图 1.
ATP竞争性抑制剂的代表结构
Figure 1.
Representative structures of ATP competitive inhibitors

阿尔茨海默病(Alzheimer’s disease, AD), 是在1906年首次由德国精神病学家和神经发病学家Alzheimer Alois发现, 并以其名字命名的中枢神经系统退行性疾病. AD患者主要临床表现为进行性认知功能障碍、记忆损害、智能下降、语言模糊和行为怪异等[1].目前由AD导致的痴呆占总痴呆病例的60%~80%[2].关于AD发病机制, 普遍认为衰老是最大的诱因[3].在我国60岁以上的人口中, 有70%~80%的老年人伴有不同程度的痴呆症状.由于AD患者日常行为能力完全受损, 不仅给患者家属带来情感上的强烈冲击, 而且对家庭经济造成沉重负担. 2015年在国际AD大会上发布的《痴呆病对全球的影响》报告显示[4], 全世界的老年痴呆患者超过4600万, 目前, 全球用于治疗痴呆的费用高达818亿美元, 而到2050年患病人数估计会增加到1.135亿, 这必然造成巨大的社会负担和经济损失.因此, 找到安全可靠的AD预防及治疗手段是目前的迫切需求.
AD的发病机制至今尚不明确, Alzheimer博士报告中描述的聚集在神经元周围的淀粉样斑块和神经元内的纤维状物质, 至今仍被认为是AD的经典病理特征.为了解释AD的发病机制, 研究工作者们提出了一系列的假说, 主要包括淀粉样蛋白级联假说、金属离子假说、氧化应激假说、遗传假说、胆碱能假说和GSK-3假说, 其中GSK-3假说是最被认可的一种机制.之前很多研究认为[5], 大脑中的淀粉样斑块是导致AD的主要因素, 但现在研究发现[6], 在健康人的大脑中添加淀粉样蛋白, 并不会导致受试者出现痴呆的临床症状; 而在少数AD患者的大脑中没有发现淀粉样斑块, 这些研究表明淀粉样斑块与AD的发生没有必然的联系.进一步研究表明[7], 由β分泌酶和γ分泌酶剪切淀粉样前体蛋白(APP)产生的可溶性β-淀粉样蛋白(Aβ)寡聚体或微型Aβ聚积具有神经毒性. Aβ可能在AD早期, 即淀粉样斑块、神经元纤维缠结和神经元死亡出现之前, 通过引起兴奋性或抑制性神经传递系统的失衡而导致突触功能的失调.然而, β分泌酶和γ分泌酶的水解功能对于多个生理过程至关重要, 抑制其活性或许会引起严重的毒副作用, 因而β和γ分泌酶可能不是治疗AD的最佳靶标[8].最近Underwood等[9]发现导致AD的驱动因素不是淀粉样蛋白, 而是tau蛋白. GSK-3同时参与tau蛋白过度磷酸化和Aβ水解过程, 因此成为研发抗老年痴呆药物的重要靶点和研究热点.
GSK-3是一种高度保守的苏氨酸/丝氨酸激酶, 在人体中参与细胞分裂、干细胞增殖、细胞凋亡(程序化细胞死亡)、细胞分化、昼夜节律(日常节奏活性)、转录和胰岛素作用等重要的生命活动[10]. GSK-3在体内主要以两种形式存在, 即GSK-3α和GSK-3β.这两种亚型虽然由不同的基因编码, 但二者的结构极其相似, 在激酶结构域中有98%的相似性, 而差异主要位于N-末端和C-末端的氨基酸序列, GSK-3α的N端富含甘氨酸的延伸使其具有较大的质量[11]. GSK-3α和GSK-3β虽然具有高度的结构相似性, 但两种异构体的功能并不完全相同. GSK-3β纯合子失活的小鼠由于肝脏和心脏缺陷是胚胎致死的, 然而GSK-3α缺失的小鼠是可以存活的[12].目前研究结果证实GSK-3β无论在生理学还是病理学上都是占优势的亚型[13], 因此关于GSK-3的研究主要集中在β亚型. GSK-3作为生物体内重要的蛋白激酶, 对体内100多种底物发生磷酸化作用[14].过表达的GSK-3使原本结合在微管上的tau蛋白过度磷酸化, 致使其脱离并累积形成成对的螺旋丝, 随后生成的神经元纤维缠结(NFTs)减弱神经元之间的突触的作用, 最终导致神经元死亡[15].另一方面, GSK-3还通过其对γ-分泌酶的作用以独特的方式诱导Aβ形成[16].目前, 已经有报道证实[17], GSK-3抑制剂在体外和动物模型中可以促进成年海马神经发生, 诱导神经干细胞增殖、迁移和分化. GSK-3的失调可以解释AD中一些早期认知缺陷.这些结果和观察表明GSK-3抑制剂的成功开发有望用于治疗早期和已经有明显病理特征的AD以及其他神经退行性疾病.
迄今为止, GSK-3抑制剂主要是利用化学方法合成得到的, 另外一些是从天然产物中提取的小分子化合物, 但大部分都处于早期研究阶段, 只有极少数进入临床试验.目前报道的GSK-3抑制剂按照作用机制分类主要包括ATP竞争性抑制剂、非ATP竞争性抑制剂、底物竞争性抑制剂、变构抑制剂、不可逆抑制剂和天然产物来源的GSK-3抑制剂.
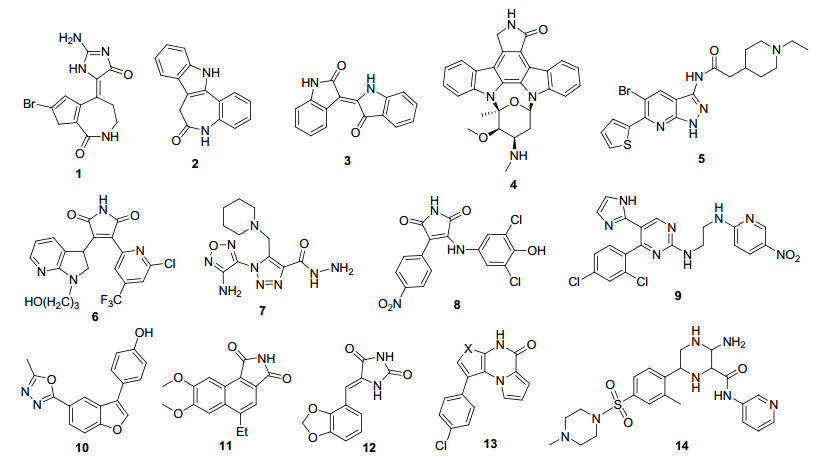
ATP竞争性抑制剂是目前研究最广泛的GSK-3抑制剂, 具有代表性的化合物结构如图 1所示. Meijer等[18]报道的Hymenialdisine (1)是从海绵中提取的小分子化合物, 可以在体内阻断GKS-3对微管结合蛋白——tau蛋白的过度磷酸化, 但Hymenialdisine对细胞周期蛋白依赖性激酶(CDKs)和酪蛋白激酶同时也存在抑制作用. Leost等[19]报道的Paullones (2)属于苯并氮杂酮类, 是有效的GSK-3抑制剂, IC50在4~80 nmol/L. Meijer等[20]发现靛玉红(3)具有抑制GSK-3的潜力, 其6位溴代的衍生物选择性抑制GSK-3. Toledo等[21]报道星形孢菌素(4)在纳摩尔范围对GSK-3有抑制作用.靛玉红和星形孢菌素是第一代GSK-3抑制剂, 共同的缺点是选择性低, 对体内其他激酶存在抑制作用. Witherington[22]、Zhang[23]、Olesen[24]、Smith[25]等课题组报道的一系列杂环化合物5~9是第二代ATP竞争性抑制剂, 相对于第一代抑制剂活性和选择性显著提高. 1, 3, 4-噁二唑[26] (10)、苯并异吲哚-1, 3-二酮[27](11)、苯基亚甲基乙内酰脲[28](12)、二吡咯并吡嗪酮或呋喃并吡咯并吡嗪酮[29](13)、2-哌嗪甲酰胺衍生物(14)是具有新型骨架的GSK-3抑制剂.然而所列举的化合物由于缺乏足够的选择性, 都在临床前阶段遭遇失败, 到目前为止, 没有作为治疗AD药物的ATP竞争性抑制剂获美国食品药品监督管理局(FDA)批准上市.对ATP竞争性抑制剂的研究, 主要是对GSK-3有抑制作用的活性化合物进行结构改造, 以提高激酶选择性和抑制活性.在此主要综述了近五年取得重大进展的ATP竞争性抑制剂.
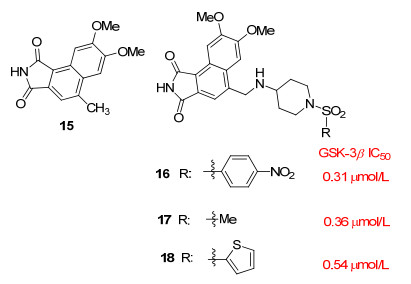
Zou等[27]研究显示, 化合物11可以抑制GSK-3β活性, 但同时对CDK2产生抑制作用. Yue等[30]基于前人的报道做进一步探索, 以提高该类化合物对GSK-3β的选择性. Yue等[30]发现GSK-3β和CDK2之间的显著差异位于ATP区域外紧接铰链区之后的螺旋D处的残基, 在这个区域GSK-3β是带正电的残基(Arg141和Arg144), 而CDK2是带负电的残基(Asp86和Asp92).螺旋D处的残基静电电位的差异, 使得设计针对GSK-3β而不是CDK2的选择性抑制剂成为可能.基于螺旋D处的残基静电电位的差异, 以与化合物11结构相似的15为先导化合物, 引入磺酰基得到磺酰基衍生物16~18(图 2), 该类化合物与GSK-3β螺旋D上带正电的残基Arg141和Arg144相互作用, 提高了对GSK-3β的选择性.化合物16~18抑制GSK-3β的IC50值在微摩尔范围(IC50分别为0.31、0.36、0.54 μmol/L), 并且对CDK2没有显著抑制作用(在100 μmol/L时的抑制效率分别为12.3%、3.4%、2.7%), 与具有末端甲基的化合物17相比, 具有较大疏水性末端官能团的衍生物对GSK-3β的抑制作用明显减弱, 这可能是由于疏水性官能团与GSK-3β高度极性表面之间的不利作用引起的.在细胞水平上使用双荧光素酶报告测定系统, 显示化合物17激活Wnt/β-连环蛋白信号传导的能力是化合物15的三倍.化合物17同时具有良好的抑制活性和选择性, 并且显著激活Wnt/β-连环蛋白信号传导通路, 是研发抑制GSK-3β药物的主要候选药物.
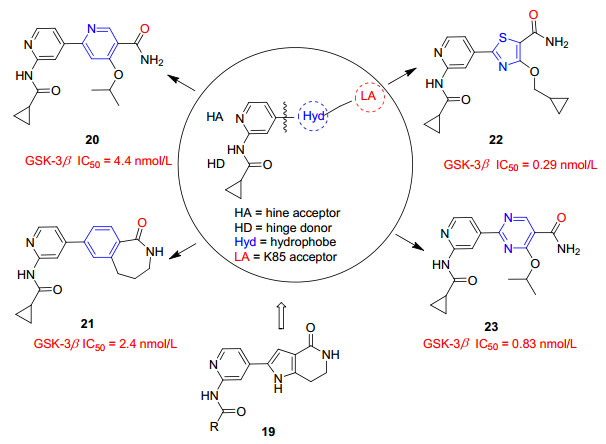
GSK-3β中K85 (Lys85)的ε-氨基基团, 在大部分结构中与配体氢键受体的N和O原子形成关键的氢键. Sivaprakasam等[31]利用这个关键氢键设计了一系列吡咯并哌啶酮衍生物, 并设想吡咯并哌啶酮核心提供三个关键片段结合GSK-3β活性位点: (1)铰链结合头部——吡啶甲酰胺中吡啶结合于GSK-3的铰链区、(2)间隔基团——吡咯并哌啶酮环、(3) K85氢键受体原子——双环中的羰基O与K85形成氢键.以化合物19为先导化合物, 将铰链头部固定为环丙基甲酰胺基吡啶之后, 探索了许多可能的中心间隔物来取代吡咯并哌啶酮核心, 以适当地补充外围受体药效基团, 其中活性最好的化合物结构如图 3所示.中心间隔为吡啶或苯环的化合物20、21在体外显示出较好的抑制活性, IC50分别为4.4、2.4 nmol/L.以噻唑或嘧啶作为中心间隔的22、23活性进一步提高, IC50分别为0.29、0.83 nmol/L.以上活性化合物在体外展现优良的抑制活性, 相关的体内活性测试有待展开.
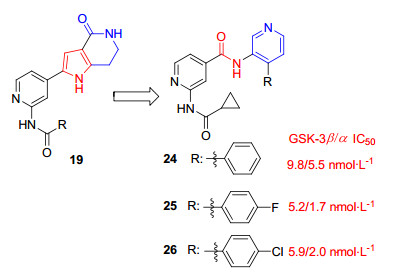
最近Luo等[32]报道的异烟酰胺衍生物也是基于吡咯并哌啶酮19的结构改造:用吡啶代替内酰胺, 吡啶环上的氮作为氢键受体可能会改善中枢神经系统的渗透, 并用酰胺基团取代了吡咯部分, 得到一系列异烟酰胺衍生物(图 4).其中活性最好的化合物24~26在纳摩尔范围抑制GSK-3β活性, IC50值分别为9.8、5.2、5.9 nmol/L.用近400种激酶对化合物24、25、26进行Ambit筛选, 结果显示这些化合物仅对GSK-3两种亚型表现出明显的抑制活性.这种高度选择性可能是由中心酰胺参与ATP口袋背面水介导的氢键结合以及强烈倾斜的4-苯基占据GSK-3β中的大糖袋两种相互作用引起的.在30 mg/kg口服剂量下, 化合物24显著降低三重转基因AD小鼠的细胞磷酸化水平(pTau), 这与LiCl在最大耐受浓度(250 mg/kg)下达到的抑制程度相当, 并且作用时间长达24 h.化合物25、26口服活性不如24, 但化合物26较24有更好的大脑渗透性.异烟酰胺衍生物具有高度的选择性、可渗透性和口服活性, 并且合成方法简便, 有希望成为工具化合物或进一步开发为上市药物.
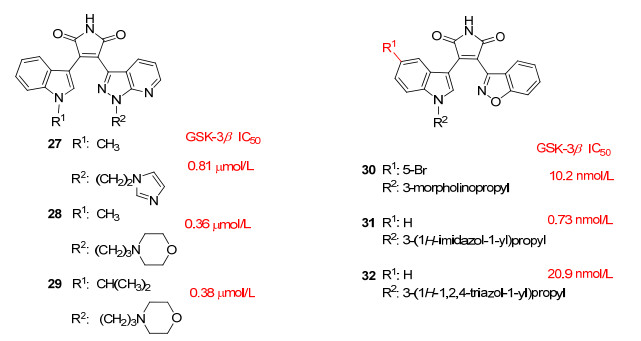
Smith等[25]报道的马来酰亚胺衍生物8有抑制GSK-3的潜在活性和良好的选择性.之后Zhang等[23]报道化合物6抑制GSK-3β的IC50=26 nmol/L, 并在包括68种蛋白激酶的选择性研究中, 选择性抑制GSK-3β.马来酰亚胺衍生物普遍具有良好的抑制活性, 这是因为马来酰亚胺环氮原子上的氢和羰基可以分别与GSK-3β的Asp133和Val135残基形成氢键结合.目前, 马来酰亚胺衍生物是研发GSK-3β抑制剂的热点. Ye等[33]对该系列化合物的结构做进一步改造, 报道了一系列具有GSK-3抑制活性的马来酰亚胺衍生物(图 5).以7-氮杂吲唑基-吲哚基-马来酰亚胺为母核的化合物27、28、29的IC50值分别为0.81、0.36、0.38 μmol/L.在包括六种激酶的选择性测试中, 27~29对GSK-3β的抑制活性明显高于其他五种激酶.此外, 27~29都显著降低了Aβ诱导的tau蛋白过度磷酸化, 在细胞水平也显示出对GSK-3β的抑制作用. Ye等[33]随后报道的一系列3-苯并异噁唑基-4-吲哚基-马来酰亚胺衍生物, 抑制活性显著提升.化合物30~32的IC50值都在纳摩尔范围, 其中31对GSK-3有显著的抑制活性, IC50=0.73 nmol/L.化合物30~32不仅可以降低Aβ诱导的tau蛋白过度磷酸化, 而且软件模拟还显示具有透过血脑屏障的潜力, 该类化合物的药代动力学性质有待进一步研究.
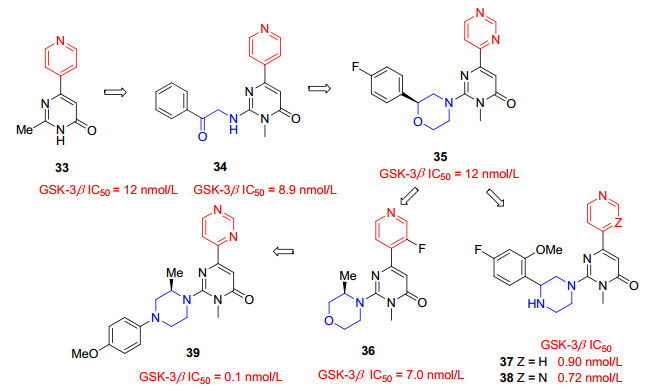
Watanabe等[34~38]在2013~2017年间报道了一系列4(3H)-嘧啶酮衍生物, 可以有效地抑制GSK-3β活性.基于4(3H)-嘧啶酮母体的结构改造主要是用酰胺基、吗啉基、哌嗪基在嘧啶酮环2位的取代以及吡啶基、嘧啶基在6-位的取代(图 6).
以33为先导化合物, 用2-氧代-乙胺在4(3H)-嘧啶酮2位取代得到的衍生物对GSK-3β有较高的抑制作用.活性较好的化合物34的IC50=8.9 nmol/L, 并且有良好的大脑渗透性.小鼠腹腔注射化合物34后, 大脑中tau蛋白磷酸化程度显著降低[34].然而, 化合物34口服后活性明显下降, 这可能是由于氨基和羰基之间形成的氢键阻碍34透过血脑屏障.用吗啉基在4(3H)-嘧啶酮2位取代得到具有口服活性的化合物35, 不仅在体内抑制小鼠tau过度磷酸化, 而且具有中等生物利用度和良好的激酶选择性[35].
基于化合物35的结构进一步优化, 在吗啉的3-位用低级烷基代替苯基改善了化合物的体外药代动力学性质, 得到有效的低分子量的GSK-3β抑制剂.化合物36的IC50=7.0 nmol/L, 相对33 (IC50=12 nmol/L), 抑制GSK-3β活性升高并且口服给药后显示体内tau磷酸化抑制活性[36].基于化合物33的另一结构改造是将吗啉部分转化成哌嗪, 哌嗪部分的氮原子与Gln185主链的氧原子之间形成新的氢键, 与33相比, 抑制活性显著增加[37].化合物37、38的IC50分别为0.90、0.72 nmol/L.然而该类化合物的药动学性质较差, 可以在苯环上引入简单的取代基以做进一步优化.
将化合物36的3-甲基吗啉部分转化成哌嗪, 得到的化合物39对GSK-3β有显著的抑制活性, IC50=0.1 nmol/L.抑制活性的显著提升归因于哌嗪氮原子上的苯基和哌嗪上的甲基分别与GSK-3β形成阳离子-π和CH-π相互作用[38].化合物39具有高细胞渗透性、口服活性和良好的脑/血浆浓度比, 是最具潜力的候选药物.
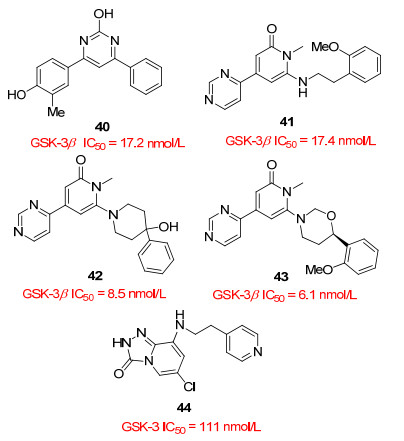
高通量筛选得到的化合物40具有抑制GSK-3β的潜在活性, 并且有良好的细胞渗透性, 但在包括50种激酶的选择性实验中, 化合物40对CDC样激酶-1 (CLK1)和酪蛋白激酶2 (CK2)有显著的抑制作用. Coffman等[39]以40为先导化合物进行结构改造, 得到选择性显著提高的吡啶衍生物(图 7).其中化合物41抑制活性良好, 体外酶实验测定IC50=17.4 nmol/L, 初步的构效关系(SAR)表明, 甲氧基在邻位取代比其在间位或对位取代的类似物, 抑制活性高30倍以上.化合物41具有良好的选择性, 然而该化合物在体外肝微粒体实验中极易被清除, 引入哌啶(42)或吗啉环(43)后, 代谢稳定性显著升高.然而, 化合物42对CLK1仍然有抑制作用, 选择性不如41、43.化合物41~43都具有良好的中枢神经系统渗透性, 可以作为先导化合物做进一步改造.
Chun等[40]报道化合物44可以穿透血脑屏障, 口服后具有良好的药代动力学性质和水溶性.体外酶实验和细胞实验测定IC50和EC50值分别为111 nmol/L、1.78 μmol/L.该类化合物具有很好的穿透血脑屏障的能力, 可以进一步优化以提高GSK-3β抑制活性, 探索成为AD治疗药物的可能性.
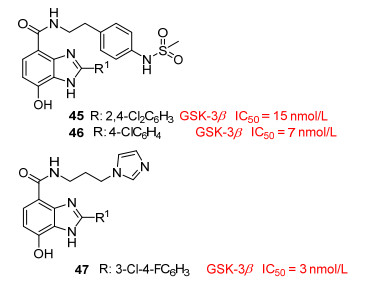
Shin等[41]利用计算辅助技术设计合成的苯并咪唑衍生物45对GSK-3β有潜在的抑制活性, 预估其IC50=15 nmol/L.利用对接模拟技术, 预测化合物45咪唑环氮上的氢原子与Val135的羰基可能形成氢键, 苯环上的羟基与Val135的NH以及Aap133的羰基也可能形成氢键. Lee等[42]在Shin等工作的基础上进一步研究, 以化合物45为先导, 设计合成了7-羟基-苯并咪唑衍生物46、47(图 8), 在酶实验和细胞实验中有较好的抑制活性, IC50分别为7和3 nmol/L.化合物47在包括七种激酶的选择性实验中, 特异性的抑制GSK-3β.苯并咪唑衍生物有良好的抑制活性和选择性, 相关的药代动力学性质有待进一步研究.
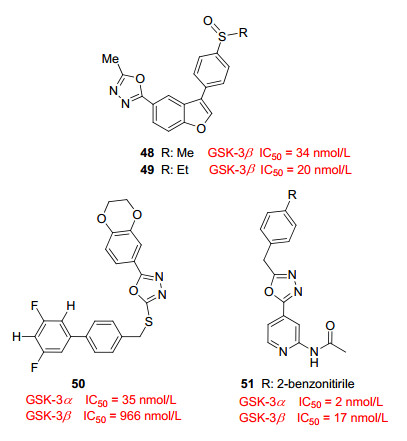
Saitoh等[43]报道的噁二唑衍生物是一类能够穿透血脑屏障的GSK-3β抑制剂(图 9).化合物48、49不仅有较好的抑制活性(IC50分别为34和20 nmol/L)和激酶选择性, 而且可以穿透血脑屏障, 具有良好的药代动力学性质.在冷水胁迫模型(CWS)中测定化合物48、49的口服活性, CWS模型中GSK-3β在S199, Thr205, Thr231和Ser396位点造成tau蛋白产生过度磷酸化, 小鼠口服48、49可以显著降低CWS小鼠模型的tau蛋白磷酸化水平. Monte等[44]报道的化合物50、51选择性地抑制GSK-3α, 而对GSK-3β产生较弱的抑制活性.尤其是化合物50选择性抑制GSK-3α (IC50=35 nmol/L), 相比GSK-3β (IC50=966 nmol/L), 选择性高达27倍; 化合物51的对GSK-3α的IC50为2 nmol/L, GSK-3β的IC50为17 nmol/L, 选择性为8.5倍.该类化合物为研发高度选择性的GSK-3α抑制剂提供了基本骨架.
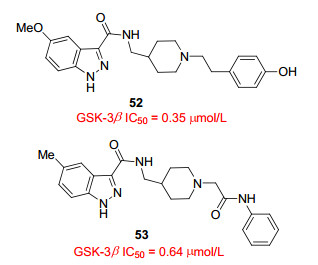
Ombrato等[45]将虚拟筛选应用于专有化合物文库, 发现1H-吲唑-3-甲酰胺结构可以作为GKS-3抑制剂的新型化学骨架.筛选出的8个含有1H-吲唑-3-甲酰胺结构的化合物, 均在低微摩尔范围对酶(GSK-3β)产生抑制作用, 其中化合物52、53具有较好的活性, IC50值分别为0.35、0.64 μmol/L. X射线晶体结构表明, 化合物53与GSK-3β的ATP口袋相结合, 其中吲唑环位于腺嘌呤结合位点, 环上的两个氮原子分别与Val135的主链NH和Asp133的主链羰基形成氢键.此外, 吲唑环上的甲基指向ATP结合口袋的内部, 形成重要的极性作用; 酰胺基团中NH与Val135主链羰基形成氢键, 这使得哌啶环直接面对Arg144的胍基, 从而形成强烈的亲水接触. X射线晶体结果证实该类化合物是以与ATP竞争的方式对GSK-3β产生抑制作用, 是一类ATP竞争性抑制剂.化合物53、54的活性有待提高, 可以作为先导化合物做进一步的构效研究.
非ATP竞争性抑制剂多为一些小分子化合物, 分子量低, 容易透过血脑屏障, 对GSK-3β具有中等抑制活性和高度的选择性, 有望在达到疗效的同时避免或减少副作用.
阳离子锂是发现的第一个“天然”GSK-3抑制剂.据报道[15], Li可以显著减少神经元细胞和非神经元细胞中tau蛋白磷酸化, 甚至在新生大鼠脑中也具有相同作用.锂也是一种情绪稳定剂, 已被用于治疗双相情感障碍.研究证实[46], Li通过抑制GSK-3β, 可能会改变AD特定的病理过程, 如tau蛋白的过度磷酸化、Aβ的沉积和Aβ诱导的神经毒性.锂通过与镁离子竞争直接抑制GSK-3, 间接通过增强的丝氨酸磷酸化和自身调节来抑制GSK-3活性.然而, 一项为期10周的、随机、单盲、多中心、安慰剂对照临床实验结果显示, Li并不能显著降低tau蛋白的过度磷酸化水平[47].另外, Frisch[48]和Macdonald[49]等发现, Li作为AD治疗剂可能会产生严重的毒副作用.以上研究结果并不能否认Li对AD的治疗作用, 但考虑到Li的毒副作用, 作为AD治疗剂, 其有效性和安全性还需要良好的长期试验来确认.
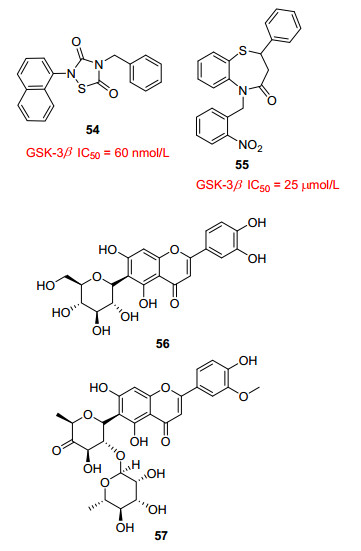
Tideglusib(图 11, 54)是噻二唑烷酮类化合物, 是GSK-3β的非ATP竞争性抑制剂, IC50值为60 nmol/L.据报道[50], tideglusib能够不可逆地抑制GSK-3β的活性, 阻止tau蛋白的过度磷酸化, 降低β淀粉样蛋白的聚集, 提高学习和记忆的能力, 延缓神经元的丢失和凋亡.临床试验数据显示[51], tideglusib可以剂量依赖性地降低脑萎缩和脑脊液中tau磷酸化和BACE-1水平, 并且证实了tideglusib能够穿过血脑屏障.一项为期26周的Ⅱ期临床研究证实[52], tideglusib的安全性是可以接受的, 但是在这项实验中没有产生任何临床益处.考虑到非线性剂量反应, 在疾病早期阶段和持续时间更长的剂量研究有待进一步展开.

Zhang等[53]报道的以苯并硫氮杂
最近, Liang等[54]对玉米穗丝提取物的活性追踪分离发现了两种6-C-糖基黄酮(图 11, 56和57)可以作为GSK-3β抑制剂.化合物56对GSK-3β显示出中等的抑制活性, 在激酶测定谱系统中显示良好的选择性.体外酶实验和分子模拟对接揭示, 化合物56以非ATP竞争抑制的方式阻断GSK-3β磷酸化tau蛋白进程, 是一种非ATP竞争性抑制剂.化合物56在神经母细胞瘤的神经保护作用, 同时可阻止tau蛋白过度磷酸化, 有(SH-SY5Y)细胞模型中对Aβ诱导的细胞毒性产生有效预防和治疗AD潜力, 值得进一步深入研究.
一直以来, 底物竞争性抑制剂与GSK-3的低亲和作用被认为是该类抑制剂最大的缺点.然而, 现在研究发现[55], 生物系统中蛋白激酶的强烈、持续性抑制作用常常导致不良反应.适度的抑制激酶活性可以在提供足够的疗效同时降低毒副作用.对于GSK-3抑制剂来说, 底物竞争性抑制剂为实现高选择性和低毒性提供了机会.
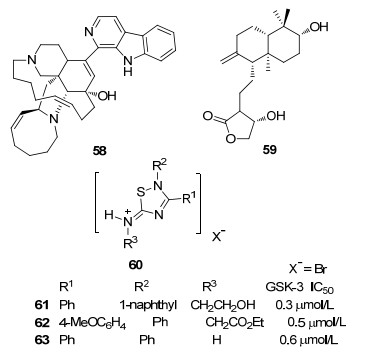
Manzamines A(图 12, 58)是一种从海绵中分离出来的生物碱, 对GSK-3β有中等的抑制活性, IC50=10 μmol/L. Manzamines A可以降低人类神经母细胞瘤细胞系中tau蛋白过度磷酸化, 具有良好的细胞渗透性.在包括六种激酶的选择性实验中, Manzamines A选择性的抑制GSK-3β和CDK-5[56]. Manzamines A的选择性有待进一步提高, 但由于化学结构独特, 改造难度较大.
Wnt信号传导在生物体内起重要作用, Wnt信号紊乱将会导致一系列神经退行性疾病如精神分裂症和阿尔茨海默病. Tapia-Rojas等[57]研究证实穿心莲内酯(图 12, 59)可以通过抑制GSK-3β的活性, 激活Wnt信号传导通路.穿心莲内酯阻止Wnt配体与受体相结合, 诱导Wnt靶基因转录, 激活Wnt信号通路.穿心莲内酯以与底物竞争的方式抑制GSK-3β活性, 是一种底物竞争性抑制剂.
Palomo等[58]报道了以5-氨基-1, 2, 4-噻二唑(图 12, 60)为母核的衍生物, 该系列化合物在微摩尔范围对GSK-3产生抑制作用, 是底物竞争性抑制剂.对接模拟显示, 5-氨基-1, 2, 4-噻二唑骨架主要是通过亚胺基带电基团的质子与Phe67的主链氧之间的氢键以及噻二唑杂环与GSK-3残基Phe67的芳香环之间的芳香S-π相互作用产生的.此外, 在噻二唑环上引入不同的取代基, 与GSK-3残基结合以增加抑制活性.化合物61~63的IC50值在亚微摩尔范围, 分别为0.3、0.5、0.6 μmol/L.该类小分子化合物具有细胞渗透性, 能够阻止炎症活化并选择性地分化神经干细胞, 减少神经元细胞死亡, 是一类具有潜力的GSK-3抑制剂, 体内活性研究有待进一步展开.
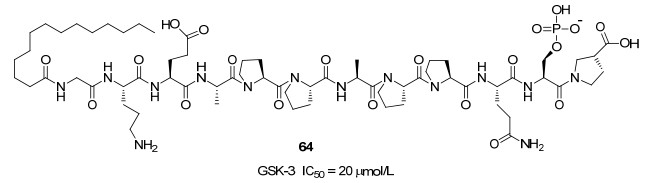
Avrahami等[55]报道的短肽L803来自热休克因子-1 (HSF-1), 序列为KEAPPAPPQS(p)P, 在体外激酶测定中发现是有效的GSK-3抑制剂.在L803的N端连接肉豆蔻酸产生的衍生物L803mts(图 13, 64)具有细胞渗透性, 是活性良好的底物竞争性抑制剂, IC50=20 μmol/L. L803-mts在阿尔茨海默病小鼠模型中, 显示出改善神经元内淀粉样β肽负荷和认知缺陷的潜力.模拟对接显示, 与底物一样, L803mts通过磷酸化的Ser10对接到磷酸口袋中; 与底物不同的是, L803-mts不与Gln 89或Asn 95发生相互作用. L803mts在89-QDKRFKN-95环内与Phe93形成紧密接触, 并且还与位于激酶的C端叶中的“疏水斑”(Val214, Ile217和Tyr216)相互作用.因此, 底物和底物竞争性抑制剂与底物口袋中的不同几何形状相互作用, 从而允许竞争性抑制.尽管L803-mts已经在神经疾病的不同动物模型中显示出效力, 但与肽化学支架相关的低药代动力学特征阻止其发展到临床试验.
Licht-Murava等[59]报道了一种新型的GSK-3抑制剂L807, 序列为KEAPPSPPQS(p)P, 与L803相比, 保留了热休克因子中的Ser6.在L807的N端连接肉豆蔻酸产生的衍生物L807mts, 通过在酶的催化位点内发生底物-抑制剂转化对GSK-3起抑制作用.与L803mts相比, L807mts是更有效的GSK-3抑制剂, IC50=1 μmol/L.使用包括139种蛋白激酶的体外激酶实验测定L807mts对GSK-3的选择性, 实验结果表明L807mts高度特异性地结合GSK-3的两种亚型.分子模拟对接结果证实, L807主要结合到激酶C端叶上用于磷酸化的残基Ser6, 与环63~69只有很少的接触(ATP结合位点), 而与环89-QDKRFKN-95没有接触(环89~95与环63~69一起限制混杂底物结合).在啮齿动物中进行测试时具有合理的药理学性质, 包括稳定性、大脑透过性、药代动力学和安全性. L807mts促进5XFAD AD小鼠模型中β淀粉样蛋白的清除, 减少了炎症反应, 提高了自噬流量, 并且改善了认知和社交能力.这种底物-抑制剂转化的肽类抑制剂为GSK-3抑制剂进入临床带来了希望.
变构抑制剂提供了一种新的GSK-3β活性调节机制, 变构抑制剂通过诱导酶结合位点的构象发生变化来调节酶活性. GSK-3β的变构抑制剂可能更具选择性, 因为它们与激酶的独特区域结合, 并且可以克服针对与ATP竞争的药物产生的抗性.此外, 与ATP竞争性抑制剂相比, 变构抑制剂的抑制作用更精细, 对GSK-3β有适度的抑制作用, 大大降低了副作用.迄今为止, 关于GSK-3β的变构调节剂报道很少.目前只报道了天然的倍半萜palinurin和喹啉衍生物.
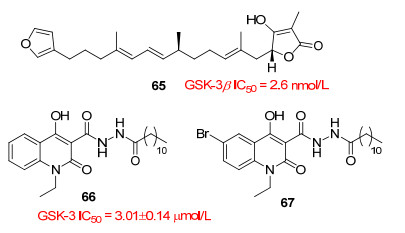
Bidon-Chanal等[60]报道的palinurin(图 14, 65)是一种天然倍半萜类, 是从海绵I. variabilis中提取的GSK-3β的高度选择性抑制剂, IC50=2.6 nmol/L, 其全合成路线已经被报道[61].分子动力学模拟显示, palinurin通过变构机制对GSK-3β产生抑制作用, 它是发现的第一个针对变构位点的化合物. Palinurin通过结合位于酶N-末端叶片上的空腔来发挥其抑制活性.值得注意的是, 这个空腔已经被认为是一个有潜力的变构结合位点.在该空腔里, Lys86与tetronic环(羟基丁酸内酯环)的去质子化羟基形成了一个盐桥和一个氢键, Tyr56的羟基与tetronic环的羰基形成了氢键.此外, 整个分子重构, 其中倍半萜部分朝向由蛋白质链形成的β-环对面的疏水性凹槽. Palinurin与空腔的结合, 限制了富含甘氨酸的环的构象从而阻止ATP结合.这种新颖的变构作用机制赋予palinurin高度选择性, 使其成为抑制GSK-3β的重要先导化合物.
Palomo等[62]报道喹啉衍生物VP0.7(图 14, 66)是一种GSK-3变构抑制剂, 抑制GSK-3的IC50值为3.01± 0.14 μmol/L.对接研究揭示VP0.7结合到酶的C端叶片, 芳香环夹在残基Arg209和Thr235之间.在这个方向上, 有利于与Arg209形成阳离子-π相互作用, 并与Thr235的甲基形成疏水相互作用.此外, VP0.7的氧与Ser236的主链NH基之间形成氢键. VP0.7的脂肪链延伸到由残基Leu169、Pro331和Thr330确定的疏水性区域周围.最近发现VP0.7和其喹啉6位溴代衍生物(67)是治疗先天性强直性营养不良Ⅰ型和脊髓型肌萎缩的先导化合物[63].
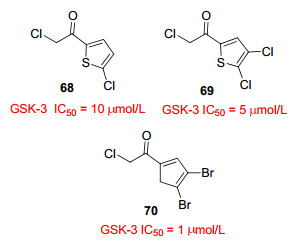
卤代甲基酮衍生物(图 15)是一类针对GSK-3的不可逆抑制剂, 并且具有高选择性.该类抑制剂的作用机制可能是阻断了GSK-3对ATP的磷酸化进程, Cys199残基位于GSK-3的ATP结合位点上, 这个残基与α-卤代甲基酮部分形成不可逆的共价结合, 使得GSK-3不能发挥磷酸化作用.据报道[64], 化合物68~70对GSK-3有中等的抑制作用, IC50值分别为10、5、1 μmol/L.在包括八种激酶(不包括GSK-3)的选择性实验中, 化合物68~70没有明显的抑制作用, 对GSK-3的选择性较好.此外, 该类化合物在不同的细胞系均具有良好的细胞渗透性.进一步研究发现[65], 在可逆抑制剂的结构中引入卤代甲基酮部分, 可以将可逆抑制剂转变为不可逆抑制剂, 并显著提高抑制活性.因此卤代甲基酮部分可以作为修饰工具, 在GSK-3抑制剂的修饰中发挥重要作用.
自然界是珍贵的分子宝库, 科研工作者从未停止过对天然产物的探索.天然产物独特的化学结构为GSK-3抑制剂的研究提供了新的化合物骨架, 靛玉红、生物碱ManzaminesA、穿心莲内酯、6-C-糖基黄酮等已经被证实是有效的GSK-3抑制剂.基于天然产物的结构修饰近来也成为GSK-3抑制剂研究的热点.因此天然产物的发现对于GSK-3抑制剂的研究意义重大.
Lu等[66]报道从川芎中提取的主要生物活性化合物——川芎嗪(图 16, 71)可以用于治疗神经血管疾病和心血管疾病.川芎嗪的神经保护特性在一些神经退行性疾病中非常明显.为了探索川芎嗪对AD大鼠的记忆保护作用, 在大鼠的侧脑室注射链脲佐菌素, 诱导具有短期或长期记忆障碍的AD大鼠模型.使用川芎嗪对这种AD大鼠治疗2周后发现, 大鼠记忆障碍得到改善, 同时恢复了空间学习和记忆保持能力.此外, 川芎嗪具有抑制GSK-3β的活性, 可以恢复胆碱能神经元的功能.目前, 川芎嗪对AD大鼠记忆的保护作用机制尚不明确, 可能是通过抑制GSK-3β活性产生的.
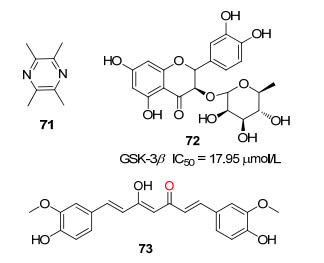
Wang等[67]发现落新妇苷(图 16, 72)具有强大的神经保护作用.对Aβ诱导的与AD有关的认知障碍具有预防作用, 但这些效应的潜在机制尚不清晰.用落新妇苷治疗三重转基因小鼠(40 mg/kg/d), 8周后观察到了落新妇苷的有益作用, 包括减轻学习和记忆缺陷, 减少斑块负荷和Aβ水平.此外, 落新妇苷(40 mg/kg/d)组动物海马区蛋白激酶B (AKT)/GSK-3β信号通路紊乱显著改善, 这可能是落新妇苷促进脑源性神经营养因子(BDNF)的表达, 而BDNF在激活AKT的同时抑制GSK-3β.落新妇苷阻止老年斑形成的有益作用可能是通过抑制GSK-3β活性产生的.因此落新妇苷具有治疗AD的潜力.
据报道, 姜黄素(图 16, 73)可以预防和治疗AD, Tang等[68]发现姜黄素抑制Aβ淀粉样斑块的形成并且促进其分解, 阻止tau蛋白过度磷酸化并增强其清除.同时姜黄素结合铜离子, 降低胆固醇, 改变小胶质细胞活性, 抑制乙酰胆碱酯酶活性, 介导胰岛素信号通路.对接模拟显示[68], 姜黄素的β-烯醇酮部分与GSK-3β的Tyr134和Val135形成氢键作用, 两个芳基分别与Lys85、Glu97、Arg141和Glu173接触, 其IC50值为17.95 μmol/L.与乙酰胆碱酯酶抑制剂相比, 姜黄素作用于多个靶点, 并且最重要的是具有改变疾病进程的能力.然而由于其生物利用度低, 目前还没有进入临床研究.
2018年1月, 辉瑞宣布停止对阿尔茨海默病新药的开发[69].在此之前, 默克等制药巨头研发的BACE-1抑制剂在临床实验中也没有达到主要疗效终点[70].一系列BACE-1抑制剂在临床试验阶段遭受失败, 使得科研人员将注意力转向tau蛋白, GSK-3作为tau蛋白磷酸化的主要激酶, 预计在未来几年会受到更广泛的研究.
GSK-3作为体内重要的蛋白激酶, 对包括糖原合成酶、tau蛋白和β-蛋白在内的一百多种底物发生磷酸化作用, 参与胰岛素信号传导、肿瘤发生、脑源性神经营养因子信号传导等过程.异常表达的GSK-3是引起神经系统疾病的主要原因.过度表达的GSK-3与AD的两大经典病理特征—淀粉样蛋白沉积和tau蛋白过度磷酸化密切相关, 是研发AD治疗药物最具希望的靶标.未来GSK-3抑制剂的研究可以从两方面进行.一方面, 利用计算机辅助技术探索GSK-3其他的可能与配体化合物结合的位点, 提高配体药物的选择性, 从而减少各种不良反应.另一方面, 发现新结构化合物, 如从以下三个方向进行: (1)以天然活性肽为先导化合物, 对其进行结构修饰, 提高肽类抑制剂的生物利用度; (2)基于目前发现的活性化合物进行结构改造, 注重提高化合物透过血脑屏障等药理学性质; (3)自然界是一个巨大的分子宝库, 许多具有新型骨架的化合物分子有待进一步发现, 可以进行广泛深入的研究, 以从自然界寻找活性良好的GSK-3抑制剂.
叶勤, 李海林, 李箕君, 临床精神医学杂志, 2015, 25, 46. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=lcjs201501027&dbname=CJFD&dbcode=CJFQYe, Q.; Li, H. L.; Li, J. J. J. Clin. Psychiatry 2015, 25, 46 (in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=lcjs201501027&dbname=CJFD&dbcode=CJFQ
张旻, 黄晓琳, 张凤霞, 内科急危重症杂志, 2017, 23, 177. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=nkjw201703001&dbname=CJFD&dbcode=CJFQZhang, W.; Huang, X. L.; Zhang, F. X. J. Int. Intensive Med. 2017, 23, 177 (in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=nkjw201703001&dbname=CJFD&dbcode=CJFQ
冀开元, 马文丽, 郑文岭, 解剖学报, 2015, 46, 164. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jpxb201502006&dbname=CJFD&dbcode=CJFQJi, K.Y.; Ma, W. L.; Zheng, W. L. Acta Anat. Sin. 2015, 46, 164 (in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jpxb201502006&dbname=CJFD&dbcode=CJFQ
Prince, M. ; Wimo, A. ; Guerchet, M. ; Ali, G. C. ; Wu, Y. T. ; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends, Alzheimer's Disease International, London, 2015, Chapter 1.
Jarrett, J. T.; Berger, E. P.; Lansbury Jr, P. T. Biochemistry 1993, 32, 4693. doi: 10.1021/bi00069a001
Herrup, K. Nat. Neurosci. 2015, 18, 794. doi: 10.1038/nn.4017
Walsh, D. M.; Klyubin, I.; Fadeeva, J. V.; Cullen, W. K.; Anwyl, R.; Wolfe, M. S.; Rowan, M. J.; Selkoe, D. J. Nature 2002, 416, 535. doi: 10.1038/416535a
Ricciarelli, R.; Fedele, E. Curr. Neuropharmacol. 2017, 15, 926. http://europepmc.org/articles/PMC5652035/
Mccubrey, J. A.; Steelman, L. S.; Bertrand, F. E.; Davis, N. M.; Sokolosky, M.; Abrams, S. L.; Montalto, G.; D'Assoro, A. B.; Libra, M.; Nicoletti, F. Oncotarget 2014, 5, 2881. https://www.mendeley.com/research-papers/regulation-ubiquitinationmediated-protein-megradation-survival-kinases-cancer/
Cormier, K. W.; Woodgett, J. R. F1000Research 2017, 6, 167. doi: 10.12688/f1000research
Hoeflich, K. P.; Luo, J.; Rubie, E. A.; Tsao, M.-S.; Jin, O.; Woodgett, J. R. Nature 2000, 406, 86. doi: 10.1038/35017574
Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Am. J. Pathol. 2008, 172, 786. doi: 10.2353/ajpath.2008.070904
Beurel, E.; Grieco, S. F.; Jope, R. S. Pharmacol. Ther. 2015, 148, 114. doi: 10.1016/j.pharmthera.2014.11.016
Hong, M.; Chen, D. C.; Klein, P. S.; Lee, V. M. J. Biol. Chem. 1997, 272, 25326. doi: 10.1074/jbc.272.40.25326
Cohen, P.; Goedert, M. Nat. Rev. Drug Discovery 2004, 3, 479. doi: 10.1038/nrd1415
Moralesgarcia, J. A.; Lunamedina, R.; Alonsogil, S.; Sanzsancristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perezcastillo, A. ACS Chem. Neurosci. 2012, 3, 963. doi: 10.1021/cn300110c
Meijer, L.; Thunnissen, A.-M. W. H.; White, A. W.; Garnier, M.; Nikolic, M.; Tasi, L.-H.; Walter, J.; Cleverley, K. E.; Salinas, P. C.; Wu, Y.-Z.; Biernat, J.; Mandelkow, E.-M.; Kim, S.-H.; Pettie, G. R. Chem. Biol. 2000, 7, 51. doi: 10.1016/S1074-5521(00)00063-6
Leost, M.; Schultz, C.; Link, A.; Wu, Y. Z.; Biernat, J.; Mandelkow, E. M.; Bibb, J. A.; Snyder, G. L.; Greengard, P.; Zaharevitz, D. W. Eur. J. Biochem. 2000, 267, 5983. doi: 10.1046/j.1432-1327.2000.01673.x
Meijer, L.; Skaltsounis, A. L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X. P.; Vonica, C. A.; Brivanlou, A.; Dajani, R. Chem. Biol. 2003, 10, 1255. doi: 10.1016/j.chembiol.2003.11.010
Toledo, L. M.; Lydon, N. B. Structure 1997, 5, 1551. doi: 10.1016/S0969-2126(97)00304-3
Witherington, J.; Bordas, V.; Gaiba, A.; Naylor, A.; Rawlings, A. D.; Slingsby, B. P.; Smith, D. G.; Takle, A. K.; Ward, R. W. Bioorg. Med. Chem. Lett. 2003, 13, 3059. doi: 10.1016/S0960-894X(03)00646-2
Zhang, H. C.; Ye, H.; Conway, B. R.; Derian, C. K.; Addo, M. F.; Kuo, G. H.; Hecker, L. R.; Croll, D. R.; Li, J.; Westover, L. ChemInform 2004, 35, 3245.
Olesen, P. H.; Sørensen, A. R.; Ursø, B.; Kurtzhals, P.; Bowler, A. N.; Ehrbar, U.; Hansen, B. F. J. Med. Chem. 2003, 46, 3333. doi: 10.1021/jm021095d
Smith, D. G.; Buffet, M.; Fenwick, A. E.; Haigh, D.; Ife, R. J.; Saunders, M.; Slingsby, B. P.; Stacey, R.; Ward, R. W. Bioorg. Med. Chem. Lett. 2001, 11, 635. doi: 10.1016/S0960-894X(00)00721-6
Monte, F. L.; Kramer, T.; Gu, J.; Anumala, U. R.; Marinelli, L.; La, P. V.; Novellino, E.; Franco, B.; Demedts, D.; Van, L. F. J. Med. Chem. 2012, 55, 4407. doi: 10.1021/jm300309a
Zou, H.; Zhou, L.; Li, Y.; Cui, Y.; Zhong, H.; Pan, Z.; Yang, Z.; Quan, J. J. Med. Chem. 2010, 53, 994. doi: 10.1021/jm9013373
Khanfar, M. A.; Hill, R. A.; Kaddoumi, A.; Sayed, K. A. E. J. Med. Chem. 2010, 53, 8534. doi: 10.1021/jm100941j
Rochais, C.; Duc, N. V.; Lescot, E.; Sopkova-de Oliveira Santos, J.; Bureau, R.; Meijer, L.; Dallemagne, P.; Rault, S. Eur. J. Med. Chem. 2009, 44, 708. doi: 10.1016/j.ejmech.2008.05.011
Yue, H.; Lu, F.; Shen, C.; Quan, J.-M. Bioorg. Chem. 2015, 61, 21. doi: 10.1016/j.bioorg.2015.05.009
Sivaprakasam, P.; Han, X.; Civiello, R. L.; Jacutin-Porte, S.; Kish, K.; Pokross, M.; Lewis, H. A.; Ahmed, N.; Szapiel, N.; Newitt, J. A. Bioorg. Med. Chem. Lett. 2015, 25, 1856. doi: 10.1016/j.bmcl.2015.03.046
Luo, G.; Chen, L.; Burton, C. R.; Xiao, H.; Sivaprakasam, P.; Krause, C. M.; Cao, Y.; Liu, N.; Lippy, J.; Clarke, W. J. J. Med. Chem. 2016, 59, 1041. doi: 10.1021/acs.jmedchem.5b01550
(a) Ye, Q. ; Li, M. ; Zhou, Y. ; Pang, T. ; Xu, L. ; Cao, J. ; Han, L. ; Li, Y. ; Wang, W. ; Gao, J. Molecules 2013, 18, 5498.
(b) Ye, Q. ; Shen, Y. ; Zhou, Y. ; Lv, D. ; Gao, J. ; Li, J. ; Hu, Y. Eur. J. Med. Chem. 2013, 68, 361.
Uehara, F.; Shoda, A.; Aritomo, K.; Fukunaga, K.; Watanabe, K.; Ando, R.; Shinoda, M.; Ueno, H.; Kubodera, H.; Sunada, S.; Saito, K.-I.; Kaji, T.; Asano, S.; Eguchi, J.; Yuki, S.; Tanaka, S.; Yoneyama, Y.; Niwa, T. Bioorg. Med. Chem. Lett. 2013, 23, 6928. doi: 10.1016/j.bmcl.2013.09.021
Fukunaga, K.; Uehara, F.; Aritomo, K.; Shoda, A.; Hiki, S.; Okuyama, M.; Usui, Y.; Watanabe, K.; Yamakoshi, K.; Kohara, T.; Hanano, T.; Tanaka, H.; Tsuchiya, S.; Sunada, S.; Saito, K.-I.; Eguchi, J.; Yuki, S.; Asano, S.; Tanaka, S.; Mori, A.; Yamagami, K.; Baba, H.; Horikawa, T.; Fujimura, M. Bioorg. Med. Chem. Lett. 2013, 23, 6933. doi: 10.1016/j.bmcl.2013.09.020
Fukunaga, K.; Sakai, D.; Watanabe, K.; Nakayama, K.; Kohara, T.; Tanaka, H.; Sunada, S.; Nabeno, M.; Okamoto, M.; Saito, K.-I.; Eguchi, J.; Mori, A.; Tanaka, S.; Inazawa, K.; Horikawa, T. Bioorg. Med. Chem. Lett. 2015, 25, 1086. doi: 10.1016/j.bmcl.2015.01.005
Usui, Y.; Uehara, F.; Hiki, S.; Watanabe, K.; Tanaka, H.; Shouda, A.; Yokoshima, S.; Aritomo, K.; Adachi, T.; Fukunaga, K.; Sunada, S.; Nabeno, M.; Saito, K.-I.; Eguchi, J.; Yamagami, K.; Asano, S.; Tanaka, S.; Yuki, S.; Yoshii, N.; Fujimura, M.; Horikawa, T. Bioorg. Med. Chem. Lett. 2017, 27, 3726. doi: 10.1016/j.bmcl.2017.06.078
Kohara, T.; Nakayama, K.; Watanabe, K.; Kusaka, S.; Sakai, D.; Tanaka, H.; Fukunaga, K.; Sunada, S.; Nabeno, M.; Saito, K.-I.; Eguchi, J.; Mori, A.; Tanaka, S.; Bessho, T.; Takiguchi-Hayashi, K.; Horikawa, T. Bioorg. Med. Chem. Lett. 2017, 27, 3733. doi: 10.1016/j.bmcl.2017.06.077
Coffman, K.; Brodney, M.; Cook, J.; Lanyon, L.; Pandit, J.; Sakya, S.; Schachter, J.; Tsenglovering, E.; Wessel, M. Bioorg. Med. Chem. Lett. 2011, 21, 1429. doi: 10.1016/j.bmcl.2011.01.017
Chun, K.; Park, J. S.; Lee, H. C.; Kim, Y. H.; Ye, I. H.; Kim, K. J.; Ku, I. W.; Noh, M. Y.; Cho, G. W.; Kim, H. Bioorg. Med. Chem. Lett. 2013, 23, 3983. doi: 10.1016/j.bmcl.2013.03.119
Shin, D.; Lee, S.-C.; Heo, Y.-S.; Lee, W.-Y.; Cho, Y.-S.; Kim, Y. E.; Hyun, Y.-L.; Cho, J. M.; Lee, Y. S.; Ro, S. Bioorg. Med. Chem. Lett. 2007, 17, 5686. doi: 10.1016/j.bmcl.2007.07.056
Lee, S.-C.; Shin, D.; Cho, J. M.; Ro, S.; Suh, Y.-G. Bioorg. Med. Chem. Lett. 2012, 22, 1891. doi: 10.1016/j.bmcl.2012.01.065
Saitoh, M.; Kunitomo, J.; Kimura, E.; Iwashita, H.; Uno, Y.; Onishi, T.; Uchiyama, N.; Kawamoto, T.; Tanaka, T.; Mol, C. D.; Dougan, D. R.; Textor, G. P.; Snell, G. P.; Takizawa, M.; Itoh, F.; Kori, M. J. Med. Chem. 2009, 52, 6270. doi: 10.1021/jm900647e
Monte, F. L.; Kramer, T.; Gu, J.; Brodrecht, M.; Pilakowski, J.; Fuertes, A.; Dominguez, J. M.; Plotkin, B.; Eldar-Finkelman, H.; Schmidt, B. Eur. J. Med. Chem. 2013, 61, 26. doi: 10.1016/j.ejmech.2012.06.006
Ombrato, R.; Cazzolla, N.; Mancini, F.; Mangano, G. J. Chem. Inf. Model. 2015, 55, 2540. doi: 10.1021/acs.jcim.5b00486
Ghribi, O.; Herman, M. M.; Savory, J. J. Neurosci. Res. 2003, 71, 853. doi: 10.1002/(ISSN)1097-4547
Hampel, H.; Ewers, M.; Bürger, K.; Annas, P.; Mörtberg, A.; Bogstedt, A.; Frölich, L.; Schröder, J.; Schönknecht, P.; Riepe, M. W. J. Clin. Psychiatry 2009, 70, 922. doi: 10.4088/JCP.08m04606
Frisch, S.; Grünwald, F.; Friedrichs, B. Int. Psychogeriatr. 2017, 29, 1747. doi: 10.1017/S1041610217000540
Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. Int. J. Geriatr. Psychiatry 2008, 23, 704. doi: 10.1002/gps.v23:7
Tolosa, E.; Litvan, I.; Höglinger, G. U.; Burn, D.; Lees, A.; Andrés, M. V.; Gómez-Carrillo, B.; León, T.; del Ser, T. Mov. Disord. 2014, 29, 470. doi: 10.1002/mds.25824
Del, S. T.; Steinwachs, K. C.; Gertz, H. J.; Andrés, M. V.; Gómezcarrillo, B.; Medina, M.; Vericat, J. A.; Redondo, P.; Fleet, D.; León, T. J. Alzheimer's Dis. 2013, 33, 205. http://www.ncbi.nlm.nih.gov/pubmed/22936007
Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J. O.; Huppertz, H. J.; Calero, M.; Andrés, M. V.; Gómezcarrillo, B.; León, T. J. Alzheimer's Dis. 2015, 45, 75. doi: 10.3233/JAD-141959
Zhang, P.; Hu, H.-R.; Huang, Z.-H.; Lei, J.-Y.; Chu, Y.; Ye, D.-Y. Bioorg. Med. Chem. Lett. 2012, 22, 7232. doi: 10.1016/j.bmcl.2012.09.043
Liang, Z.; Zhang, B.; Su, W. W.; Williams, P. G.; Li, Q. X. ACS Chem. Neurosci. 2016, 7, 912. doi: 10.1021/acschemneuro.6b00059
Avrahami, L.; Licht-Murava, A.; Eisenstein, M.; Eldar-Finkelman, H. Biochim. Biophys. Acta 2013, 1834, 1410. doi: 10.1016/j.bbapap.2013.01.016
Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M. J.; Castro, A.; Morales, S.; Navarro, M. L.; del Monte-Millán, M.; Medina, M.; Pennaka, H.; Balaiah, A.; Peng, J.; Cook, J.; Wahyuono, S.; Martínez, A. J. Nat. Prod. 2007, 70, 1397. doi: 10.1021/np060092r
Tapia-Rojas, C.; Schüller, A.; Lindsay, Carolina B.; Ureta, Roxana C.; Mejías-Reyes, C.; Hancke, J.; Melo, F.; Inestrosa, Nibaldo, C. Biochem. J. 2015, 466, 415. doi: 10.1042/BJ20140207
Palomo, V.; Perez, D. I.; Perez, C.; Morales-Garcia, J. A.; Soteras, I.; Alonso-Gil, S.; Encinas, A.; Castro, A.; Campillo, N. E.; Perez-Castillo, A.; Gil, C.; Martinez, A. J. Med. Chem. 2012, 55, 1645. doi: 10.1021/jm201463v
Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. Sci. Signaling 2016, 9, ra110. doi: 10.1126/scisignal.aah7102
Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D. I.; Martínez, A.; Luque, F. J.; Medina, M. Eur. J. Med. Chem. 2013, 60, 479. doi: 10.1016/j.ejmech.2012.12.014
Pérez, M.; Pérez, D. I.; Martínez, A.; Castro, A.; Gómez, G.; Fall, Y. Chem. Commun. 2009, 3252. http://www.ncbi.nlm.nih.gov/pubmed/19587930
Palomo, V.; Soteras, I.; Perez, D. I.; Perez, C.; Gil, C.; Campillo, N. E.; Martinez, A. J. Med. Chem. 2011, 54, 8461. doi: 10.1021/jm200996g
Palomo, V.; Perez, D. I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J. A.; Reyes, J. A.; Campillo, N. E.; Perez-Castillo, A. M.; Rubin, L. L.; Timchenko, L.; Gil, C.; Martinez, A. J. Med. Chem. 2017, 60, 4983. doi: 10.1021/acs.jmedchem.7b00395
Perez, D. I.; Conde, S.; Pérez, C.; Gil, C.; Simon, D.; Wandosell, F.; Moreno, F. J.; Gelpí, J. L.; Luque, F. J.; Martínez, A. Bioorg. Med. Chem. 2009, 17, 6914. doi: 10.1016/j.bmc.2009.08.042
Perez, D. I.; Palomo, V.; Pérez, C.; Gil, C.; Dans, P. D.; Luque, F. J.; Conde, S.; Martínez, A. J. Med. Chem. 2011, 54, 4042. doi: 10.1021/jm1016279
Lu, F.; Li, X.; Li, W.; Wei, K.; Yao, Y.; Zhang, Q.; Liang, X.; Zhang, J. Acta Biochim. Biophys. Sin. 2017, 49, 722. doi: 10.1093/abbs/gmx059
Wang, D.; Li, S.; Chen, J.; Liu, L.; Zhu, X. Cell. Mol. Neurobiol. 2017, 37, 695. doi: 10.1007/s10571-016-0405-9
(a) Di Martino, R. M. C. ; De Simone, A. ; Andrisano, V. ; Bisignano, P. ; Bisi, A. ; Gobbi, S. ; Rampa, A. ; Fato, R. ; Bergamini, C. ; Perez, D. I. ; Martinez, A. ; Bottegoni, G. ; Cavalli, A. ; Belluti, F. J. Med. Chem. 2016, 59, 531.
(b) Tang, M. ; Taghibiglou, C. J. Alzheimer's Dis. 2017, 58, 1003.
Hawkes, N. BMJ[Br. Med. J.] 2018, 360, DOI: 10.1136/bmj.k122.
Hawkes, N. BMJ[Br. Med. J.] 2017, 356, DOI: 10.1136/bmj.j845.

图 2 苯并[e]异吲哚-1, 3-二酮衍生物的结构
Figure 2 Structures of benzo[e]isoindole-1, 3-dione derivatives

图 13 底物竞争性抑制剂L803mts的结构
Figure 13 Structure of substrate competitive inhibitor L803mts

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:












