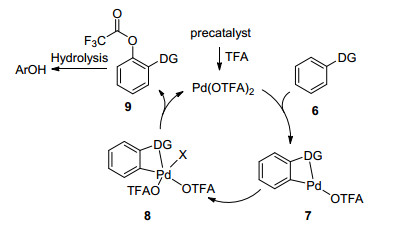
三氟乙酸根(OTFA)作为金属Pd的配体, 由于具有较强的吸电子能力使得芳香环更容易进攻Pd金属形成环Pd化合物, 类似Scheme 2所示的化合物7.随后Pd化合物7发生氧化加成还原消除可得到OTFA取代的芳香环化合物9.后处理水解即可得到羟基化的产物.因此, 使用三氟乙酸(TFA)或含三氟乙酸根的化合物作为羟基源的报道相对较多.在这一过程中, 往往需要一个导向基团(DG)首先配位过渡金属最终实现羟基化反应.
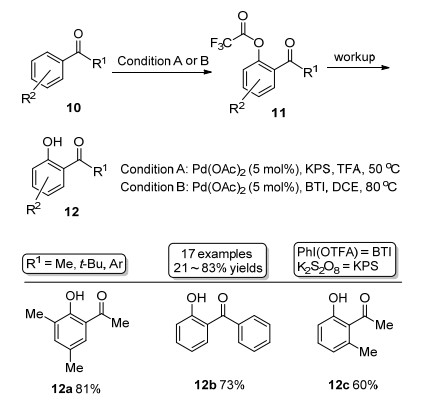
2012年, 董广彬课题组[4]报道羰基导向的Pd催化邻位羟基化, 使用的底物都是芳基酮衍生物.反应以过硫酸钾(KPS)作为氧化剂, TFA作为羟基源试剂, 同时也作为反应的溶剂(Scheme 3).作者还使用了双(三氟乙酰氧基)碘苯[PhI(OTFA)2, BTI]作为羟基源试剂在三氟乙酸钯催化剂作用下, 实现了芳基酮邻位的羟基化.该反应条件弥补了个别以TFA作为羟基源时不发生羟基化的问题, 提高了该方法的普适性.类似使用BTI为羟基源方法还可见Kwong等[5]的报道.
同年, 饶燏课题组[6]以Pd(OAc)2为催化剂, Selectfluor(四氟硼酸盐)作为氧化剂, TFA/TFAA (三氟乙酸酸酐)的混合液作为反应溶剂, 在温和条件下可实现不同芳基酮的邻位羟基化(Eq. 1).经研究, TFA和TFAA的最佳比例为9:1, 过多的TFAA会降低反应速率.值得注意的是, 作者还将方法拓展到苯甲酸酯、苯甲酰胺、乙酰苯胺和芳基磺酰胺等衍生物的邻位羟基化.作者还将布洛芬的羧基转化为酯基, 然后导向实现了布洛芬分子的邻位羟基化修饰(Eq. 2).随后, 饶燏课题组[7]以更为廉价的KPS替换Selectfluor作为氧化剂, 同样实现了芳基酮、苯甲酸酯、苯甲酰胺、乙酰苯胺和芳基磺酰胺等衍生物的邻位羟基化反应.
|
|
(1) |
|
|
(2) |
Kornienko等[8]在饶燏课题组的基础上, 以哒嗪衍生物作为导向基, 在Pd催化下以TFA/TFAA混合液为反应溶剂实现邻位的羟基化反应(Eq. 3). Batra等[9]则以吡唑作为导向基完成了邻位的羟基化, 以Pd(OAc)2为催化剂, KPS为氧化剂, TFA/TFAA为溶剂得到酚类化合物(Eq. 4).有趣的是当TFA/TFAA的用量较多时, 即反应底物溶度较低时才能得到邻位羟基化产物.而如果TFA/TFAA的用量较少时, 即反应物溶度较大的情况下, 反应主要生成吡唑邻位的自偶联的产物.冀亚飞课题组[10]报道了类似的条件下3-芳基苯并异噁唑的C—H羟基化(Eq. 5). Youn等[11]报道了N-苄基苯磺胺衍生物的邻位C—H羟基化(Eq. 6).上述反应的机理均类似Scheme 2所示, TFA的羟基源, KPS为氧化剂.
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
杨尚东[12]和Joseph等[13]分别报道了氧化磷导向基和偶氮苯导向基导向的邻位羟基化(Eqs. 7, 8).在这两个反应中双(三氟乙酰氧基)碘苯既作为羟基源又作为氧化剂.
|
|
(7) |
|
|
(8) |
最近, Sorensen等[14]利用瞬态导向基的策略实现了苯甲醛的邻位C—H羟基化制备水杨醛衍生物(Eq. 9).反应中的羟基源为p-TsOH(对甲苯磺酸).瞬态导向基为2-氨基-4-氯苯甲酸, 氨基可以与醛基原位反应生成亚胺中间体, 金属Pd与亚胺氮原子以及羧基氧原子配位从而导向活化邻位C—H键生成环钯中间体34, 类似Scheme 2所示:对甲苯磺酸根配位Pd金属, 氧化加成和还原消除后得到对甲苯磺酰氧化的产物, 经水解后处理得到水杨醛衍生物.
|
|
(9) |
饶燏课题组[15]不仅开发了Pd催化的羟基化, 同时还发现了Ru金属同样可以高效地催化苯甲酸酯的邻位羟基化(Eq. 10).该方法同样使用TFA/TFAA的混合液作为反应溶剂, 同时也作为羟基源, KPS作为氧化剂.尽管该反应的机理并不明确, 但是作者认为可能的反应机理类似Pd催化的羟基化, 即二价Ru配位导向基并活化邻位C—H键, 随后经历氧化加成和还原消除得到三氟乙酸根取代的芳环, 后处理得到苯酚衍生物(Scheme 2).整个催化循环过程中经历的是Ru(Ⅱ)/Ru(Ⅳ)循环而非Ru(0)/Ru(Ⅱ)循环.
|
|
(10) |
邻苯二酚骨架普遍存在于天然化合物和药物分子中, 通过苯酚邻位羟基化可以快速有效地制备邻苯二酚.但是由于酚羟基与过渡金属配位活化邻位C—H键形成的环金属过渡态为不稳定的四元环, 因此往往需要对酚羟基进行修饰, 再进一步导向邻位羟基化.饶燏课题组[16]通过筛选弱配位的导向基保护酚羟基, 并发现当使用二甲基氨基甲酸酯作为导向基时, 在Ru催化下, TFA/TFAA的混合液作为反应溶剂, KPS作为氧化剂可高效地实现邻位羟基化(Eq. 11).作者还通过半一锅法的方法, 以化合物39为原料, 首先在Ru催化下邻位羟基化, 混合物用水合肼处理氨基甲酸酯可水解从而生成邻二酚化合物40 (Eq. 12).
|
|
(11) |
|
|
(12) |
随后, 饶燏等[17]在尝试芳基酰胺类化合物羟基化的时候发现当使用Ru催化剂时, 羟基化发生在与羰基相连的芳环上; 而使用Pd作为催化剂时, 羟基化发生在与氮原子相连的芳环上(Eqs. 13, 14).作者通过动力学同位素效应(KIE)以及密度泛函理论计算(DFT)均验证, 当催化剂为Ru时, C—H键的活化更易发生在与羰基相连的芳环上, 形成的环钌过渡态46更为稳定; 而使用Pd催化剂时, C—H键则更易发生在与氮原子相连的芳环上, 形成的环钯过渡态47则更为稳定.
|
|
(13) |
|
|
(14) |
2015年, Hong课题组[18]报道了利用羟基化的方法合成极光激酶抑制剂的骨架结构(Eq. 15).反应使用BTI作为氧化剂, 在Ru催化下实现黄酮或色酮衍生物邻位羟基化.作者通过生物活性测试对比发现:羟基化产物的半数致死量均远低于未羟基化原料的半数致死量.
|
|
(15) |
TFA的酸性很强, 且价格相对昂贵. 2013年, Patel等[19]使用了价格相对便宜且酸性相对较弱的AcOH(乙酸)作为羟基化试剂, 在Pd(OAc)2催化下, 以双醋酸根碘苯(DIB)为氧化剂实现了2-芳基苯并噻唑邻位的C—H羟基化(Eq. 16).该方法相比使用TFA的方法需要更高的反应温度.当反应条件改为TFA作为羟基源, 使用过硫酸氢钾复合盐(Oxone)作为氧化剂依然有效.作者认为反应可能的机理类似Scheme 2所示的历程(TFA改为AcOH).
|
|
(16) |
2014年, 史炳锋等[20]报道了Cu(OAc)2作用下的邻位C—H羟基化反应(Eq. 17).该反应的羟基源来自Cu(OAc)2中的醋酸根, 我们将该类归纳在AcOH为羟基源的章节里.该底物中的导向基易脱除转变为羧基得到水杨酸衍生物.作者认为可能的催化循环如Scheme 4所示.首先二价Cu与底物52配位形成化合物54.化合物54可以发生歧化反应生成三价Cu化合物56和一价Cu催化剂.另一方面, 化合物54也可以Cu催化邻位C—H键活化, 然后再发生歧化反应生成三价Cu化合物56和一价Cu催化剂.三价Cu化合物还原消除可得到乙酰氧基化产物57和一价Cu催化剂.化合物57后处理水解即得到羟基化产物53, 一价Cu催化剂可氧化重新得到二价铜催化剂.
|
|
(17) |
2016年, 孙逊课题组[21]使用8-氨基喹啉作为双齿导向基, 使用Cu(OAc)2作为催化剂实现邻位羟基化(Eq. 18).反应可能的机理类似图 4所示, 羟基的氧原子来自醋酸铜. Jana等[22]发现当反应中加入吡啶作为配体, 可以提高该反应的产率.余金权、戴辉雄和林海霞等[23]使用邻噁唑苯胺作为双齿导向基实现邻位羟基化(Eq. 19).作者认为可能的机理是Cu(OAc)2催化乙酰氧基化, 因此羟基的氧原子来自醋酸铜.最近, 周兵等[24]报道了三价Rh催化的邻位C—H羟基化反应(Eq. 20).导向基团可以为吡啶、偶氮苯, 亚胺、嘧啶和二氮唑.作者提出可能的机理是Rh催化乙酰氧基化, 乙酰基来源于双醋酸基碘苯, 反应经历Rh(Ⅲ)/Rh(Ⅴ)循环.
|
|
(18) |
|
|
(19) |
|
|
(20) |
Oxone(过硫酸氢钾复合盐)往往在TFA或者AcOH参与的C—H羟基化反应中用作氧化剂. Kim等[25]报道了以Oxone为羟基源的C—H羟基化反应(Eq. 21).作者尝试的底物都为多取代的吡啶作为导向基.可能的反应机理如Scheme 5所示:首先金属Pd插入Oxone的O—O键之间生成68, 芳环氧化加成得到化合物69, 然后还原消除可得到苯酚产物(ArOH).
|
|
(21) |
2014年, Kamal和Nagesh等[26]报道了Pd催化2-芳基苯并咪唑邻位C—H羟基化(Eq. 22), 尽管没有给出可能的反应机理, 作者认为羟基中的氧原子来自Oxone.作者还发现羟基化的产物可以插入和稳定DNA(脱氧核糖核酸).焦宁课题组[27]发现了三苯基膦(PPh3)配体可以促进Pd催化肟醚导向的邻位C—H羟基化(Eq. 23).反应可能的机理类似Scheme 5所示, 但作者认为反应首先发生C—H键的活化, 然后Pd插入Oxone的O—O键中, 最后还原消除得到苯酚产物.
|
|
(22) |
|
|
(23) |
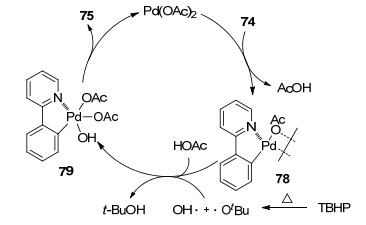
叔丁基过氧化氢(TBHP)和双氧水(H2O2)广泛应用在氧化反应中, 然而在C—H羟基化领域很少有报道[28]. 2015年, 孙培培课题组[29]报道了Pd催化的2-芳基吡啶邻位的C—H羟基化反应(Eq. 24).范学森和张新迎等[30]以酚羟基为导向基, 在TBHP存在下实现C—H羟基化(Eq. 25).该反应可快速制备重要的配体以及配体合成中间体联苯双酚化合物.上述反应中羟基的氧原子均来自TBHP.反应的机理(以芳基吡啶化合物为例)如Scheme 6所示. Pd催化剂在氮原子导向下活化邻位C—H键形成Pd二聚体78. THBP均裂生成羟基自由基HO•和叔丁氧自由基t-BuO•, t-BuO•与HOAc反应生成醋酸根自由基AcO•和叔丁醇t-BuO.随后AcO•和HO•氧化加成Pd形成四价钯化合物79.最后, 79还原消除可得到羟基化产物75, 并重新生成Pd催化剂.
|
|
(24) |
|
|
(25) |
2015年, Itoh等[31]使用更为经济的H2O2为羟基源试剂, 实现了Pd催化2-芳基吡啶邻位的C—H羟基化反应(Eq. 26).该反应的机理类似TBHP参与的羟基化反应, 但是形成的羟基配位的四价Pd化合物中的羟基来自H2O2.
|
|
(26) |
2013年, 郭灿城课题组[32]使用四(五氟苯)卟啉氯化锰(F20TPPMnCl)作为催化剂, 间氯过氧苯甲酸(m-CP- BA)作为氧化剂实现了β-取代萘的α位C—H羟基化(Eq. 27).反应中Mn催化剂首先与m-CPBA作用生成过氧酰基锰化合物82, 该化合物中的氧原子可以亲电进攻萘环的α位, 最后离去Mn金属得到萘酚化合物.
|
|
(27) |
氧气(O2)廉价易得, 为可再生资源, 因此, 使用氧气作为羟基源备受有机化学家的关注. 2009年, 余金权等[33]报道了Pd催化苯甲酸邻位的羟基化反应制备水杨酸衍生物(Eq. 28).反应使用对苯醌(BQ)作为氧化剂.尽管作者没有提出详细的反应机理, 但是通过同位素标记实验证实了羟基中的氧原子来自于氧气分子.
|
|
(28) |
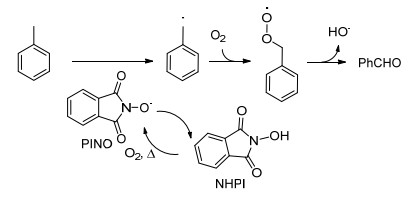
2013年, 焦宁课题组[34]报道了在吡啶导向下, Pd和N-羟基邻苯二甲酰亚胺(NHPI)共同催化的C—H羟基化反应(Eq. 29).作者认为NHPI加热均裂形成邻苯二甲酰亚胺-N-氧自由基(PINO)并触发苄基自由基的形成, 随后苄基自由基在氧气存在下形成过氧化自由基, 进一步分解形成羟基自由基和苯甲醛.类似Scheme 7所示羟基自由基可氧化加成二价Pd化合物, 还原消除得到羟基化产物.
|
|
(29) |
2017年, Faessler等[35]报道了Cu催化的串联羟基化和N—N键生成反应制备吲唑衍生物(Eq. 30).该反应的机理尚不明确, 但是作者通过一系列的对照实验, 证实了羟基中的氧原子来自氧气分子.
|
|
(30) |
2017年, Kim等[36]报道了Rh催化2-芳基吡啶邻位C—H羟基化反应(Eq. 31).反应使用双(特戊酰基)碘苯(PhI(OPiv)2)作为氧化剂.作者认为该反应的羟基氧原子来自水中.首先Rh催化剂在氟化银(AgF)活化下与底物74作用生成环铑化合物87.在氧化剂PhI(OPiv)2存在下, 水分子可以插入Rh—C键形成化合物89.作者认为过程中可能经历中间体88, 最后化合物89水解即可得到羟基化产物75和重新生成Rh催化剂(Scheme 8).
|
|
(31) |
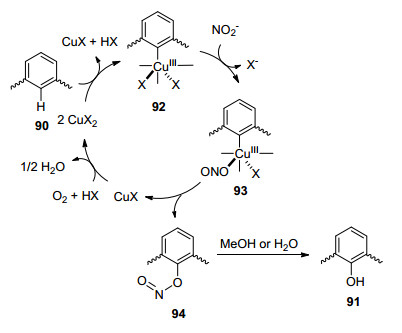
2013年, 王梅祥课题组[37]报道了Cu催化、亚硝酸钾(KNO2)参与的选择性羟基化反应(Eq. 32).值得注意的是, 该反应只有当使用甲醇作为溶剂时, 反应才能选择性的进行羟基化; 而当使用乙腈和二甲基亚砜(DMSO)的混合液作为溶剂时反应选择性的进行硝基化.反应首先是Cu催化活化底物90的C—H键生成三价Cu化合物92, 这一步可能也涉及歧化反应.亚硝基负离子与92相互作用生成化合物93, 93还原消除可得到一价Cu催化剂和化合物94.一价Cu氧化和重新得到二价Cu催化剂完成催化循环.化合物94则与水或者甲醇反应即得到羟基化产物91 (Scheme 9).
|
|
(32) |
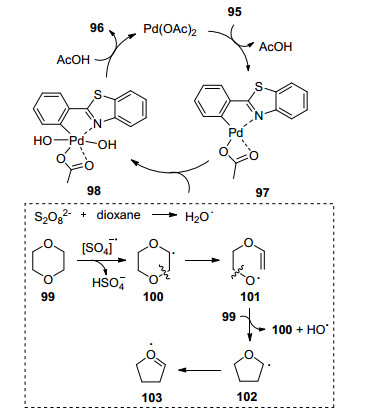
2015年, Chakraborti等[38]报道了1, 4-dioxane(1, 4-二氧六环)作为羟基的C—H羟基化反应(Eq. 33), 反应使用Pd作为催化剂, Na2S2O8为氧化剂.该反应适用于非常多的导向基团, 比如苯并噁唑、偶氮苯、酰胺、氨基甲酸酯和不对称尿素.在这个反应中, 首先催化剂Pd在导向基导向下活化邻位C—H键生成环钯化合物97.过硫酸阴离子降级1, 4-二氧六环反应可生成羟基自由基.羟基自由基氧化加成化合物97得到98.最后98还原消除生成羟基化产物96, 并得到Pd催化剂, 配体交换重新得到初始的Pd催化剂(Scheme 10).
|
|
(33) |
综上所述, 近几年通过C—H键活化实现直接的羟基化反应取得了快速的发展, 为苯酚的制备提供了一条高效的途径.目前, 酸或者酸根参与的过渡金属催化C—H键羟基化反应的研究相对较为成熟, 大量优秀的工作实现了以TFA或者PhI(TFA)2为羟基源的C—H羟基化反应, 尤其是Pd和Ru催化下的反应, 但是强酸的使用限制了该方法的应用.与此同时, 也有不少文献报道了过氧化物(Oxone、TBHP、H2O2和m-CPBA)参与的C—H羟基化反应, 尽管此类方法避免了强酸的使用, 且具有较高的原子经济性, 但是反应的底物普适性较低.
另一方面, 氧气和水廉价易得, 为可再生资源, 作为羟基源具有非常大的吸引力, 但是相关研究很少.因此, 使用绿色羟基, 设计新型的催化剂体系、发展反应条件温和、反应普适性高的C-H羟基化反应仍是该领域研究的长期目标.丰富反应底物类型, 使之能应用于复杂的天然产物和药物分子的合成中, 这无疑具有极其重要的学术和应用价值.
(a) Tyman, J. H. P. Synthetic and Natural Phenols, Elsevier, Netherlands, 1996.
(b) Luo, J. ; Preciado, S. ; Larrosa, I. J. Am. Chem. Soc. 2014, 136, 4109.
(c) Cui, N. ; Zhao, Y. ; Wang, Y. Chin. J. Org. Chem. 2017, 37, 20(in Chinese).
(崔娜, 赵宇, 王云侠, 有机化学, 2017, 37, 20.) http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract345716.shtml
马兰萍, 蔡育军, 杨立, 刘中立, 有机化学, 2001, 21, 518. doi: 10.3321/j.issn:0253-2786.2001.07.006Ma, L.; Cai, Y.; Yang.L.; Liu, Z.Chin.J.Org.Chem.2001, 21, 518(in Chinese). doi: 10.3321/j.issn:0253-2786.2001.07.006
刘向前, 张慧, 蒋咏文, 有机化学, 2012, 32, 1434. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract341430.shtmlLiu, X; Zhang, H.; Jiang, Y.Chin.J.Org.Chem.2012, 32, 1434(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract341430.shtml
Mo, F.; Trzepkowski, L.J.; Dong, G.Angew.Chem.Int.Ed.2012, 51, 13075. doi: 10.1002/anie.201207479
Choy, P.Y.; Kwong, F.Y.Org.Lett.2013, 15, 270. doi: 10.1021/ol303088z
Shan, G.; Yang, X.; Ma, L.; Rao, Y.Angew.Chem.Int.Ed.2012, 51, 13070. doi: 10.1002/anie.201207458
Rao, Y.Synlett.2013, 24, 2472. doi: 10.1055/s-00000083
Rastogi, S.K.; Medellin, D.C.; Kornienko, A.Org.Biomol.Chem.2014, 12, 410. doi: 10.1039/C3OB42255K
Batchu, H.; Bhattacharyya, S.; Kant, R.; Batra, S J.Org.Chem.2015, 80, 7360. doi: 10.1021/acs.joc.5b00733
Guo, Y.; Yu, K.-K.; Xing, L.-H.; Liu, H.-W.; Wang, W.; Ji, Y.-F.Adv.Synth.Catal.2017, 359, 410. doi: 10.1002/adsc.201600853
Jeong, E.J.; Jo, Y.H; Jang, M.J.; Youn, S.W.Bull.Korean Chem.Soc.2015, 36, 453.
Zhang, H.-Y.; Yi, H.-M.; Wang, G.-W.; Yang, B.; Yang, S.-D.Org.Lett.2013, 15, 6186. doi: 10.1021/ol403028a
Nguyen, T.H.L.; Gigant, N.; Delarue-Cochin, S.; Joseph, D.J.Org.Chem.2016, 81, 1850. doi: 10.1021/acs.joc.5b02614
Chen, X.-Y.; Ozturk, S.; Sorensen, E.J.Org.Lett.2017, 19, 6280. doi: 10.1021/acs.orglett.7b02906
Yang, Y.; Lin, Y.; Rao, Y.Org.Lett.2012, 14, 2874. doi: 10.1021/ol301104n
Yang, X.; Sun, Y.; Chen, Z.; Rao, Y.Adv.Synth.Catal.2014, 356, 1625. doi: 10.1002/adsc.v356.7
Sun, Y.-H.; Sun, T.-Y.; Wu, Y.-D.; Zhang, X.; Rao, Y.Chem.Sci.2016, 7, 2229. doi: 10.1039/C5SC03905C
Kim, K.; Choe, H.; Jeong, Y.; Lee, J.H.; Hong, S.Org.Lett.2015, 17, 2550. doi: 10.1021/acs.orglett.5b01138
Banerjee, A.; Bera, A.; Guin, S.;.Rout, S.K.; Patel, B.K Tetrahedron.2013, 69, 2175. doi: 10.1016/j.tet.2012.12.067
Li, X.; Liu, Y.-H.; Gu, W.-J.; Li, B.; Chen, F.-J.; Shi, B.-F.Org.Lett.2014, 16, 3904. doi: 10.1021/ol5016064
Wang, M.; Hu, Y.; Jiang, Z.; Shenb, H.C.; Sun, X.Org.Biomol.Chem.2016, 14, 4239. doi: 10.1039/C6OB00392C
Singh, B.K.; Jana, R.J.Org.Chem.2016, 81, 831. doi: 10.1021/acs.joc.5b02302
Sun, S.-Z.; Shang, M; Wang, H.-L.; Lin, H.-X.; Dai, H.-X.; Yu, J.-Q.J.Org.Chem.2015, 80, 8843. doi: 10.1021/acs.joc.5b01351
Wu, Y.; Zhou, B.Org.Lett.2017, 19, 3532. doi: 10.1021/acs.orglett.7b01494
Kim, S.H.; Lee, H.S.; Kim, S.H.; Kim, J.N.Tetrahedron.Lett.2008, 49, 5863. doi: 10.1016/j.tetlet.2008.07.141
Kamal, A.; Srinivasulu, V.; Sathish, M.; Tangella, Y.; Nayak, V.L.; Rao, M.P.N.; Shankaraiah, N.; Nagesh, N.Asian J.Org.Chem.2014, 3, 68. doi: 10.1002/ajoc.201300214
Liang, Y.-F.; Wang, X.; Yuan, Y.; Liang, Y.; Li, X.; Jiao, N.ACS Catal.2015.
朱文明, 应炜炜, 魏文廷, 有机化学, 2017, 37, 2841. http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346179.shtmlZhu, W.M.; Ying, W.W.; Wei, W.T.Chin.J.Org.Chem.2017, 37, 2841(in Chinese). http://manu19.magtech.com.cn/Jwk_yjhx/CN/abstract/abstract346179.shtml
Dong, J.; Liu, P.; Sun, P.J.Org.Chem.2015, 80, 2925. doi: 10.1021/acs.joc.5b00167
Duan, S.; Xu, Y.; Zhang, X.; Fan, X.Chem.Commun.2016, 52, 10529 doi: 10.1039/C6CC04756D
Yamaguchi, T.; Yamaguchi, E.; Tada, N.; Itoh, A.Adv.Synth.Catal.2015, 357, 2017. doi: 10.1002/adsc.v357.9
Chena, C.-D.; Shenga, W.-B.; Shia, G.-J.; Guo, C.-C.J.Phys.Org.Chem.2013, 26, 23. doi: 10.1002/poc.v26.1
Zhang, Y.-H.; Yu, J.-Q.J.Am.Chem.Soc.2009, 131, 14654. doi: 10.1021/ja907198n
Yan, Y.P.; Feng, P.; Zheng, Q.Z.; Liang, Y.F.; Lu, J.F.; Cui, Y.X.; Jiao.N.Angew.Chem., Int.Ed.2013, 52, 5827. doi: 10.1002/anie.201300957
Chen, C.-Y.; He, F.; Tang, G.; Ding, H.; Wang, Z.; Li, D.; Deng, L.; Faessler, R.Eur.J.Org.Chem.2017, 6604.
Kim, K.; Hyun, J.; Kim, J.; Kim, H Asian J.Org.Chem.2017, 6, 907. doi: 10.1002/ajoc.v6.7
Zhang, H; Zhao, L.; Wang, D.-X.; Wang, M.-X.Org.Lett.2013, 15, 3836. doi: 10.1021/ol401453f
Seth, K.; Nautiyal, M.; Purohit, P.; Parikh, N.; Chakraborti, A.K.Chem.Commun.2015, 51, 191. doi: 10.1039/C4CC06864E

Scheme 3 羰基导向的Pd催化邻位羟基化反应
Scheme 3 Carboxyl groups direct Pd-catalyzed ortho hydroxylation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





































 下载:
下载:








