Received Date:
15 March 2018 Revised Date:
23 April 2018 Available Online:
01 August 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 51502162, 21501002, 21576159), the Open Foundation of State Key Laboratory of Coordination Chemistry of Nanjing University (Nos. SKLCC1613, SKLCC1604), the Open Fund of Key Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, the Chinese Academy of Sciences (No. PCOM201704), the National Students' Innovation Training Program (No. 201710433179) and the Young Teacher Supporting Fund of Shandong University of Technology
Abstract:
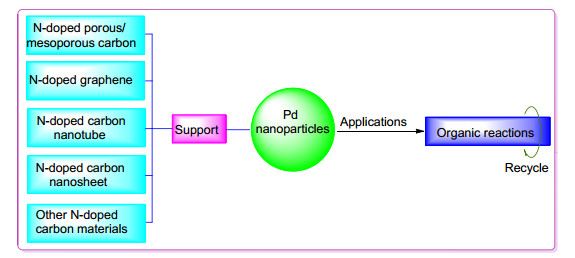
The N-doped carbon materials supported Pd nanocatalysts have attracted extensive attention in the catalysis field due to its unique advantages such as high efficiency, easy separation, purification and recyclability. The recent progress in the synthesis and application of supported Pd nanocatalysts based on different types of carbon materials supports including N-doped porous/mesoporous carbon, N-doped graphene, N-doped carbon nanotube and N-doped carbon nanosheet is reviewed. And the research trends of N-doped carbon materials supported Pd nanocatalysts are also prospected.
Ghosh, R.; Adarsh, N. N.; Sarkar, A. J. Org. Chem. 2010, 75, 5320. doi: 10.1021/jo100643j
[2]
Chung, K. H.; So, C. M.; Wong, S. M.; Luk, C. H.; Zhou, Z. Y.; Lau, C. P.; Kwong, F. Y. Chem. Commun. 2012, 48, 1967. doi: 10.1039/c2cc15972d
[3]
Micksh, M.; Tenne, M.; Strassner, T. Organometallics 2014, 33, 3966. doi: 10.1021/om5004336
[4]
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612. doi: 10.1021/cr900074m
[5]
Valente, C.; Çalimsiz, S.; Hoi, K. H.; Mallik, D.; Sayah, M.; Organ, M. G. Angew. Chem., Int. Ed. 2012, 51, 3314. doi: 10.1002/anie.v51.14
[6]
Li, X. W.; Chen, F.; Xu, W. F.; Li, Y. Z.; Chen, X. T.; Xue, Z. L. Inorg. Chem. Commun. 2011, 14, 1673. doi: 10.1016/j.inoche.2011.07.004
[7]
李晓微, 周晋, 禚淑萍, 有机化学, 2014, 34, 2063.Li, X. W.; Zhou, J.; Zhuo, S. P. Chin. J. Org. Chem. 2014, 34, 2063(in Chinese).
[8]
Nelson, D. J. Eur. J. Inorg. Chem. 2015, 2012.
[9]
(a) Fihri, A. ; Bouhrara, M. ; Nekoueishahraki, B. ; Basset, J. M. ; Polshettiwar, V. Chem. Soc. Rev. 2011, 40, 5181. (b) Feng, C. L. ; Hei, L. Y. ; Li, Z. ; Liu, L. T. Chin. J. Org. Chem. 2016, 36, 179(in Chinese). (冯翠兰, 黑莉楹, 李珍, 刘澜涛, 有机化学, 2016, 36, 179. ) (c) Kong, S. N. ; Malik, A. U. ; Qian, X. F. ; Shu, M. H. ; Xiao, W. D. Chin. J. Org. Chem. 2018, 38, 432(in Chinese). (孔胜男, Abaid Ullah Malik, 钱雪峰, 舒谋海, 肖文德, 有机化学, 2018, 38, 432. )
[10]
Guerra, J.; Herrero, M. A. Nanoscale 2010, 2, 1390. doi: 10.1039/c0nr00085j
[11]
Georgakilas, V.; Perman, J. A.; Tucek, J.; Zboril, R. Chem. Rev. 2015, 115, 4744. doi: 10.1021/cr500304f
Scheuermann, G. M.; Rumi, L.; Steurer, P.; Bannwarth, W.; Mülhaupt, R. J. Am. Chem. Soc. 2009, 131, 8262. doi: 10.1021/ja901105a
[14]
Georgakilas, V.; Tiwari, J. N.; Kemp, K. C.; Perman, J. A.; Bourlinos, A. B.; Kim, K. S.; Zboril, R. Chem. Rev. 2016, 116, 5464. doi: 10.1021/acs.chemrev.5b00620
Pikna, L.; Milkovič, O.; Saksl, K.; Heželová, M.; Smrčová, M.; Puliš, P.; Michalik, Š.; Gamcová, J. J. Solid State Chem. 2014, 212, 197. doi: 10.1016/j.jssc.2014.01.032
Li, Z. L.; Liu, J. H.; Huang, Z. W.; Yang, Y.; Xia, C. G.; Li, F. W. ACS Catal. 2013, 3, 839. doi: 10.1021/cs400077r
[24]
Li, Z. L.; Liu, J. H.; Xia, C. G.; Li, F. W. ACS Catal. 2013, 3, 2440. doi: 10.1021/cs400506q
[25]
Ding, S. S.; Zhang, C. H.; Liu, Y. F.; Jiang, H.; Chen, R. Z. Appl. Surf. Sci. 2017, 425, 484. doi: 10.1016/j.apsusc.2017.07.068
[26]
Ding, S. S.; Zhang, C. H.; Liu, Y. F.; Jiang, H.; Chen, R. Z. J. Ind. Eng. Chem. 2017, 46, 258. doi: 10.1016/j.jiec.2016.10.037
[27]
Hu, S.; Zhang, X.; Qu, Z. Y.; Jiang, H.; Liu, Y. F.; Huang, J.; Xing, W. H.; Chen, R. Z. J. Ind. Eng. Chem. 2017, 53, 333. doi: 10.1016/j.jiec.2017.05.004
[28]
Xu, X.; Tang, M. H.; Li, M. M.; Li, H. R.; Wang, Y. ACS Catal. 2014, 4, 3132. doi: 10.1021/cs500859n
[29]
Tang, M. H.; Mao, S. J.; Li, M. M.; Wei, Z. Z.; Xu, F.; Li, H. R.; Wang, Y. ACS Catal. 2015, 5, 3100. doi: 10.1021/acscatal.5b00037
[30]
Jiang, H. Z.; Yu, X. L.; Nie, R. F.; Lu, X. H.; Zhou, D.; Xia, Q. H. Appl. Catal., A:Gen. 2016, 520, 73. doi: 10.1016/j.apcata.2016.04.009
[31]
Dong, Z. P.; Dong, C. X.; Liu, Y. S.; Le, X. D.; Jin, Z. C.; Ma, J. T. Chem. Eng. J. 2015, 270, 215. doi: 10.1016/j.cej.2015.02.045
[32]
Zhang, L.; Feng, C.; Gao, S. T.; Wang, Z.; Wang, C. Catal. Commun. 2015, 61, 21. doi: 10.1016/j.catcom.2014.12.004
[33]
Zhang, L.; Dong, W. H.; Shang, N. Z.; Feng, C.; Gao, S. T.; Wang, C. Chin. Chem. Lett. 2016, 27, 149. doi: 10.1016/j.cclet.2015.08.007
[34]
Zeng, M. F.; Wang, Y. D.; Liu, Q.; Yuan, X.; Feng, R. K.; Yang, Z.; Qi, C. Z. Int. J. Biol. Macromol. 2016, 89, 449. doi: 10.1016/j.ijbiomac.2016.05.011
[35]
Wu, X. X.; Zhou, H. New J. Chem. 2017, 41, 10245. doi: 10.1039/C7NJ01947E
[36]
Long, Y.; Liu, Y. S.; Zhao, Z. M.; Luo, S.; Wu, W.; Wu, L.; Wen, H.; Wang, R. Q.; Ma, J. T. J. Colloid Interface Sci. 2017, 496, 465. doi: 10.1016/j.jcis.2017.02.051
[37]
Wei, Z. Z.; Gong, Y. T.; Xiong, T. Y.; Zhang, P. F.; Li, H. R.; Wang, Y. Catal. Sci. Technol. 2015, 5, 397. doi: 10.1039/C4CY00946K
[38]
Deng, D. S.; Han, G. Q.; Zhu, X.; Xu, X.; Gong, Y. T.; Wang, Y. Chin. Chem. Lett. 2015, 26, 277. doi: 10.1016/j.cclet.2014.12.001
Jin, Y. X.; Zhao, J.; Li, F.; Jia, W. P.; Liang, D. X.; Chen, H.; Li, R. R.; Hu, J. J.; Ni, J. M.; Wu, T. Q.; Zhong, D. P. Electrochim. Acta 2016, 220, 83. doi: 10.1016/j.electacta.2016.10.087
[48]
Chao, L.; Qin, Y.; He, J. J.; Ding, D.; Chu, F. Q. Int. J. Hydrogen Energy 2017, 42, 15107. doi: 10.1016/j.ijhydene.2017.04.245
[49]
Xu, H.; Yan, B.; Zhang, K.; Wang, J.; Li, S. M.; Wang, C. Q.; Shiraishi, Y.; Du, Y. K.; Yang, P. Electrochim. Acta 2017, 245, 227. doi: 10.1016/j.electacta.2017.05.146
[50]
Xu, H.; Yan, B.; Zhang, K.; Wang, J.; Li, S. M.; Wang, C. Q.; Du, Y. K.; Yang, P.; Jiang, S. J.; Song, S. Q. Appl. Surf. Sci. 2017, 416, 191. doi: 10.1016/j.apsusc.2017.04.160
[51]
Li, Z. P.; Ruan, M. N.; Du, L. Q.; Wen, G. M.; Dong, C.; Li, H. W. J. Electroanal. Chem. 2017, 805, 47. doi: 10.1016/j.jelechem.2017.10.015
Vanyorek, L.; Halasi, G.; Pekker, P.; Kristály, F.; Kónya, Z. Catal. Lett. 2016, 146, 2268. doi: 10.1007/s10562-016-1857-8
[59]
Wang, L. L.; Zhu, L. P.; Bing, N. C.; Wang, L. J. J. Phys. Chem. Solids 2017, 107, 125. doi: 10.1016/j.jpcs.2017.03.025
[60]
Dong, B. Q.; Li, Y. H.; Ning, X. M.; Wang, H. J.; Yu, H.; Peng, F. Appl. Catal., A:Gen. 2017, 545, 54. doi: 10.1016/j.apcata.2017.07.035
[61]
Duan, X. M.; Xiao, M. C.; Liang, S.; Zhang, Z. Y.; Zeng, Y.; Xi, J. B.; Wang, S. Carbon 2017, 119, 326. doi: 10.1016/j.carbon.2017.04.039
[62]
Zhang, H.; Yan, X. H.; Huang, Y. D.; Zhang, M. R.; Tang, Y. W.; Sun, D. M.; Xu, L.; Wei, S. H. Appl. Surf. Sci. 2017, 396, 812. doi: 10.1016/j.apsusc.2016.11.035
[63]
Zuo, P. P.; Duan, J. Q.; Fan, H. L.; Qu, S. J.; Shen, W. Z. Appl. Surf. Sci. 2018, 435, 1020. doi: 10.1016/j.apsusc.2017.11.179

 下载:
下载:






















































 下载:
下载:












