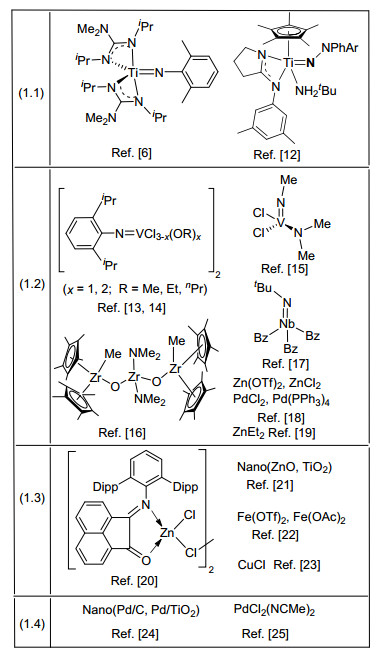
图式 1.
基于不同成胍机理的过渡金属催化剂分类
Scheme 1.
Transition metal catalysts for guanylation classified on the basis of different mechanisms

胍具有特殊的'CN3'结构, 常以盐的形式存在, 具有很多特殊的性能[1].最为重要的是其碱性很强(pKa=13.5), 与氢氧化钠相近, 在生理条件下处于质子化状态, 这是形成配体和受体、酶和底物之间氢键、静电等相互作用的基础, 因此, 胍可以作为有机碱催化剂[1d, 2].胍很容易手性化, 是一类很好的不对称手性催化剂[2].胍是多种药物、天然产物、农药、消毒剂、杀虫剂、抗菌整理剂, 甜味剂及炸药中重要的结构单元[1].另外, 胍也可用作配体, 稳定多种金属配合物[3].
胍及其衍生物的经典合成方法是利用胺与亲电的胍化试剂反应而制备[4], 但这种方法存在合成路线长, 副产物多, 分离提纯过程复杂等缺点.研究人员一直在探索新的合成路径, 如胺与碳二亚胺的成胍反应原子经济性地构建胍.早期研究表明, 氟化四丁基铵(TBAF)可以促进某些芳香胺与双芳基碳二亚胺的成胍反应[5]. 2003年, Richeson等[6]报道了第一例金属钛氮宾配合物催化芳香伯胺与碳二亚胺的成胍反应. 2006年, Hou等[7]报道了首例半夹心稀土金属烷基配合物催化的仲胺与碳二亚胺的成胍反应.基于这两个开拓性的工作, 在近10多年里, 胺与碳二亚胺催化直接成胍取得了飞速的发展, 新的催化剂及催化体系不断涌现[8~11].国内有几个课题组, 如我们组[8]、苏州大学沈琪等[9]、安徽师范大学王绍武等[10]、香港中文大学谢作伟等[11], 在过渡、稀土和主族金属催化成胍反应方面开展了系统研究.基于上面提到的胍的性能及应用情况, 几篇综述对此进行了概括.如Taylor[2c]和Coles等[1d]分别综述了胍作为有机碱催化剂; Carrillo-Hermosilla等[4]综述了胍的经典及高效催化合成方法; 我们课题组[8]综述了催化成胍反应的类型和胍类化合物的合成.本文将系统综述近年来过渡金属催化胺与碳二亚胺的成胍反应, 直接构建非环胍和环状胍的催化机理、反应体系、底物范围及成胍产物的结构特点等.
用于催化成胍的过渡金属催化剂很多(Scheme 1), 其中涉及的过渡金属主要有Ti、Zr、V、Nb、Mo、Fe、Pd、Cu、Ag、Zn、Hg等, 但不同的催化剂由于其结构不同, 催化成胍机理和性能也不同.下面通过四种不同的成胍反应机理, 分类阐述.
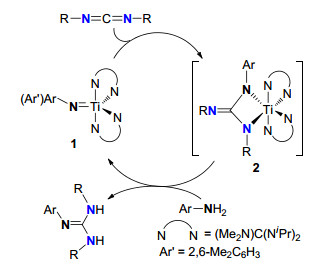
胺与碳二亚胺反应直接成胍是一种原子经济的直接构筑多取代胍及其衍生物的方法, 具有重要的理论意义和实际应用价值.研究表明脂肪伯胺与碳二亚胺在非常苛刻的条件下可以低产率生成相应的胍类化合物, 但高效合成胍类化合物需要相应的催化剂. Richeson等[6]开创性地利用Ti=N氮宾配合物(Me2N)C(NiPr)2]2Ti=N(2, 6-Me2C6H3) (1)作为催化剂, 实现了芳香伯胺与碳二亚胺的成胍反应高效合成胍.催化循环机理如Scheme 2.配合物1与碳二亚胺分子之间通过[2+2]环加成生成中间体2, 2与胺发生质子化反应生成胍, 并释放催化剂活性中间体.但同族金属(如Zr)配合物却没有与Ti配合物相似的催化活性.由于仲胺在催化循环中不能再生“Ti=N”亚胺活性物种, 这种催化循环体系不适合于二级胺.
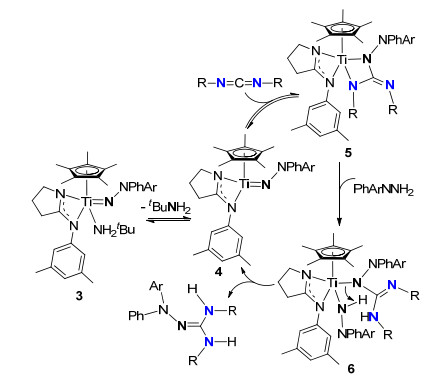
2013年, Gade等[12]合成了系列Ti-肼亚胺基配合物, 并将它们用作成胍反应催化剂, 通过1H NMR, 13C NMR, 15N NMR谱表征及动力学对成胍反应机理进行了深入的研究, 结果表明这种成胍反应机理是[2+2]环加成/质子化机理(Scheme 3).催化循环的第一步是配合物4的Ti=N部分与碳二亚胺[2+2]环加成生成胍中间体5, 5与肼反应转化为中间体6, 最后发生分子内质子化而释放胺基胍及再生中间体4完成催化循环.
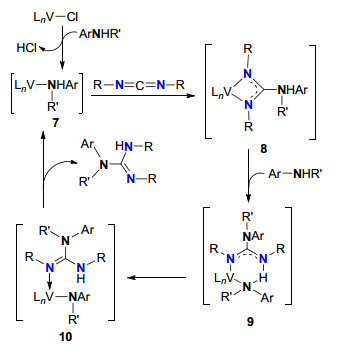
Hou等[7]于2006年报道了首例半夹心稀土金属烷基配合物催化的伯胺/仲胺与碳二亚胺的成胍反应.研究表明, 该成胍反应经历插入/质子化机理.该类催化剂和插入/质子化机理的发现, 大幅扩展了胺类底物的范围, 是仲胺与碳二亚胺成胍反应的首例. Galindo及其合作者[15]运用理论计算对胺与碳二亚胺[2+2]环加成进行了深入的研究, 在随后的研究中, 他们采用V(N-2, 6-iPr2C6H3)Cl3作为成胍反应催化剂前体, 实现了伯胺/仲胺与碳二亚胺的催化成胍反应.有可能经历两种路径:一种是碳二亚胺对V=N键的加成, 与Richeson等提出的机理一致, 但是该机理与动力学研究结果不一致; 第二种是首先胺与钒配合物反应形成配合物7, 其次是碳二亚胺插入到7的[V—N]键中形成胍中间体8, 8再与一分子胺反应生成中间体9和10, 进一步释放胍产物和[V—N]中间体7(Scheme 4).这与DFT计算结果非常吻合, 因此, 采用V(N-2, 6-iPr2C6H3)Cl3作催化剂催化成胍的是一种插入/质子化机理[15].不但VB族的钒(V)配合物对胺与碳二亚胺成胍有很好的催化活性, 同族的Nb-烷基等亚胺配合物[17]、氧桥联的Ti和Zr配合物及O—Zr配合物[14]也有比较好的催化活性.
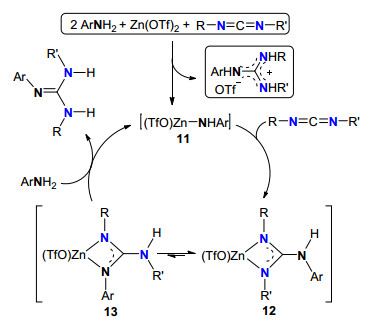
2010年, 我们组[18]以简单易得的Zn(OTf)2为催化剂前体, 以一级芳胺和各种碳二亚胺为原料, 高效地合成了一系列多取代胍.其催化机理(Scheme 5)包括如下几个过程: (1)一级芳胺、Zn(OTf)2与碳二亚胺反应生成活性“Zn-N”物种11, 同时放出三氟甲磺酸胍; (2)碳二亚胺首先配位到11的锌金属中心, 锌-氮键对碳二亚胺亲核加成而生成胍中间体12, 12异构化形成中间体13; (3) 12或13与另一分子胺质子解重新生成11并生成最终产物胍.
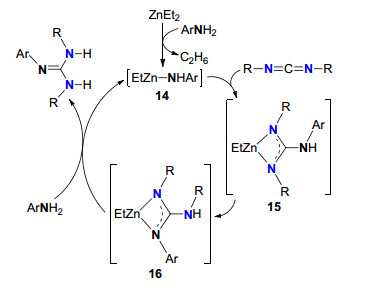
Alonso-Moreno等[19]以简单易得的ZnEt2为催化剂, 实现了胺与碳二亚胺的成胍反应构建胍(Scheme 6). ZnEt2与胺首先生成关键中间体[EtZn-NHAr]14, 随后碳二亚胺插入到Zn—N键中形成中间体15, 15异构化形成中间体16, 进而发生质子化反应生成胍.反应底物适用范围广, 带有不同取代基的芳胺、仲胺和杂环胺均可以与多种碳二亚胺参与成胍反应.
2013年, Mandal等[16]在室温下通过Cp2*(Me)-Zr(OH) (Cp*=η5-C5Me5)和Zr(NMe2)4的原位反应合成了两个氧原子桥联三个Zr的配合物[Cp2*(Me)Zr(μ-O)Zr(NMe2)2(μ-O)Zr(Me)Cp2*] (Eq. 1).以该配合物为催化剂, 实现了胺与碳二亚胺直接成胍反应.研究表明该成胍反应也是一种插入/质子化机理, 该类金属配合物催化活性很高, 经历五次循环利用, 其催化活性基本保持不变.
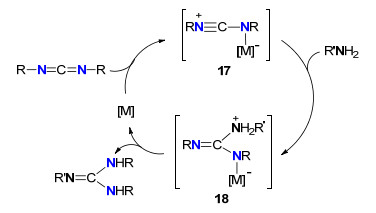
许多过渡金属路易斯酸(如CuCl、FeCl3、FeCl2)、纳米氧化物(如ZnO、TiO2、NiO)及羧基化物[Fe(OAc)2、Zn(OTf)2、Fe(OTf)2]等是好的成胍催化剂[20~23, 26~28]. 2012年, Royo等[22]发现便宜易得的铁及亚铁氯化物、乙酸基化合物、三氟甲磺酸化合物以及羰基化合物是一种好的成胍催化剂, 能在温和的条件下催化胺与碳二亚胺成胍, 这是一种典型的碳二亚胺活化、亲核加成及分子内质子化机理, 其可能的催化循环机理如Scheme 7.铁或亚铁化合物对碳二亚胺进行活化形成中间体17, 胺对17亲核形成中间体18, 最后进行分子内质子化形成胍及重新生成铁或亚铁化合物.
|
|
(1) |
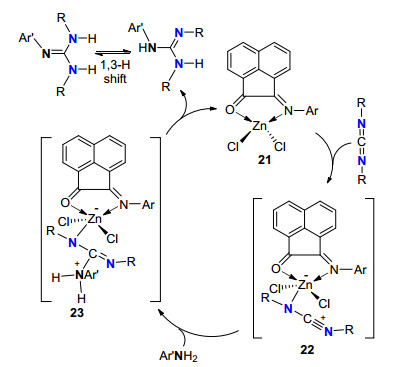
2016年, Panda等[20]以锌配合物21作为催化剂前体(Eq. 2), 在溶液中首先解离成单体21', 21'能催化各种芳胺与碳二亚胺成胍.该反应底物范围广, 反应活性高.可能反应机理如Scheme 8.锌配合物21'首先活化碳二亚胺中形成五配位中间体22, 然后胺对22亲核加成形成五配位中间体23; 最后发生分子内质子转移, 释放出最终产物胍和催化剂活性中间体21'而完成催化循环.
|
|
(2) |
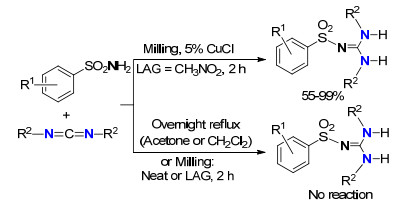
2014年, Friščić等[23]以5 mol%的CuCl为催化剂, 将经过研磨的苯磺酰胺类化合物与碳二亚胺反应可高效合成苯磺酰胍(Scheme 9).值得指出的是, 反应物研磨时不足化学计量的液体是必要的, 假如将苯磺酰胺类化合物与碳二亚胺混合物溶解在CH2Cl2或丙酮中, 即使回流反应24 h, 也没有产物形成.当没有催化剂CuCl参与的液体辅助研磨反应混合物时, 其成胍转化率很低.这些研究表明, CuCl通过与碳二亚胺配位起到活化碳二亚胺的作用, 从而有利于苯磺酰胺对碳二亚胺的亲核进攻.
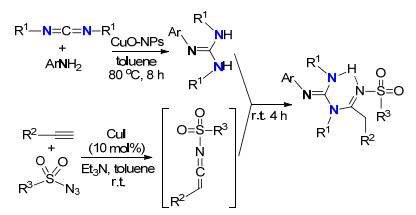
2015年, Yavari等[27]以苯胺和N, N'-二环己烷基碳二亚胺(DCC)为原料, 考察了各种铜催化剂的催化成胍性能, 包括CuI、CuBr、Cu (OAc)2、CuCl2、纳米Cu2O、纳米CuO等, 发现以纳米CuO粒子为催化剂, 以甲苯为溶剂是最佳的成胍反应体系, 同时所合成的胍可以进一步功能化合成一系列亚氨基功能化的多取代胍(Scheme 10).
2016年, García等[28]以石墨烯支持的铜纳米粒子为非均相催化剂, 详细研究了取代苯胺与碳二亚胺成胍反应.他们发现, 该类型的纳米粒子由于作用面积大, 吸附能力强, 而且Cu-石墨烯之间的相互作用等因素使得催化活性很高, 其催化活性远大于其它支持配体, 如碳纳米管、MgO、CeO2和Al2O3等.
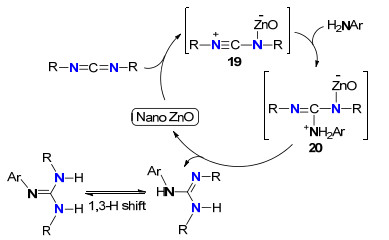
Kantam等[21]以纳米ZnO为催化剂, 以甲苯为溶剂, 80 ℃下高产率合成了一系列三取代胍.各种脂肪胺, 芳胺以及杂环伯胺、仲胺与各种碳二亚胺均适合该反应体系, 其催化成胍机理见Scheme 11.首先是纳米ZnO对碳二亚胺进行活化形成中间体19, 19的中心碳原子亲电性增强, 有利于胺的亲核进攻形成中间体20, 最后发生分子内质子化生成胍以及重新释放纳米ZnO而完成整个催化循环.
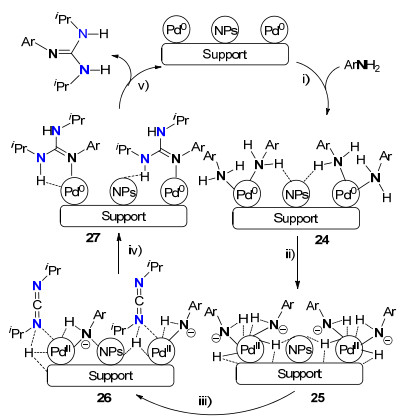
2012年, Garcia等[24]利用负载的钯纳米粒子, 如纳米Pd/MgO、Pd/C、Pd/CeO2和Pd/TiO2, 作为成胍反应催化剂, 高产率合成了一系列多取代胍, 并对成胍机理进行了深入的研究.他们发现, 该类成胍反应涉及两种过程, 一种是非均相催化成胍, 另一种是均相催化成胍.非均相成胍机理主要包括如下五步(Scheme 12): (ⅰ)苯胺与Pd(0)配位形成中间体24; (ⅱ) N—H键氧化加成产生Pd(Ⅱ)中间体25; (ⅲ)碳二亚胺与二价Pd配位形成中间体26; (ⅳ) Pd—N键对碳二亚胺亲核加成后伴随还原消除形成Pd(0)中间体27; (ⅴ)胍从Pd(0)物种上解离成最终产物及形成Pd(0)纳米粒子而实现催化循环.
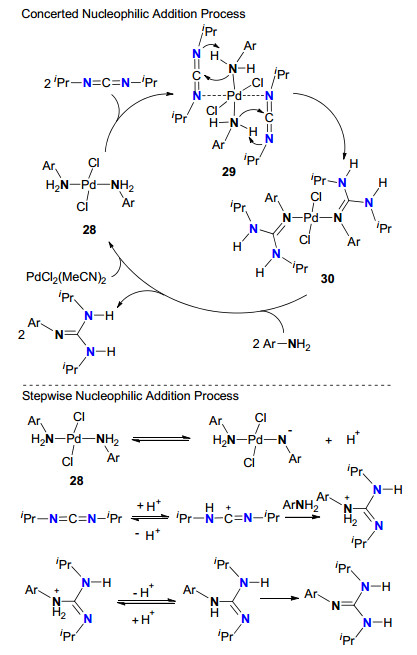
2013年, Garcia等[25]又以PdCl2(MeCN)2为均相催化剂研究了胺与碳二亚胺的成胍反应.基于分离的几种Pd(Ⅱ)配合物中间体, 他们提出了两种可能的均相催化成胍机理(Scheme 13).第一种机理可能经历协同的过程:两分子苯胺与PdCl2(MeCN)2配位形成Pd(Ⅱ)配合物28, 两分子N, N'-二异丙基碳二亚胺(DIC)与Pd(Ⅱ)形成中间体29.在29中, 由于DIC与Pd(Ⅱ)的配位, 从而使胺的亲核性得到提高, 有利于胺与碳二亚胺的亲核加成, 因此, 29经过亲核加成和分子内质子化形成中间体30. 30与两分子胺配位, 释放两分子胍的同时再生28.第二种机理可能经历分步的过程.该过程主要决定于中间体28的酸性, 也决定了中间体上氢原子对碳二亚胺的质子化, 从而活化碳二亚胺, 使其容易接受苯胺的亲核进攻, 再经分子内质子转移而完成整个催化循环.
由于胍分子中含有2个氨基, 它可作为咪唑、三嗪和嘧啶等杂环的起始原料[29].胍与双官能团化合物如二酮、乙酰丙酮酸酯和丙二腈缩合, 消除氨基或水, 生成α-氨基杂环化合物, 如以不同的芳香基取代胍中间体同烯胺酮通过Bredereck缩合可得到种类众多的2-胺基取代嘧啶类环状胍化合物[30].这些环状胍类化合物因其稳定性高, 生物活性良好等特性, 在医药、农药和材料等行业中得到了广泛的应用[31].
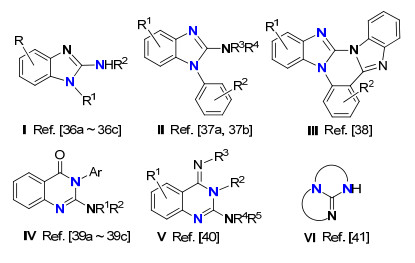
环状胍一般通过联二胺与胍类化合物成环、氮杂环丙烷和碳二亚胺环加成反应以及三苯基膦等磷试剂和叠氮化合物发生氮杂维蒂希反应制备[32].胺与碳二亚胺通过过渡金属催化直接成环状胍一般是通过多组分一锅反应而制备.胺与碳二亚胺通过这种串联反应所合成的典型环状胍如Figure 1所示, 涉及到的成胍机理也有所不同[33~39].
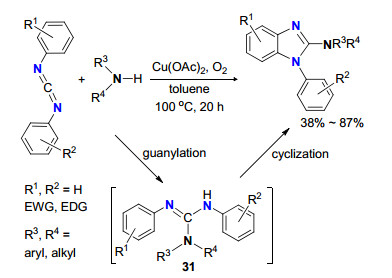
Bao等[33a, 33b]以碳二亚胺和胺为起始原料, 醋酸铜为催化剂, 通过串联成胍反应和C—H活化构筑C—N键直接合成2-氨基苯并咪唑环状胍(Scheme 14).碳二亚胺与胺首先进行成胍反应生成中间体31, 31发生C—H活化/分子内C—N偶联反应生成环状胍化合物.该反应条件能兼容脂肪族和芳香族, 具有产率高、原料易得等优点.
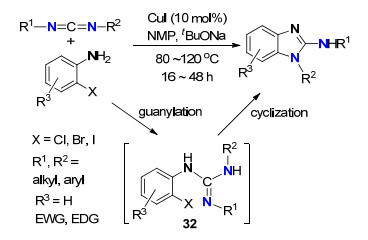
经由胍中间体的C—H或C—X键胺化已经被报道[34]. 2011年, Xi等[35]发展了CuI催化的邻卤代苯胺和碳二亚胺的串联反应高效合成了2-氨基苯并咪唑化合物(Scheme 15).胺和碳二亚胺经成胍反应生成中间体32, 32在铜催化下经分子内乌尔曼偶联生成2-氨基苯并咪唑.该反应产率高, 底物邻卤苯胺类化合物普适性广, 对称和不对称的碳二亚胺均可使用, 并具有好的立体选择性.
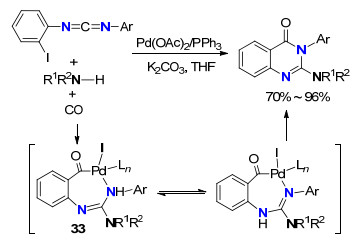
Alper等[36]报道了采用醋酸钯作催化剂, 邻卤代芳基碳二亚胺、胺和CO通过串联成胍反应、羰基插入、分子内环化合成了六元环状胍2-异喹唑啉-4(3H)-酮(Scheme 16).该反应产率高, 反应条件温和.反应历程经历: (1)胺与邻卤代芳基碳二亚胺发生成胍反应生成胍; (2)在钯催化剂作用下, 通过氧化加成/CO插入/还原消除生成最终产物, 其中33为中间体Pd配合物.进一步, 他们通过类似的反应机理, 以双-邻卤代芳基碳二亚胺、胺和CO为反应物, 有效地合成了一类双环六元胍喹唑啉类化合物.
值得指出的是, 也可以用羰基金属有机化合物代替CO合成2-异喹唑啉-4(3H)-酮化合物[37]. Wu等[38]发展了以异腈代替CO, 通过类似的机理合成了一类六元环状胍喹唑啉-4(3H)-亚氨基化合物(Eq. 3).
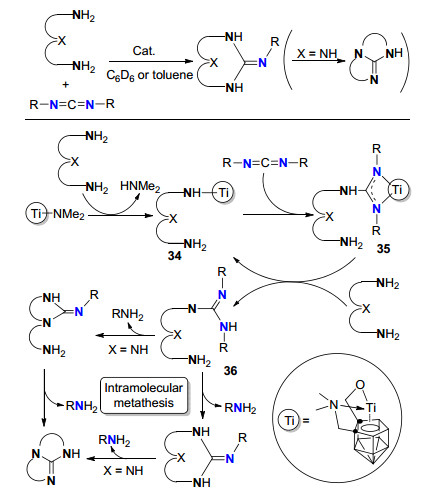
2006年, Xie等[39]报道了碳硼烷基-烷氧基钛胺基配合物催化胺与碳二亚胺成胍反应.当胺为二胺或三胺时能有效构建单环/双环胍.该类反应的可能机理如Scheme 17.首先碳二亚胺插入到中间体[Ti-N]配合物34中形成胍中间体35, 35通过质子化生成胍36, 然后36发生分子内复分解反应生成环状胍产物.
|
|
(3) |
经典的成胍方法主要是胺、醇或卤代烷与胍基化试剂通过多步反应而成, 虽然已经取得了很大的进展, 但主要存在合成路线长, 分离提纯过程繁琐, 产率低, 同时有些合成方法采用有毒的汞盐, 因此采用胺与碳二亚胺直接成胍就显得尤为重要.自从2003年Richeson教授首次报道芳香伯胺与碳二亚胺通过Ti=N氮宾配合物催化直接成胍以来, 成胍的方法与手段越来越多, 特别是在催化剂设计、溶剂、反应温度和时间的选择、底物范围的筛选以及相应的机理的探讨等方面, 很多课题组做出了突出的贡献.本文以过渡金属催化胺与碳二亚胺成胍为主线, 综述了成胍的研究进展, 特别是近十多年来所取得的成就.
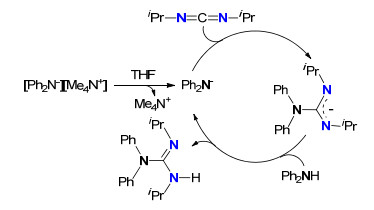
通过文献梳理发现, 虽然直接成胍已经取得了很大的进展, 但仍然存在一些问题.一些高效的催化剂, 存在对水、氧特别敏感, 成胍反应必须在无水无氧条件下进行; 过渡金属催化剂虽然能基本满足催化胺与碳二亚胺成胍的要求, 但过渡金属的毒性、残留等对最终产物产生不利影响, 成为一个非常棘手的问题, 开发无金属催化剂将是一个新的课题, 这方面的工作已经有所报道[40].最近, Harder等[41]使用[Ph2N-][Me4N+]作为胺与碳二亚胺成胍催化剂, 发现虽然催化效率比金属催化剂低, 但这是第一例无金属参与的成胍反应.其提出的反应机理如Scheme 18所示, 其反应机理与金属参与的成胍反应机理不同.反应的第一步是[Ph2N-][Me4N+]在溶剂THF的作用下, 解离成阳离子Me4N+和阴离子Ph2N-, 阴离子Ph2N-对碳二亚胺亲核加成形成胍阴离子, 然后与Ph2NH质子化, 释放产物胍的同时再生阴离子Ph2N-. DFT计算表明, 阴离子Ph2N-对碳二亚胺亲核加成经历的过渡态可能包含[Me4N]+、[Ph2N]-和碳二亚胺三种物种或者包含[Ph2N]-和碳二亚胺两种物种, 但是以后者比较合理.
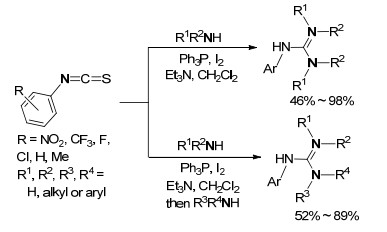
2016年, Phakhodee等[42]以异硫氰酸芳基酯和胺为原料, 以Ph3P-I2/Et3N为催化反应体系, 一锅法高效合成三取代胍(Scheme 19), 这些为以后的研究方向提供了一条全新的思路.
另外, 底物的普适性仍然是一个需要深入研究的课题, 对于亲核性较弱或有大位阻的胺以及大位阻的碳二亚胺的成胍反应对条件比较苛刻; 目前直接成胍的催化剂主要是均相催化剂, 而对于有利于回收和再利用的非均相催化剂研究不多, 因此设计和开发高活性的均相和非均相催化剂, 扩大底物范围, 从而合成出性能优异的胍类化合物仍然需要进一步深入研究.
(a) Berlinck, R. G. S. Nat. Prod. Rep. 1999, 16, 339.
(b) Blondeau, P. ; Segura, M. ; Pérez-Fernández, R. ; de Mendoza, J. Chem. Soc. Rev. 2007, 36, 198.
(c) Berlinck, R. G. S. ; Burtoloso, A. C. B. ; Kossuga, M. H. Nat. Prod. Rep. 2008, 25, 919.
(d) Coles, M. P. Chem. Commun. 2009, 3659.
(e) Berlinck, R. G. S. ; Burtoloso, A. C. B. ; Trindade-Silva, A. E. ; Romminger, S. ; Morais, R. P. ; Bandeira, K. ; Mizuno, C. M. Nat. Prod. Rep. 2010, 27, 1871.
(f) Castagnolo, D. ; Schenone, S. ; Botta, M. Chem. Rev. 2011, 111, 5247.
(g) Berlinck, R. G. S. ; Trindade-Silva, A. E. ; Santos, M. F. C. Nat. Prod. Rep. 2012, 29, 1382.
(h) Berlinck, R. G. S. ; Trindade-Silva, A. E. ; Santos, M. F. C. Nat. Prod. Rep. 2012, 29, 1382.
(i) Peng, K. ; Ding, W. ; Tu, W. ; Hu, J. ; Liu, C. ; Yang, J. Acta Chim. Sinica 2016, 74, 713(in Chinese).
(彭开美, 丁伟, 涂伟萍, 胡剑青, Liu Chao, Yang Jian, 化学学报, 2016, 74, 713. )
(j) Xu, X. ; Chen, X. ; Li, J. ; Xu, W. ; Zhang, Y. Chin. J. Org. Chem. 2016, 36, 1985(in Chinese).
(徐树英, 陈小佳, 黎吉辉, 许文茸, 张玉苍, 有机化学, 2016, 36, 1985. )
(a) Selig, P. Synthesis 2013, 45, 703.
(b) Fu, X. ; Tan, C. -H. Chem. Commun. 2011, 47, 8210.
(c) Taylor, J. E. ; Bull, S. D. ; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109.
(a) Edelmann, F. T. Coord. Chem. Rev. 1994, 137, 403.
(b) Zhou, Y. ; Yap, G. P. A. ; Richeson, D. S. Organometallics 1998, 17, 4387.
(c) Luo, Y. ; Yao, Y. ; Shen, Q. Macromolecules 2002, 35, 8670.
(d) Coles, M. P. Dalton Trans. 2006, 985.
(e) Zhang, J. ; Zhou, X. C. R. Chim. 2010, 13, 633.
(f) Zhang, J. ; Zhou, X. Dalton Trans. 2011, 40, 9637.
(a) Alonso-Moerno, C. ; Antiñolo, A. ; Carrillo-Hermosilla, F. ; Otero, A. Chem. Soc. Rev. 2014, 43, 3406.
Molina, P.; Aller, E.; Lorenzo, A. Synlett 2003, 714. http://www.arkat-usa.org/get-file/19748
Ong, T.-G.; Yap, G. P. A.; Richeson, D. S. J. Am.Chem.Soc. 2003, 125, 8100. doi: 10.1021/ja035716j
(a) Zhang, W. -X. ; Nishiura, M. ; Hou, Z. Synlett 2006, 1213.
(b) Zhang, W. -X. ; Nishiura, M. ; Hou, Z. Chem. Eur. J. 2007, 13, 4307.
(c) Zhang, W. -X. ; Hou, Z. Org. Biomol. Chem. 2008, 6, 1720.
(d) Suzuki, T. ; Zhang, W. -X. ; Nishiura, M. ; Hou, Z. J. Synth. Org. Chem. Jpn. 2009, 67, 451.
(e) Nishiura, M. ; Hou, Z. Bull. Chem. Soc. Jpn. 2010, 83, 595.
(a) Zhang, W. -X. ; Xu, L. ; Xi, Z. Chem. Commun. 2015, 51, 254.
(b) Xu, L. ; Zhang, W. -X. ; Xi, Z. Organometallics 2015, 34, 1787.
(c) Chi, Y. ; Xu, L. ; Du, S. ; Yan, H. ; Zhang, W. -X. ; Xi, Z. Chem. Eur. J. 2015, 21, 10369.
(d) Zhang, W. -X. ; Li, D. ; Wang, Z. ; Xi, Z. Organometallics 2009, 28, 882.
(a) Du, Z. ; Li, W. ; Zhu, X. ; Xu, F. ; Shen, Q. J. Org. Chem. 2008, 73, 8966.
(b) Zhu, X. ; Du, Z. ; Xu, F. ; Shen, Q. J. Org. Chem. 2009, 74, 6347.
(c) Cao, Y. ; Du, Z. ; Li, W. ; Li, J. ; Zhang, Y. ; Xu, F. ; Shen, Q. Inorg. Chem. 2011, 50, 3729.
(d) Li, Z. ; Xue, M. ; Yao, H. Sun, H. ; Zhang, Y. Shen, Q. J. Organomet. Chem. 2012, 713, 27.
(e) Cai, L. ; Yao, Y. ; Xue, M. ; Zhang, Y. ; Shen, Q. Appl. Organomet. Chem. 2013, 27, 366.
(f) Tu, J. ; Li, W. ; Xue, M. ; Zhang, Y. ; Shen, Q. Dalton Trans. 2013, 42, 5890.
(a) Liu, C. ; Zhou, S. ; Wang, S. ; Zhang, L. ; Yang, G. Dalton Trans. 2010, 39, 8994.
(b) Wu, Y. ; Wang, S. ; Zhang, L. ; Yang, G. ; Zhu, X. ; Zhou, Z. ; Zhu, H. ; Wu, S. Eur. J. Org. Chem. 2010, 326.
(a) Shen, H. ; Chan, H. -S. ; Xie, Z. Organometallics 2006, 25, 5515.
(b) Shen, H. ; Xie, Z. J. Organomet. Chem. 2009, 694, 1652.
(c) Shen, H. ; Wang, Y. ; Xie, Z. Org. Lett. 2011, 13, 4562.
Schweizer, P. D.; Wadepohl, H.; Gade, L. H. Oganometallics 2013, 32, 3697. doi: 10.1021/om400323p
Montilla, F.; Pastor, A.; Galindo, A. J.Organomet.Chem. 2004, 689, 993. doi: 10.1016/j.jorganchem.2004.01.005
Romero-Fernández, J.; Carrillo-Hermosilla, F.; Antiñolo, A.; Alonso-Moreno, C.; Rodríguez, A. M.; López-Solera, I.; Otero, A. Dalton Trans. 2010, 39, 6419. doi: 10.1039/c0dt00064g
Montilla, F.; del Río, D.; Pastor, A.; Galindo, A. Organometallics 2006, 25, 4996. doi: 10.1021/om060535m
Mukherjee, A.; Sen, T. K.; Mandal, S. K.; Maity, B.; Koley, D. RSC Adv. 2013, 3, 1255. doi: 10.1039/C2RA21778C
Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; Suά;rea, F. J.; López-Solera, I.; Fernά;ndez-Galά;n, R.; Villaenño, E. Dalton Trans. 2013, 42, 8223. doi: 10.1039/c3dt50477h
(a) Li, D. ; Guang, J. ; Zhang, W. -X. ; Wang, Y. ; Xi, Z. Org. Biomol. Chem. 2010, 8, 1816.
(b) Li, D. ; Wang, Y. ; Zhang, W. -X. ; Zhang, S. ; Guang, J. ; Xi, Z. Organometallics 2011, 30, 5278.
(a) Alonso-Moreno, C. ; Carrillo-Hermosilla, F. ; Garcés, A. ; Otero, A. ; López-Solera, I. ; Rodríguez, A. M. ; Antiñolo, A. Organometallics 2010, 29, 2789.
(b) Bravo, I. ; Alonso-Moreno, C. ; Posadas, I. ; Albaladejo, J. ; Carrillo-Hermosilla, F. ; Ceña, V. ; Garzón, A. ; López-Solera, I. ; Romero-Castillo, L. RSC Adv. 2016, 6, 8267.
Bhattacharjee, J.; Sachdeva, M.; Banerjee, I.; Panda, T. K. J. Chem.Sci. 2016, 128, 875. doi: 10.1007/s12039-016-1096-y
Kantam, M. L.; Priyadarshini, S.; Joseph, P. J. A.; Srinivas, P.; Vinu, A.; Klabunde, K. J.; Nishina, Y. Tetrahedron 2012, 68, 5730. doi: 10.1016/j.tet.2012.05.044
Pottabathula, S.; Royo, B. Tetrahedron Lett. 2012, 53, 5156. doi: 10.1016/j.tetlet.2012.07.065
Tan, D.; Mottillo, C.; Katsenis, A. D.; Štrukil, V.; Friščić, T. Angew. Chem., Int.Ed. 2014, 126, 9475. doi: 10.1002/ange.201404120
Grirrane, A.; Garcia, H.; Corma, A.; Álvarez, E.Chem. Eur.J. 2012, 18, 14934. doi: 10.1002/chem.v18.47
Grirrane, A.; Garcia, H.; Álvarez, E. Beilstein J.Org. Chem. 2013, 9, 1455. doi: 10.3762/bjoc.9.165
Tayama, E.; Ishikawa, M.; Iwamoto, H.; Hasegawa, E. Tetrahedron Lett. 2012, 53, 5159. doi: 10.1016/j.tetlet.2012.07.070
Yavari, I.; Sodagar, E.; Nematpour, M.; Askarian-Amiri, M. Synlett 2015, 26, 1230. doi: 10.1055/s-00000083
Frindy, S.; Kadib, A. E.; Lahcini, M.; Primo, A.; García, H. ACS Catal. 2016, 6, 3863. doi: 10.1021/acscatal.6b00995
(a) Rauws, T. R. M. ; Maes, B. U. W. Chem. Soc. Rev. 2012, 41, 2463.
(b) Mulcahy, J. V. ; Bois, J. D. J. Am. Chem. Soc. 2008, 130, 12630.
(c) Aron, Z. D. ; Overman, L. E. Chem. Commun. 2004, 253.
(d) Gainer, M. J. ; Bennett, N. R. ; Takahashi, Y. ; Looper, R. E. Angew. Chem., Int. Ed. 2011, 50, 684.
(a) Ishikawa, T. ; Kumamoto, T. Synthesis 2006, 737.
(b) Heys, L. ; Moore, C. G. ; Murphy, P. J. Chem. Soc. Rev. 2000, 29, 57.
(a) Schmuck, C. Coord. Chem. Rev. 2006, 250, 3053.
(b) Edelmann, F. T. Chem. Soc. Rev. 2012, 41, 7657.
(c) Turočkin, A. ; Honeker, R. ; Raven, W. ; Selig, P. J. Org. Chem. 2016, 81, 4516.
(a) Isobe, T. ; Fukuda, K. ; Tokunaga, T. ; Seki, H. ; Yamaguchi, K. ; Ishikawa, T. J. Org. Chem. 2000, 65, 7774.
(b) Butler, D. C. D. ; Inman, G. A. ; Alper, H. J. Org. Chem. 2000, 65, 5887.
(c) Heinelt, U. ; Schultheis, D. ; Jäger, S. ; Lindenmaier, M. ; Pollex, A. ; Beckmann, H. S. G. Tetrahedron 2004, 60, 9883.
(d) Berlinck, R. G. S. ; Kossuga, M. H. Nat. Prod. Rep. 2005, 22, 516.
(e) Kim, M. ; Mulcahy, J. V. ; Espino, C. G. ; Bois, J. D. Org. Lett. 2006, 8, 1073.
(f) Blackburn, C. ; Achab, A. ; Elder, A. ; Ghosh, S. ; Guo, J. ; Harriman, G. ; Jones, M. J. Org. Chem. 2005, 70, 10206.
(g) Hirota, S. ; Kato, R. ; Suzuki, M. ; Soneta, Y. ; Otani, T. ; Saito, T. Eur. J. Org. Chem. 2008, 2075.
(a) Lv, X. ; Bao, W. J. Org. Chem. 2009, 74, 5618.
(b) He, H. -F. ; Wang, Z. -J. ; Bao, W. Adv. Synth. Catal. 2010, 352, 2905.
(c) Yuan, G. ; Liu, H. ; Gao, J. ; Xu, H. ; Jiang, L. ; Wang, X. ; Lv, X. RSC Adv. 2014, 4, 21904.
(d) Xu, B. ; Peng, B. ; Cai, B. ; Wang, S. ; Wang, X. ; Lv, X. Adv. Synth. Catal. 2016, 358, 653.
(a) Evindar, G. ; Batey, R. A. Org. Lett. 2003, 5, 133.
(b) Deng, X. ; McAllister, H. ; Mani, N. S. J. Org. Chem. 2009, 74, 5742.
(c) Saha, P. ; Ramana, T. ; Purkait, N. ; Ali, M. A. ; Paul, R. ; Punniyamurthy, T. J. Org. Chem. 2009, 74, 8719.
(d) Yuan, G. ; Liu, H. ; Gao, J. ; Yang, K. ; Niu, Q. ; Mao, H. ; Wang, X. ; Lv, X. J. Org. Chem. 2014, 79, 1749.
(e) Chi, Y. ; Zhang, W. -X. ; Xi, Z. Org. Lett. 2014, 16, 6274.
Wang, F.; Cai, S.; Liao, Q.; Xi, C. J.Org.Chem. 2011, 76, 3174. doi: 10.1021/jo200014v
(a) Zeng, F. ; Alper, H. Org. Lett. 2010, 12, 1188.
(b) Zeng, F. ; Alper, H. Org. Lett. 2010, 12, 3642.
Roberts, B.; Liptrot, D.; Luker, T.; Stocks, M. J.; Barber, C.; Webb, N.; Dods, R.; Martin, B. Tetrahedron Lett. 2011, 52, 3793. doi: 10.1016/j.tetlet.2011.05.052
Qiu, G.; Liu, G.; Pu, S.; Wu, J. Chem.Commun. 2012, 48, 2903. doi: 10.1039/c2cc18001d
Shen, H.; Wang, Y.; Xie, Z. Org.Lett. 2011, 13, 4562. doi: 10.1021/ol201752e
Baishya, A.; Peddarao, T.; Barman, M. K.; Nembenna, S. New J. Chem. 2015, 39, 7503. doi: 10.1039/C5NJ01612F
Penafiel, J. ; Maron, L. ; Harder, S. Angew. Chem., Int. Ed. 2015, 54, 201.
Wangngae, S. ; Pattarawarapan, M. ; Phakhodee, W. Synlett 2016, 27, 1121.

图式 1 基于不同成胍机理的过渡金属催化剂分类
Scheme 1 Transition metal catalysts for guanylation classified on the basis of different mechanisms

图式 2 芳香胺与碳二亚胺经钛Ti=N氮宾配合物催化的[2+2]成胍机理
Scheme 2 [2+2]-Cycloaddition mechanism for guanylation reaction of aromatic amines with carbodiimides by Ti=N catalysts

图式 3 钛肼亚胺配合物催化的成胍机理
Scheme 3 Aminoguanylation mechanism catalyzed by titanium hydrazinediido complexes

图式 4 钒配合物催化胺与碳二亚胺的成胍机理
Scheme 4 Guanylation mechanism of amines with carbodiimides catalyzed by vanadium compounds

图式 5 Zn(OTf)2催化胺与碳二亚胺的成胍机理
Scheme 5 Zn(OTf)2-catalyzed guanylation mechanism of amines with carbodiimides

图式 6 ZnEt2催化的胺与碳二亚胺成胍机理
Scheme 6 ZnEt2-catalyzed guanylation mechanism of amines with carbodiimides

图式 7 铁及亚铁化合物催化胺与碳二亚胺成胍机理
Scheme 7 Fe-catalyzed guanylation mechanism of amines with carbodiimides

图式 8 锌配合物催化的胺与碳二亚胺成胍机理
Scheme 8 Zn-catalyzed guanylation mechanism of amines with carbodiimides

图式 9 CuCl催化的苯磺酰胺与碳二亚胺的成胍反应
Scheme 9 Mechanochemical CuCl-catalyzed guanylation of arylsulfonamides with carbodiimides

图式 10 纳米CuO催化成胍反应及进一步转化
Scheme 10 Nano-CuO-catalyzed guanylation of amine with carbodiimides and further transformation for the synthesis of iminoguanidines

图式 11 纳米ZnO催化的胺与碳二亚胺成胍机理
Scheme 11 Nano ZnO-catalyzed guanylation mechanism of amines with carbodiimides

图式 12 非均相纳米Pd催化胺与碳二亚胺成胍机理
Scheme 12 Heterogeneous nano-Pd-catalyzed guanylation of amine with carbodiimides

图式 13 均相Pd(Ⅱ)催化胺与碳二亚胺成胍的两种可能机理
Scheme 13 Two possible guanylation mechanisms catalyzed by PdCl2(MeCN)2 in a homogeneous phase

图式 14 Cu(Ⅱ)催化胺与碳二亚胺成胍/C—N键形成反应
Scheme 14 Cu(Ⅱ)-catalyzed guanylation/N-arylation of amines with carbodiimides

图式 15 Cu(Ⅰ)催化胺与碳二亚胺成胍/C—N键形成反应
Scheme 15 Cu(Ⅰ)-catalyzed guanylation/N-arylation of amines with carbodiimides

图式 16 Pd(Ⅰ)催化胺与碳二亚胺成胍/CO插入/C—N键形成反应
Scheme 16 Palladium-catalyzed guanylation/cyclocarbonylation of amines, carbodiimides and CO

图式 17 钛氨基配合物催化胺与碳二亚胺成胍/复分解环化反应
Scheme 17 Titanium amido complex catalyzed guanylation/ metathesis cyclization

图式 18 [Ph2N-][Me4N+]催化二苯基胺与N, N'-二异丙基碳二亚胺的成胍反应机理
Scheme 18 Guanylation mechanism of diphenylamine with N, N'-diisopropylcarbodiimide catalyzed by [Ph2N-][Me4N+]

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:

