Citation:
Sun Hongshun, Li Yulong, Jiang Hong, Guo Cheng, Shen Linjiang. Two Binuclear Nonionic Magnetic Resonance Contrast Agents with High Relaxivity[J]. Chinese Journal of Organic Chemistry,
2018, 38(7): 1779-1785.
doi:
10.6023/cjoc201801044
Received Date:
29 January 2018 Revised Date:
04 March 2018 Available Online:
01 July 2018
Fund Project:
Project supported by the Natural Science Foundation in Jiangsu Province (No. 17KJB320001), the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (No. PPZY2015B179), the Training Program of Students Innovation and Entrepreneurship in Jiangsu Province (No. 201712920001Y), and the Qing Lan Project of Jiangsu Province
Abstract:
Magnetic resonance imaging (MRI) is widely used in diagnostic medicine and soft tissue imaging. Contrast agents (CAs) can improve the specificity of MRI enhancement. Herein, the design, synthesis and relaxivity of two novel binuclear nonionic Gd-based DOTA-hydrazide derived MRI contrast agents, (Gd-DOTAH)2-DYMB and (Gd-DOTAH)2-DYMBP are reported. They have improved longitudinal relaxivity values of 11.4 and 11.7 L·mmol-1·s-1 per molecule or 5.7 and 5.9 L·mmol-1·Gd-1·s-1 at 0.5 T, respectively, which are higher than that of the mononuclear clinical macrocyclic agent Gd-DOTA. And in vitro MRI studies suggest the potential of these two agents as MRI contrast agents in improving the diagnostic sensitivity and accuracy.
Magnetic resonance imaging (MRI) has become a routinely used clinical diagnostic tool in modern medicine. Compared to other diagnostic imaging techniques, MRI has many advantages. It is noninvasive and without radiation burden, and has excellent spatial resolution and permits deep tissue penetration leading to superb soft tissue contrast.[1, 2] In order to improve the specificity and acquire higher-intensity MR singal, contrast agents (CAs) are widely used to assist in the acquisition and interpretation of MRI images. In clinical practice, more than 35 percent of MRI examinations are performed with the administration of CAs to further enhance the image contrast.[3, 4]
Up to now, US Food and Drug Administration (FDA) has approved nine gadolinium-based contrast agents (GBCAs) for clinical application, which are all T1-weighted MRI images.[5] However, because of the disadvantages such as rapid clearance, poor cell uptake, non-specific binding and especially low relaxivity of these GBCAs, the development of higher relaxivity CAs is necessary.[6] According to Solomon-Bloembergen-Morgan (SBM) equation, the relaxivity of GBCAs is mainly determined by residence time (τM), the number of first-shell (inner) water molecules (q), rotational correlation time (τR), and electron spin relaxation time (T1e). For small molecular weight GBCAs, either increasing the parameter q or lengthening τR could enhance the relaxivity.[7~9]
Since the increase of inner coordination water molecules often accompanying the decrease of thermodynamic and kinetic stability, all currently approved GBCAs have only one coordinated water molecule (q=1).[10, 11] As a result, another strategy to slow down the rotational correlation time of the agents was widely used. For this purpose, one method is to connect small molecule weight CAs with macromolecules such as proteins, dendrimers, nanoparticles, or liposomes.[12, 13] However, the macromolecules usually have relatively slow clearance rates in human body, which is disadvantageous to the practical application.[14, 15] Construction of multiple-nuclear agents by fusing several small-molecule agents together is another approach to elongate the rotational correlation time. Some examples have been reported with greatly improved relaxivity.[4, 16~20]
The current GBCAs usually use linear DTPA (diethylenetriaminepentaacetic acid) or macrocyclic DOTA (1, 4, 7, 10-tetraazacyclododecan-N, N', $N''$, $N'''$-tetraacetic acid) as chelators for gadolinium ion. Both types of them have been applied to construct multinuclear agents. However, some investigations have proved that linear GBCAs have a risk of nephrogenic systemic fibrosis (NSF) in certain patients with kidney dysfunction, whereas macrocyclic GBCAs have a lower risk for the development of NSF.[21, 22] Because the macrocyclic agents are more stable than the linear agents.[23] For this reason, we preferentially choose macrocyclic DOTA as the chelator to construct multinuclear agents.
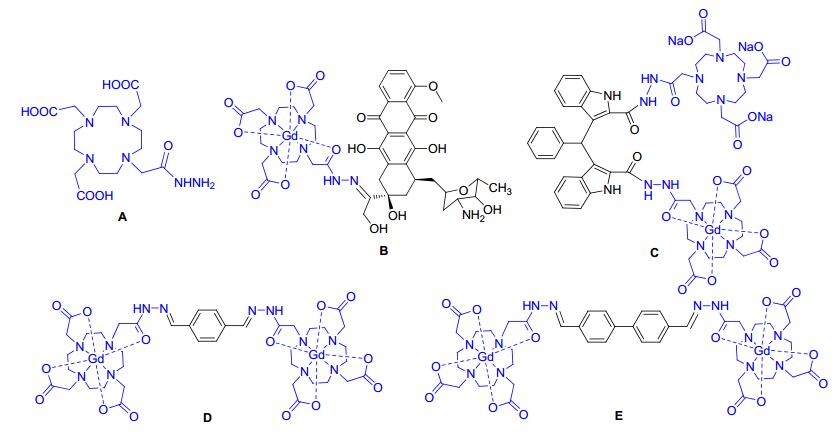
DOTA-hydrazide (DOTAH, A) is a good chelator for Gd3+ and has been researched as the first MRI prodrug- procontrast complex (B) and necrosis avid contrast agent (C).[24~27] In this study, we designed and synthesized two novel dimeric nonionic CAs, (Gd-DOTAH)2-DYMB (DYMB=1, 4-diylidenemethylbenzene, D) and (Gd- DOTAH)2-DYMBP (DYMBP=1, 4-diylidenemethylbi-phenyl, E), in which two DOTAH units that functioned as Gd3+ chelators, were affixed onto benzene and biphenyl, respectively (Chart 1). The relaxometric studies and T1-weighted MR images have been performed for the two complexes.
Figure Chart 1
Figure Chart 1.
Structures of A~E
The chelator DOTAH (A), the first MRI prodrug-procontrast complex (B), a necrosis avid contrast agent (C) and two novel binuclear CAs designed and synthesized in this study (D, E)
In this study, DOTA-hydrazide was chosen as the chelator for Gd3+ to build the binuclear MRI contrast agengs. Herein, the coordination oxygen from hydrazide group provided an additional donor for the chelated Gd3+, which compensated for the missing acetic chelating arm from DOTA to DOTAH. In turn, it improved the thermodynamic stability of the resulting complexes. As far as it goes, DOTAH as a macrocyclic ligand has higher thermodynamic stability and slower dissociation kinetics to avoid transmetallation in vivo compared to the linear ligand like DTPA. At the same time, the Gd3+ binding neutralized the charges of the three acetic groups of DOTAH, leading to nonionic CAs, which is beneficial to low osmotic pressure. Diylidenemethylbenzene and diylidenemethylbiphenyl were chosen as the linkages of Gd-DOTAH moieties, the rigidity of the aromatic nucleus would restrict the internal motion of the molecules, resulting in improving the relaxivity compared to the flexible alkyl chains.[28]
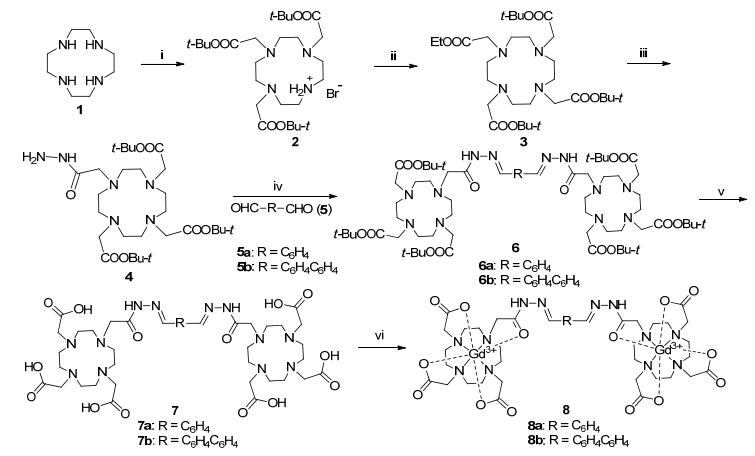
The desired complexes (Gd-DOTAH)2-DYMB and (Gd- DOTAH)2-DYMBP and their precursors were prepared following the procedures in Scheme 1. The first step begins with a threefold alkylation of cyclen with tert-butylbromoacetate to generate 1, 4, 7-tris(tert-butoxycarbonylmethyl)-1, 4, 7, 10-tetraazacyclododecane hydrobromide (2). Alkylation of the secondary amino group with ethyl bromoacetate (EBA) afforded 1, 4, 7-tris(tert-butoxycarbonylmethyl)-10-ethoxycarbonylmethyl-1, 4, 7, 10-tetraazacyclododecane (3) in 90% yield and good purity. For introducing hydrazide moieties, the ethyl ester (3) was brought to react with a large excess of hydrazine monohydrate to get compound DOTA-hydrazide (4) as a white power solid which is different from some reported references in which the product 4 is a glassy solid. The condensation reactions were complished using intermediate 4 and terephthalaldehyde (5a) or 4, 4'-biphenyldicarboxalde- hyde (5b) in absolute ethanol refluxing for 4 h. Deprotection of the compouds 6a~6b were achieved within 24 h by reaction with trifluoroacetic acid (TFA). The last complexes 8a~8b were obtained by combining 7a~7b and gadolinium chloride in an aqueous solution at 50 ℃, and the purification of 8a~8b were accomplished by C-18 SPE cartridge.
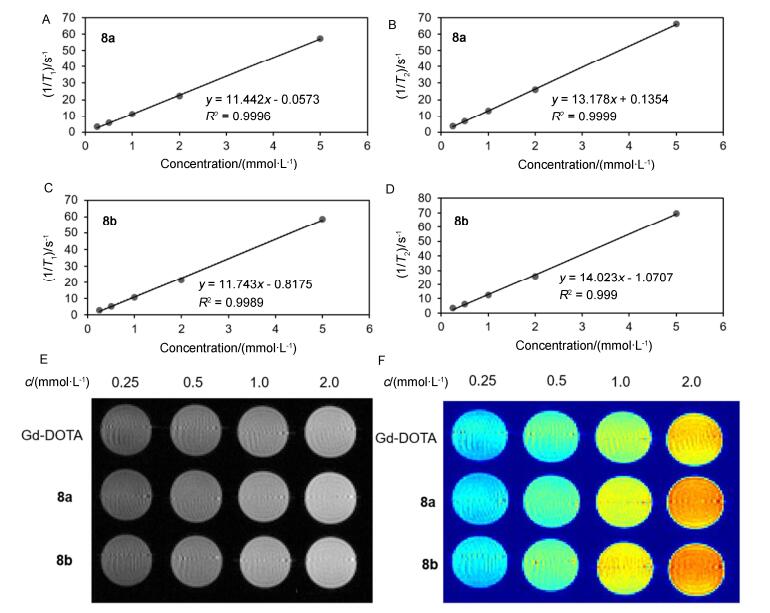
To evaluate the contrast enhancement of the new agents, longitudinal relaxivity r1 and transverse relaxivity r2 of the new agents were calculated from the linear fitting of 1/T1 and 1/T2 plot verse complexes concentraions (Figures 1A~1D). Commercially available Gd-DOTA was used as compare. The longitudinal relaxivities (r1) of (Gd- DOTAH)2-DYMB (MW=1244) and (Gd-DOTAH)2- DYMBP (MW=1319) were 11.4 and 11.7 L•mmol-1•s-1 per molecule or 5.7 and 5.9 L•mmol-1•s-1 per Gd at 0.5 T, respectively (Table 1). The slightly greater molecular weight favors a slightly higher relaxivity of (Gd-DOTAH)2- DYMBP than (Gd-DOTAH)2-DYMB. And their relaxivities are higher than that of the currently used clinical mononuclear macrocyclic CAs Gd(DOTA) (3.6 L•mmol-1• Gd-1•s-1, MW=563, Table 1). The improvement of relaxivities was mainly attributed to the increase in molecular weight and the slower rotational correlation time (τR) due to rigidity of molecular. At the same time, the ratios of r2/r1 are calculated to be 1.15 and 1.19 (Table 1), the low ratios of r2/r1 indicate that all the new agents can be used as T1-weighted MRI CAs. As shown in Figures 1E and 1F, complexes 8a and 8b present excellent positive T1 contrast enhancement, and the brightness of MR images enhance with the increasing concentrations of solutions, showing a clear dose-dependent color change, due to the relaxation of water proton increase with the increase of concentration dose. The higher relaxivities and brighter MR images mean that these two binuclear macrocyclic contrast agents may be used at lower dosage of agents and thus reducing the clinical toxicity.
Figure 1
Figure 1.
MR relaxivities and T1-weighted MR images of 8a~8b at 0.5 T
r1 and r2 relaxivity curves of 8a (A, B), r1 and r2 relaxivity curves of 8b (C, D), T1-weighted MR images of 8a~8b and Gd-DOTA at different concentrations from 0.25 to 2.0 mmol/L (E, F)
In summary, two nonionic binuclear MRI CAs, (Gd- DOTAH)2-DYMB and (Gd-DOTAH)2-DYMBP have been designed and synthesized using diylidenemethylbenzene or diylidenemethyl-biphenyl as the linker centers. Their longitudinal relaxivity values are 11.4 and 11.7 L•mmol-1•s-1 per molecule or 5.7 and 5.9 L•mmol-1• Gd-1•s-1 at 0.5 T, respectively. The relaxivities are higher than that of the FDA approved mononuclear macrocyclic agent Gd(DOTA). The relaxivity improvement can be mainly accounted for the increase of the molecular weight and the slower rotational correlation time due to rigidity of linkers. Furthermore, the low r2/r1 ratios and good positive imaging effect showed that the two new contrast agents are suitable as T1-weighted MRI CAs. And they may be used at lower dosage of agents and thus reduce the clinical toxicity.
4.
Experimental section
4.1
General procedures
Flash column chromatography was performed using 200~400 mesh silica gel (Fisher). NMR spectra were recorded on a Varian Inova 300 spectrometer and a 500 MHz Bruker Ascend Avance Ⅲ HD at room temperature. Chemical shifts for 1H NMR and 13C NMR were reported as δ, and referenced to an internal deuterated solvent central line. Multiplicity and coupling constants (J) were calculated automatically on MestReNova 10.0, a NMR processing software from Mestrelab Research. MS (ESI) was determined by using a MicroTOF-Q Ⅱ HRMS/MS instrument (Bruker). Element analysis was determined by using a Perkin-Elmer 240c elemental analysis instrument. The MRI testing and T1 relaxation time measurements were tested at a 0.5 T NMR120-Analyst NMR Analyzing and Imaging system (Niuman Corporation, Shanghai, China). Melting points were determined in open capillaries and were uncorrected. All chemicals and reagents were used as received without further purification. Glassware was dried in an oven at 130 ℃ and purged with a dry atmosphere prior to use. Unless otherwise mentioned, reactions were performed open to air. Reactions were monitored by thin-layer chromatography (TLC) and visualized by a dual short/long wavelength UV lamp.
To a suspension of cyclen (1.72 g, 10 mmol) and NaHCO3 (2.52 g, 30 mmol) in dry acetonitrile (100 mL) at 0 ℃ was added a solution of t-butyl bromoacetate (5.85 g, 30 mmol) in dry acetonitrile (100 mL) dropwise over a period of 1 h. The temperature was maintained at 0 ℃ during the addition, after which the reaction mixture was allowed to come to room temperature for 24 h and then refluxed for 4 h [TLC, V(H2O):V(CH3CN)=5:95]. After cooled, the solid was filtered, the filtrate was evaporated under reduced pressure and yellow oil solid was got. The crude product was dissolved in a small amount of methanol and then added moderate methylene dichloride until fluffy white solid appeared. After filtration, 4.88 g of white solid was got, yield 82%. m.p. 189.7~191.6 ℃(lit.[29] m.p. 190~191 ℃); 1H NMR (300 MHz, CDCl3) δ: 10.00 (s, 2H), 3.36 (s, 4H), 3.28 (s, 2H), 3.09 (s, 4H), 2.91 (s, 7H), 2.87 (s, 5H), 1.45 (s, 27H); 13C NMR (75 MHz, CDCl3) δ: 170.43, 169.55, 81.74, 81.58, 58.13, 51.31, 49.18, 48.85, 47.43, 28.15, 28.12; HRMS (ESI+) calcd for C26H51N4O6 [M+H]+ 515.3809, found 515.3867.
Compound 2 (2.93 g, 5 mmol) was stirred under nitrogen with ethyl bromoacetate (1.25 g, 7.5 mmol) and potassium carbonate (1.0 g, 7.5 mmol) in dry acetonitrile. The mixture was heated at 80 ℃ and stirred for further 12 h and monitored by TLC [V(H2O):V(CH3CN)=5:95]. After cooled, the solid was filtered, the filtrate was removed under reduced pressure. The residue was dissolved in a minimum volume of dichloromethane and then purified by flash column chromatography (SiO2, CH2Cl2 with an increasing gradient of methanol from 0 to 5%). The solvent was evaporated to give 3 (2.7 g) as a yellow glassy solid, yield 90%. 1H NMR (300 MHz, CDCl3) δ: 4.14 (q, J=6.9 Hz, 2H), 3.14 (br, 10H), 2.32 (br, 8H), 2.12 (s, 6H), 1.43 (s, 27H), 1.24 (t, J=7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 172.08, 171.72, 170.56, 81.53, 81.46, 55.40, 54.35, 54.31, 49.85, 27.68, 27.55; ESI-MS m/z: 601.4 [M+H]+, 623.4 [M+Na]+. Anal. calcd for C30H56N4O8: C 59.98, H 9.40, N 9.33; found C 59.89, H 9.42, N 9.35.
Compound 3 (1.2 g, 3.0 mmol) was dissolved in absolute ethanol (20 mL) and added to a solution of hydrazine monohydrate (12.5 mL, 200 mmol). The reaction mixture was stirred at reflux under an atmosphere of nitrogen for 12 h following the reaction TLC [V(H2O):V(CH3CN)=5:95]. The solution was evaporated under reduced pressure and the residue was recrystallized with ethyl acetate and ether to obtain 4. White solid, 0.82 g, yield 47%. m.p. 120.9~123.2 ℃; 1H NMR (300 MHz, CDCl3) δ: 9.53 (s, 1H), 4.25 (s, 2H), 3.34 (s, 6H), 3.05 (br, 6H), 2.50 (s, 6H), 2.26 (br, 6H), 1.43 (s, 27H); 13C NMR (75 MHz, CDCl3) δ: 172.34, 171.98, 170.81, 81.82, 81.75, 55.68, 55.64, 54.60, 50.19, 27.96, 27.83; ESI-MS m/z: 587.5 [M+H]+, 609.5 [M+Na]+. Anal. calcd for C28H54N6O7: C 57.31, H 9.28, N 14.32; found C 57.26, H 9.30, N 14.35.
4.2.4
Synthesis of (t-Bu-DOTAH)2-DYMB (6a)
To a 100 mL round-bottom flask fitted with a magnetic stir bar were added compound 4 (1.17 g, 2.0 mmol), 1, 4-phthalaldehyde (5a, 0.14 g, 1.0 mmol) and 30 mL of absolute ethanol. The mixture was stirred at 75 ℃ for 4 h. After completion of the reaction, ethanol was removed in vacuo, and the resulting residue was recrystallized with ethyl acetate. Filtered, and washed with cold ethyl acetate to obtain 6a. White solid, 1.08 g, yield 85%. The product was used without further purification. m.p. 145.7~147.8 ℃; 1H NMR (300 MHz, CDCl3) δ: 12.75 (s, 2H), 8.62 (s, 2H), 7.69 (s, 3H), 7.58 (t, J=8.7 Hz, 1H), 3.51 (s, 4H), 3.05 (s, 12H), 2.72~2.12 (br, 32H), 1.45 (s, 27H), 1.42 (s, 27H); 13C NMR (75 MHz, CDCl3) δ: 172.14, 171.84, 168.53, 147.81, 135.81, 127.40, 82.19, 81.95, 81.85, 55.84, 55.63, 55.40, 50.22, 27.95, 27.82; ESI-MS m/z: 1271.9 [M+H]+. Anal. calcd for C64H110N12O14: C 60.45, H 8.72, N 13.22; found C 60.38, H 8.74, N 13.25.
4.2.5
Synthesis of (t-Bu-DOTAH)2-DYMBP (6b)
To a 100 mL round-bottom flask fitted with a magnetic stir bar were added compound 4 (1.17 g, 2.0 mmol), 4, 4'-Biphenyldicarboxaldehyde (5b, 0.21 g, 1.0 mmol) and 30 mL absolute ethanol. The mixture was stirred at 75 ℃ for 4 h. After completion of the reaction, ethanol was removed in vacuo, and the resulting residue was recrystallized with ethyl acetate. The mixture was filtered, and washed with cold ethyl acetate to get 1.13 g of light yellow solid (yield 84%). The product was used without further purification. m.p. 161.5~163.7 ℃; 1H NMR (300 MHz, CDCl3) δ: 12.85 (s, 2H), 8.72 (s, 2H), 7.79 (d, J=8.0 Hz, 3H), 7.68 (d, J=7.9 Hz, 1H), 7.54 (d, J=8.0 Hz, 4H), 3.57 (s, 4H), 3.07 (s, 12H), 2.71~2.11 (br, 32H), 1.47 (s, 27H), 1.43 (s, 27H); 13C NMR (75 MHz, CDCl3) δ: 172.10, 171.87, 168.56, 147.96, 141.37, 133.98, 127.99, 126.65, 82.05, 81.94, 55.91, 55.66, 55.43, 50.27, 27.98, 27.86. HRMS (ESI+) calcd for C70H114N12O14Na [M+Na]+ 1369.8456, found 1369.8459.
4.2.6
Synthesis of (DOTAH)2-DYMB (7a)
A solution of 6a (0.64 g, 0.5 mmol) in TFA (5 mL) was stirred at room temperature for 24 h. TFA was removed under reduced pressure and the residue was dissolved in ethyl acetate (5 mL). After freezing for 12 h, the mixture was filtered to obtain white solid 7a (quantitative). m.p. 201.6~203.2 ℃; 1H NMR (300 MHz, D2O) δ: 8.09 (s, 1H), 7.93 (s, 1H), 7.69 (d, J= 9.8 Hz, 4H), 4.05~2.70 (m, 48H); 13C NMR (75 MHz, D2O) δ: 202.08, 198.30, 152.71, 137.91, 132.97, 130.78, 130.36, 120.88, 117.01, 58.55, 57.37, 56.55, 52.95, 51.84; ESI-MS m/z: 935.5 [M+H]+. Anal. calcd for C40H62N12O14: C 51.38, H 6.68, N 17.98; found C 51.31, H 6.70, N 18.02.
4.2.7
Synthesis of (DOTAH)2-DYMBP (7b)
A solution of 6b (0.67 g, 0.5 mmol) in TFA (5 mL) was stirred at room temperature for 24 h. TFA was removed under reduced pressure and the residue was dissolved in ethyl acetate (5 mL). After freezing for 12 h, the mixture was filtered to obtain yellow solid 7b (quantitative). m.p. 212.2~214.7 ℃; 1H NMR (300 MHz, D2O) δ: 7.94 (s, 1H), 7.76 (s, 1H), 6.84~7.67 (br, 8H), 4.00~2.52 (br, 48H); 13C NMR (75 MHz, D2O) δ: 173.05, 165.60, 165.13, 152.58, 143.61, 143.03, 135.37, 130.97, 129.48, 120.99, 117.12, 59.08, 58.10, 56.50, 54.21, 53.45, 51.22, 50.69. HRMS (ESI+) calcd for C46H66N12O14Na [M+Na]+ 1033.4714, found 1033.4703.
4.2.8
Synthesis of (Gd-DOTAH)2-DYMB (8a)
To a solution of 7a (0.37 g, 0.4 mmol) in water (20 mL) was added a solution of GdCl3 in water (0.05 mol/L, 10 mL, 0.5 mmol) while maintaining the pH at 6.5 by addition of 1 mol/L NaOH. The solution was stirred at 50 ℃ for 24 h. The pH was raised to 8.0. The solution was filtered through a 0.2 μm filter, and the pH was lowered to 7.0 by adding 1 mol/L HCl. The solution was loaded on a Hypersep C-18 cartridge, and the stationary phase was washed extensively with water. The product was recovered after gradient elution [V(MeOH):V(H2O)=1:9~3:2]. The fractions containing the product were evaporated under reduced pressure to give 8a as light yellow solid, 0.43 g, yield 85%. ESI-MS m/z: 1242.2 [M-2H]-. A correct Gd isotope pattern was observed. Anal. calcd for C40H56Gd2- N12O14•2H2O: C 37.55, H 4.73, N 13.14; found C 37.50, H 4.74, N 13.17.
4.2.9
Synthesis of (Gd-DOTAH)2-DYMBP (8b)
To a solution of 7b (0.41 g, 0.4 mmol) in water (20 mL) was added a solution of GdCl3 in water (0.05 mol/L, 10 mL, 0.5 mmol) while maintaining the pH at 6.5 by addition of 1 mol/L NaOH. The solution was stirred at 50 ℃ for 24 h. The pH was raised to 8.0. The solution was filtered through a 0.2 μm filter, and the pH was lowered to 7.0 by adding 1 mol/L HCl. The solution was loaded on a Hypersep C-18 cartridge, and the stationary phase was washed extensively with water. The product was recovered after gradient elution [V(MeOH):V(H2O)=1:9~3:2]. The fractions containing the product were evaporated under reduced pressure to give 8b as yellow solid, 0.44 g, yield 83%. ESI-MS m/z: 1318.3 [M―H]―. A correct Gd isotope pattern was observed. Anal. calcd for C46H60Gd2N12O14• 2H2O: C 40.76, H 4.76, N 12.40; found C 40.68, H 4.77, N 12.43.
4.3
MRI measurement
To obtain T1 relaxation times of the aqueous solution of the new agents at different concentrations, following measurement parameters were used: repetition time (TR)= 6000 ms, number of data=25, and number of averages (NA)=2. While the T2 relaxation times were determined with the following parameters: TR=6000 ms, echo time (TE)=1 ms, echo count=6000 and NA=2. Both r1 and r2 relaxivities were calculated by the linear fitting of 1/T1 or 1/T2 as a function of metal concentration. For T1-weighted MR images, the instrument parameters were set as follows: TR=100 ms, TE=18.2 ms, imaging matrix=192×256, slice thickness=5 mm, field of view (FOW)=100 mm×100 mm and NA=2.
Supporting Information1H NMR and 13C NMR spectra, mass spectra of the synthesized compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
He, W.; Oliver, T. B.; Michael, G. K.; Eric, C. H.; Mariya, B.; Agata, W.; Ou, C.; Yue, C.; Nan, L.; Satoshi, O.; Jose, M. C.; Markus, H.; Christian, T. F.; Daniel, M. M.; Gerhard, A.; Harald, I.; Alan, J.; Peter, N.; Moungi, G. B. Proc. Natl. Acad. Sci. U. S. A.2017, 114, 2325. doi: 10.1073/pnas.1620145114
[22]
Huckle, J. E.; Altun, E.; Jay, M. J.; Semelka, R. C. Invest. Radiol.2016, 51, 236. doi: 10.1097/RLI.0000000000000228
[23]
Boros, E.; Gale, E. M.; Caravan, P. Dalton Trans. 2015, 44, 4804. doi: 10.1039/C4DT02958E
[24]
Frullano, L.; Tejerina, B.; Meade, T. J. Inorg. Chem.2006, 45, 8489. doi: 10.1021/ic0612045
[25]
Fuge, F.; Weiler, M.; Gätjens, J.; Lammers, T.; Kiessling, F. Tetrahedron Lett.2013, 54, 918. doi: 10.1016/j.tetlet.2012.11.151
[26]
Cresens, E. ; Ni, Y. -C. ; Adriaens, P. ; Verbruggen, A. ; Marchal, G. WO 2002038546, 2002[Chem. Abstr. 2002, 136, 379071].
[27]
Zhang, J. -D. ; Curry, K. WO 2016090491, 2016 [Chem. Abstr. 2016, 165, 112684].
Jagadish, B.; Brickertalbrecht, G. L.; Nichol, G. S.; Mash, E. A.; Raghunand, N. Tetrahedron Lett.2011, 52, 2058. doi: 10.1016/j.tetlet.2010.10.074
Figure Chart 1
Structures of A~E
The chelator DOTAH (A), the first MRI prodrug-procontrast complex (B), a necrosis avid contrast agent (C) and two novel binuclear CAs designed and synthesized in this study (D, E)
Figure 1
MR relaxivities and T1-weighted MR images of 8a~8b at 0.5 T
r1 and r2 relaxivity curves of 8a (A, B), r1 and r2 relaxivity curves of 8b (C, D), T1-weighted MR images of 8a~8b and Gd-DOTA at different concentrations from 0.25 to 2.0 mmol/L (E, F)

 下载:
下载:

 下载:
下载:




