Table 1.
Optimization of the reaction condition for the deprotection of N-Boc groupa
Citation:
Zeng Yijie, Duan Yue, Zhao Hui, Hu Xiangguo. Efficient and Chemoselective Deprotection of N-t-Butyloxycarbonyl Group Mediated by Selectfluor[J]. Chinese Journal of Organic Chemistry,
2018, 38(7): 1712-1717.
doi:
10.6023/cjoc201801036

Selectfluor介导的高效高选择性地脱除叔丁氧羰基方法研究
摘要:
商品化的selectfluor[即1-氯甲基-4-氟-1,4-重氮化二环2.2.2辛烷双(四氟硼酸)盐]具有不吸潮的特性,是一种被应用得最为广泛的氟代试剂.因此,发现并理解其新的反应特性对于应用该试剂具有非常重要的意义.本工作报道了selectflour可以用于选择性脱除双保护氨基上的一个叔丁氧羰基保护基.该方法具有条件温和、操作简单、化学选择性好等特点.跟其他方法相比,该脱保护方法有一定的应用价值,被成功应用于一系列氨基酸衍生物的脱保护,在医药上合成有潜在用途的嘌呤衍生物.以氘代乙腈为溶剂的核磁反应实验解释了为什么该反应需要等物质的量的selectfluor试剂.
-
关键词:
- selectfluor
- / 叔丁氧羰基
- / 脱保护
- / 选择性
- / 合成
English
Efficient and Chemoselective Deprotection of N-t-Butyloxycarbonyl Group Mediated by Selectfluor
Abstract:
Selectfluor, 1-chloromethyl-4-fluoro-1, 4-diazoniabicyclo-[2.2.2] octane bis(tetrafluoroborate), is among the most popular fluorinating reagents owning to its commercially availability and non-hygroscopic property. The discovery and understanding of new reactivities of selectfluor are thus important for reaction design and optimization when this popular reagent is employed. It has been found that selectfluor could selectively remove Boc group from doubly protected amines in acetonitrile. This deprotection could be of interest when compared to other reported methods, not only because selectfluor is a solid and easy-to-handle, but also because the reaction is mild, operationally simple and chemoselective. The potential usefulness of this method is demonstrated by the deprotection of a series of protected amino acids and a one-step synthesis of pharmaceutically important purine derivative. The NMR experiments conducted in CD3CN explain why stoichiometric amount of selectfluor is needed for a successful reaction.
-
Key words:
- selectfluor
- / t-butyloxycarbonyl
- / deprotection
- / selectivity
- / synthesis
-
Selectfluor, (1-chloromethyl-4-fluoro-1, 4-diazoniabi- cyclo-[2.2.2]octane bis(tetrafluoroborate), is among the most popular electrophilic fluorinating reagents owning to its commercial availability and non-hygroscopic property.[1] At the same time, the relatively weak N—F bond (homolytic bond dissociation energy: 254.9 kJ/mol in CH3CN) also renders it as a powerful radical fluorine source in the presence of photocatalysts or transition-metal catalysts.[2] Furthermore, it can also be used as an oxidant owing to its strong oxidative property.[3] The discovery and understanding of new reactivities of selectfluor are thus important for reaction design and optimization when this popular reagent is employed.
The t-butyloxycarbonyl (Boc) group is one of the most commonly used protective group of amine in organic synthesis, because of the ease of incorporation and removal of this group when necessary, as well as its chemical orthogonality with other common amine protecting groups such as carboxybenzyl (Cbz) and fluorenylmethyloxycarbonyl (Fmoc). Although it is common to protect an amine group with mono-Boc group, double protection of amine with N, N-di-Boc or N, N-Boc, carbamate group is necessary under various circumstances. For example, di-Boc protected amines are frequently used as convenient phthalimide equivalents in Mitsunobu and Gabriel reactions.[4] Double protection of amine with Boc is also required to resist strong oxidation conditions.[5] The selective deprotection of Boc group from doubly protected amine is hence desirable. Although the deprotection of Boc protecting group is well documented, [4] only a few methods are suitable for the selective removal of Boc group from a doubly protected amine.[6]
Herein, we report a new mode of reactivity of selectfluor. We demonstrate for the first time that selectfluor can selectively remove Boc group from di-protected amines in acetonitrile under mild conditions in a chemoselective fashion.
1. Results and discussion
As a continuing interest in stereoselectively fluorinated compounds, [7] recently we conducted the photo-catalyzed benzylic fluorination developed by Chen and co-workers, [8] using di-Boc protected phenyl alanine 1 as the substrate. To our surprise, no desired product 3 was observed after 24 h, but the Boc deprotected compound 2 was obtained instead in low yield (Eq. 1). We quickly realized the potential of selectfluor for the selective removal of a Boc group, and set out for further investigation.

(1) 
Initially, We chose compound 1 for the optimization of reaction condition. When 1 equiv. of selectfluor reagent in acetonitrile was used, 50 ℃ was needed to get a full conversion and the desired product 2 was obtained in 85% yield (Table 1, Entry 2). Increasing the temperature gave lower yield possibly due to the deprotection of the two Boc groups on compound 1 (Entry 3). Varying the amount of selectfluor showed that the highest yield was obtained when 1 equiv. of selectfluor was employed (Entries 4~6). Solvents screening showed that acetonitrile was the best choice for this transformation. Thus, the optimized conditions are as follows: substrates (0.25 mmol), selectfluor (1.0 equiv.), and CH3CN (5 mL), stirring for 10 h at 50 ℃.
Table 1

 下载:
导出CSV
下载:
导出CSV

Entry Selectfluor/equiv. Solvent t/℃ Yield/% 1 1.0 CH3CN 40 76 2 1.0 CH3CN 50 85 3 1.0 CH3CN 70 77 4 0.5 CH3CN 50 60 5 0.8 CH3CN 50 78 6 2.0 CH3CN 50 85 7 1.0 THF 50 trace 8 1.0 EtOAc 50 trace 9 1.0 CHCl3 50 trace 10 1.0 CH2Cl2 50 trace 11 1.0 MeOH 50 13 12 1.0 DMSO 50 23 13 1.0 DMF 50 Trace a All of the reactions were carried out on 0.25 mmol scale for 10 h. With the optimized condition in hand, we next proceeded to test of the substrate scope. As shown in Table 2, deprotection of the Boc group from di-Boc protected phenylalanine 1 proceeded well to furnish the corresponding mono N-Boc products 2 in good yield (Table 2, Entry 1). Other carbamates protected phenylalanines 6, 8 and 10 were also viable substrates. In these cases, the N-Boc group was removed selectively in high yields, leaving the N-Cbz, N-CO2Me and N-CO2Et groups intact (Entries 2~4). It is interesting to note that this condition only deprotected N-Boc from the N, N-carbamate, Boc-protected substrates. N, N-Ts, Boc- and N, N-Ac, Boc-protected substrates 12, 14 were stable under the reaction conditions (Entries 5, 6). This interesting selectivity is obviously different from the previous methods and may be useful for the design of chemoselective reactions.[6c, 6e] The reaction condition is suitable for the deprotection of other protected amino acids, and compounds 16, 18, 20, 22, 24, 26, 28, 30 and 32 all underwent the deprotection smoothly to afford the desired products in good yields (Entries 7~15). It is noteworthy that the deprotection took place without affecting the chiral center of the α-amino acids tested. The comparison experiments showed that deprotected amino acids with one chiral center such as 2, 7, 9, 11 have identical specific rotation to the samples obtained from the free phenylalanine through selective protection, [9] and the 1H NMR of crude products 25 and 33 also indicated no detectable epimerization. Although O-Boc is generally believed to be much more labile than its N-counterpart, N-Boc group can be removed selectively under this condition (Entry 16). Other common protecting groups for hydroxyl, such as benzoyl (Bz) and tert-butyloxycarbonyl (TBS), were quite compatible with the reaction conditions (Entries 17, 18). The relative low yield of TBS-substrates might be due to the presence of small amount of fluoride ion (Entry 18). N-Boc could be also selectively removed from a benzyl amine substrate in good yield (Entry 19). Finally, this conditions could be used to prepare the mono-Boc protected purine 43 in one-step in 10 h. Previously, this pharmaceutically important compound needed to be prepared from 42 in two steps and 4 d.[10] This experiment thus demonstrated the potential usefulness of this method.
Table 2
Table 2. Test of the substrate scope下载:
导出CSV

Entry Substrate Product Yield/% 1 

85 2 

80 3 

84 4 

76 5 

0 6 

0 7 

77 8 

86 9 

83 10 

81 11 

73 12 

70 13 

77 14 

89 15 

78 16 

71 17 

72 18 

62 19 

82 20 

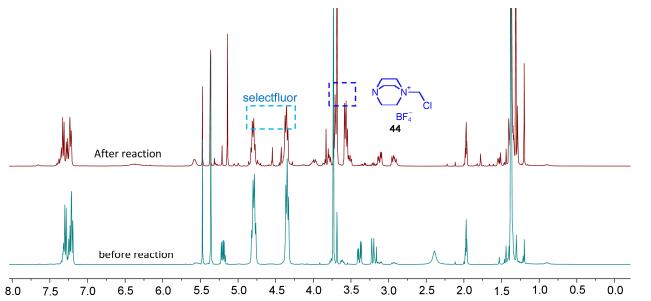
60 As for the mechanism, we postulated that selectfluor may serve as a Lewis acid to effect the deprotection of Boc group owning to the F+ nature of this reagent.[6a~6e, 11] Similar mechanism for the deprotection of doubly protected amino group with an iron catalyst has been reported previously.[6e] The need of stoichiometric amount of selectfluor may be due to the decomposition of this reagent under the reaction conditions. This was supported by the NMR experiment of 1 in CD3CN, in which significant amount of unidentified decomposed product (possibly 44, two peaks at around δ 3.5~4.0) of selectfluor (two peaks at around δ 4.4 and 4.75) was visible in the 400 MHz 1H NMR spectra (Figure 1).
Figure 1
3. Conclusions
Based on the unexpected observation when conducting the Chen’s procedure for the benzylic fluorination of 1, [8] It has been found for the first time that selectfluor could selectively remove a Boc group from doubly protected amines in acetonitrile. This deprotection could be of interest when compared to other reported methods, [6a~6e] not only because selectfluor is a solid and easy-to-handle, but also because the reaction is mild, operationally simple and chemoselective. The potential usefulness of this method is demonstrated by the deprotection of a series of proteceted amino acids and a one-step synthesis of pharmaceutically important purine 43. The NMR experiment conducted in CD3CN explains why stoichiometric amount of selectfluor is needed for a successful reaction. In addition to providing a new method for deprotection, the reactivity of selectfluor demonstrated in this work should be valuable for reaction design and optimization when this popular fluorinating reagent is employed.
4. Experimental section
4.1 General information
All reagents were used as received from commercial sources without further purification or prepared as described in the literature. All solvents were dried over 4 Å molecular sieves before use unless otherwise stated. Reactions were stirred using Teflon-coated magnetic stirring bars. Analytical thin layer chromatography (TLC) was performed with 0.20 mm silica gel 60 F plates with 254 nm fluorescent indicator. TLC plates were visualized by ultraviolet light or by treatment with a spray of Pancaldi reagent [(NH4)6MoO4, Ce(SO4)2, H2SO4, H2O] or a solution 0.5% (V/V) ninhydrin in n-butanol. Chromatographic purification of products was carried out by flash column chromatography on silica gel (230~400 mesh). Melting points were determined using a WRX-4 visual melting point apparatus. Both melting points and boiling points are uncorrected. Infrared spectra were recorded on an IR Affinity-1. NMR spectra were measured in CDCl3 (with TMS as internal standard) or D2O or MeOD on a Bruker AV400 (1H at 400 MHz, 13C at 100 MHz, 19F at 376 MHz) magnetic resonance spectrometer. High-resolution mass spectra (HRMS) were recorded on a SYNAPT G2Si High Definition MS System. The HRMS were measured under ESI model (specified in the section of characterization data).
4.2 General procedures for selective deprotection of Boc
To a stirred solution of corresponding amine (1 equiv.) in dry MeCN was added selectfluor (1 equiv.) in one portion. Then the mixture was stirred for 24 h at 50 ℃, followed by quenching with ethyl acetate and water (V:V=1:1). The organic phase was washed with brine, dried with MgSO4, and the solvent was evaporated under reduced pressure after filtering. Then the crude product was purified by flash chromatography on silica gel(hexane/EtOAc) to give the deprotected compounds. The NMR data of compounds 2, 7, 9, 11, 17, 19, 21, 23, 25, 27 are shown below. For the data of other compounds please see the Supporting Information.
(S)-Methyl 2-[(tert-butoxycarbonyl)amino]-3-phenyl- propanoate (2): Colorless liquid. [α]D25+40.8 (c 1.0, CHCl3) [lit.[6b] [α]D25+18.4 (c 1.54, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 7.25~7.04 (m, 5H), 4.90 (d, J=7.29 Hz, 1H), 4.58~4.46 (m, 1H), 3.64 (s, 3H), 3.07~2.95 (m, 2H), 1.35 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 171.3, 154.0, 135.0, 128.3, 127.5, 126.0, 78.9, 53.4, 51.2, 37.3, 27.3.
(S)-Methyl 2-(((benzyloxy)carbonyl)amino)-3-phenyl- propanoate (7):[12] Colorless liquid. [α]D25-22.3 (c 0.8, MeOH) [lit.[13] [α]D30-14.9 (c 1.0, MeOH)]; 1H NMR (400 MHz, CDCl3) δ: 7.34~7.26 (m, 10H), 5.22 (d, 1H), 5.09 (d, J=2.69 Hz, 2H), 4.66 (ddd, J=11.8, 5.9, 5.9 Hz, 1H), 3.72 (s, 3H), 3.17~3.03 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 172.0, 155.6, 129.3, 128.6, 128.5, 128.2, 128.1, 127.2, 67.0, 54.8, 52.4, 38.2.
(S)-Methyl 2-[(methoxycarbonyl)amino]-3-phenylpropa- noate (9): Colorless liquid. [α]D25+48.0 (c 1.0, CHCl3) [lit.[14] [α]D20+57.0 (c 1.0, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 7.31~7.10 (m, 5H), 5.21 (d, J=7.34 Hz, 1H), 4.67~4.62 (m, 1H), 3.71 (s, 3H), 3.65 (s, 3H), 3.15~3.04 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 172.1, 156.3, 135.8, 129.2, 128.6, 127.1, 54.8, 52.3, 52.2, 38.3.
(S)-Methyl 2-[(ethoxycarbonyl)amino]-3-phenylpropa- noate (11):[15] Colorless liquid. [α]D25+36.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.31~7.11 (m, 5H), 5.21 (s, 1H), 4.66~4.61(m, 1H), 4.09 (dd, J=14.20, 7.20 Hz, 2H), 3.70 (s, 3H), 3.15~3.04 (m, 2H), 1.21 (t, J=7.20 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 172.2, 155.9, 135.9, 129.2, 128.6, 127.1, 61.2, 54.7, 52.3, 38.3, 14.5.
Methyl 2-[(tert-butoxycarbonyl)amino]acetate (17):[16] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 5.62 (t, J=5.81 Hz, 1H), 3.68 (s, J=5.81 Hz, 2H), 3.74 (s, 3H), 1.45 (s, 9H); 13CNMR (100 MHz, CDCl3) δ: 170.8, 155.8, 79.5, 51.8, 42.0, 28.1.
(S)-Methyl 2-[(tert-butoxycarbonyl)amino]propanoate (19): Colorless liquid. [α]D25-5.2 (c 1.0, CHCl3) [lit.[6b] [α]D25-2.75 (c 1.0, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 5.08 (s, 1H), 4.33 (p, J=7.4 Hz, 1H), 3.75 (s, 3H), 1.45 (s, 9H), 1.37 (d, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 173.8, 155.1, 79.9, 52.3, 49.2, 28.3, 18.7.
(S)-Methyl 2-(((benzyloxy)carbonyl)amino)propanoate (21): Colorless liquid. [α]D25-37.5 (c 1.0, MeOH) [lit.[17] [α]D25-33.0 (c 1.0, MeOH)]; 1H NMR (400 MHz, CDCl3) δ: 7.41~7.27 (m, 5H), 5.24 (s, 1H), 5.04 (dd, J=14.22, 7.02 Hz, 1H), 3.65 (s, 3H), 1.52 (s, 3H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 171.2, 153.5, 151.1, 135.2, 128.5, 128.4, 128.3. 83.7, 68.8, 54.3, 52.2, 27.9, 15.6.
(S)-Methyl 2-[(tert-butoxycarbonyl)amino]-4-methyl- pentanoate (23):[18] Pale yellow liquid. [α]D25-7.3 (c 1.0, CHCl3) [lit.[19] [α]D25-4.21 (c 1.56, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 5.23 (d, J=8.91 Hz, 1H), 4.34~4.29 (m, 1H), 3.73 (s, 3H), 1.76~1.66 (m, 1H), 1.62~1.44 (m, 2H), 1.44 (s, 9H), 0.95 (d, J=2.32 Hz, 3H), 0.94 (d, J=2.32 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 173.8, 155.3, 79.4, 51.9, 41.5, 28.2, 24.6, 22.7, 21.7.
(2S, 3S)-Methyl 2-[(tert-butoxycarbonyl)amino]-3-me- thylpentanoate (25): Colorless liquid. [α]D25+18.3 (c 1.0, CHCl3) [lit.[20] [α]D25+16.6 (c 3.7, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 5.07 (d, J=9.1 Hz, 1H), 4.27 (dd, J=9.1, 4.9 Hz, 1H), 3.73 (s, 3H), 1.96~1.72 (m, 1H), 1.45 (s, 9H), 1.28~1.05 (m, 1H), 0.95~0.98 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 172.9, 155.5, 79.8, 57.9, 52.0, 38.0, 28.3, 25.0, 15.5, 11.6.
(S)-Dimethyl 2-[(tert-butoxycarbonyl)amino]-pentane- dioate (27):[21] Colorless oil. [α]D25+15.8 (c 1.2, CHCl3) [lit.[20] [α]D25+12.5 (c 2.0, CHCl3)]; 1H NMR (400 MHz, CDCl3) δ: 5.06 (d, J=7.74 Hz 1H), 4.26 (s, 1H), 3.68 (s, 3H), 3.61 (s, 3H), 2.41~2.27 (m, 2H), 2.20~2.06 (m, 1H), 1.95~1.82 (m, 1H), 1.37 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 172.2, 171.6, 154.3, 79.0, 51.8, 51.4, 50.8, 29.0, 27.3, 26.8.
Supporting Information General procedure for the synthesis of the substrates and 1H NMR, 13C NMR spectra of products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
-
-
[1]
(a) Banks, R. E. ; Mohialdinkhaffaf, S. N. ; Lal, G. S. ; Sharif, I. ; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595.
(b) Nyffeler, P. T. ; Duron, S. G. ; Burkart, M. D. ; Vincent, S. P. ; Wong, C. H. Angew. Chem., Int. Ed. 2005, 44, 192. -
[2]
(a) Rueda-Becerril, M. ; Sazepin, C. C. ; Leung, J. C. T. ; Okbinoglu, T. ; Kennepohl, P. ; Paquin, J. F. ; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026.
(b) Yin, F. ; Wang, Z. T. ; Li, Z. D. ; Li, C. Z. J. Am. Chem. Soc. 2012, 134, 10401.
(c) Yan, H. ; Zhu, C. Sci. China: Chem. 2017, 60, 214. -
[3]
Stavber, S. Molecules 2011, 16, 6432. doi: 10.3390/molecules16086432
-
[4]
Greene, T. W. ; Wuts, P. G. M. in Protective Groups in Organic Synthesis, Wiley, New York, 1999, pp. 518~525.
-
[5]
Deniau, G.; Slawin, A. M. Z.; Lebl, T.; Chorki, F.; Issberner, J. P.; van Mourik, T.; Heygate, J. M.; Lambert, J. J.; Etherington, L. A.; Sillar, K. T.; O'Hagan, D. Chem. Biochem. 2007, 8, 2265. http://www.thelancet.com/search/results?searchTerm=telomere&fieldName=ArticleTitleAbstractKeywords
-
[6]
(a) Yadav, J. S. ; Subba Reddy, B. V. ; Reddy, K. S. Synlett 2002, 468.
(b) Mohapatra, D. K. ; Durugkar, K. A. ARKIVOC 2005, 14, 20.
(c) Hernadez, J. N. ; Crisostomo, F. R. P. ; Martin, T. ; Martin, V. S. Eur. J. Org. Chem. 2007, 5050.
(d) Zheng, J. L. ; Yin, B. L. ; Huang, W. M. ; Li, X. P. ; Yao, H. Q. ; Liu, Z. G. ; Zhang, J. C. ; Jiang, S. Tetrahedron Lett. 2009, 50, 5094.
(e) Lopez-Soria, J. M. ; Perez, S. J. ; Hernandez, J. N. ; Ramirez, M. A. ; Martin, V. S. ; Padron, J. I. RSC. Adv. 2015, 5, 6647.
(f) Walters, M. A. ; Hoem, A. B. J. Org. Chem. 1994, 59, 2645.
(g) Hernández, J. N. ; Ramírez, M. A. ; Martín, V. S. J. Org. Chem. 2003, 68, 743.
(h) Boyle, T. P. ; Bremner, J. B. ; Coates, J. A. ; Keller, P. A. ; Pyne, S. G. Tetrahedron 2005, 61, 7271. -
[7]
(a) Hu, X. G. ; Thomas, D. S. ; Griffith, R. ; Hunter, L. Angew. Chem., Int. Ed. 2014, 53, 6176.
(b) Hu, X. G. ; Lawer, A. ; Peterson, M. B. ; Iranmanesh, H. ; Ball, G. E. ; Hunter, L. Org. Lett. 2016, 18, 662.
(c) Yan, N. ; Fang, Z. ; Liu, Q. Q. ; Guo, X. H. ; Hu, X. G. Org. Biomol. Chem. 2016, 14, 3469.
(d) Yan, N. ; Lei, Z. W. ; Su, J. K. ; Liao, W. L. ; Hu, X. G. Chin. Chem. Lett. 2017, 28, 467. -
[8]
Xia, J. B.; Zhu, C.; Chen, C. J. Am. Chem. Soc. 2013, 135, 17494. doi: 10.1021/ja410815u
-
[9]
Comparison of specific rotations was used in Ref. [11] for the same purpose.
-
[10]
Dey, S.; Garner, P. J. Org. Chem. 2000, 65, 7697. doi: 10.1021/jo000983i
-
[11]
(a) Liu, J. J. ; Wong, C. H. Tetrahedron Lett. 2002, 43, 4037.
(b) Shah, S. T. A. ; Singh, S. ; Guiry, P. J. J. Org. Chem. 2009, 74, 2179. -
[12]
Navarre, L.; Martinez, R.; Genet, J. P.; Darses, S. J. Am. Chem. Soc. 2008, 130, 6159. doi: 10.1021/ja710691p
-
[13]
Barrett, A. G. M.; Pilipauskas, D. J. Org. Chem. 1990, 55, 5170. doi: 10.1021/jo00304a035
-
[14]
Adams, H.; Bawa, R. A.; Jones, S. Org. Biomol. Chem. 2006, 4, 4206. doi: 10.1039/B610055D
-
[15]
Pandey, R. K.; Dagade, S. P.; Dongare, M. K.; Kumar, P. Synth. Commun. 2003, 33, 4019. doi: 10.1081/SCC-120026337
-
[16]
Heller, S. T.; Sarpong, R. Org. Lett. 2010, 12, 4572. doi: 10.1021/ol1018882
-
[17]
Wakasugi, K.; Iida, A.; Misaki, T.; Nishii, Y.; Tanabe, Y. Adv. Synth. Catal. 2003, 345, 1209. doi: 10.1002/(ISSN)1615-4169
-
[18]
Mandal, P. K.; McMurray, J. S. J. Org. Chem. 2007, 72, 6599. doi: 10.1021/jo0706123
-
[19]
Burk, M. J.; Allen, J. G. J. Org. Chem. 1997, 62, 7054. doi: 10.1021/jo970903j
-
[20]
Zheng, J.; Yin, B.; Huang, W.; Li, X.; Yao, H.; Liu, Z.; Zhang, J.; Jiang, S. Tetrahedron Lett. 2009, 50, 5094. doi: 10.1016/j.tetlet.2009.06.104
-
[21]
Tomita, K.; Oishi, S.; Ohno, H.; Fujii, N. Pept. Sci. 2008, 90, 503. doi: 10.1002/bip.20968
-
[1]
-
Table 1. Optimization of the reaction condition for the deprotection of N-Boc groupa
Entry Selectfluor/equiv. Solvent t/℃ Yield/% 1 1.0 CH3CN 40 76 2 1.0 CH3CN 50 85 3 1.0 CH3CN 70 77 4 0.5 CH3CN 50 60 5 0.8 CH3CN 50 78 6 2.0 CH3CN 50 85 7 1.0 THF 50 trace 8 1.0 EtOAc 50 trace 9 1.0 CHCl3 50 trace 10 1.0 CH2Cl2 50 trace 11 1.0 MeOH 50 13 12 1.0 DMSO 50 23 13 1.0 DMF 50 Trace a All of the reactions were carried out on 0.25 mmol scale for 10 h.  下载: 导出CSV
下载: 导出CSV
Table 2. Test of the substrate scope
Entry Substrate Product Yield/% 1 85 2 80 3 84 4 76 5 0 6 0 7 77 8 86 9 83 10 81 11 73 12 70 13 77 14 89 15 78 16 71 17 72 18 62 19 82 20 60
下载: 导出CSV
-


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 12
- 文章访问数: 1379
- HTML全文浏览量: 114
