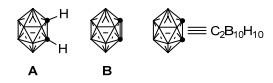
图 1.
邻碳硼烷(A)与邻碳硼炔(A)的结构
Figure 1.
Structures of o-carborane (A) and o-carboryne (A)

碳硼烷是二十面体硼氢笼分子内一个或多个B—H顶点被C—H单元取代的化合物[1~4], 其中相邻的两个B—H顶点被C—H单元取代所形成的碳硼烷称为邻碳硼烷(图 1, A)[5~7].邻碳硼烷在硼中子捕获疗法(Boron Neutron Capture Therapy, BNCT)[8~12]、超分子设计[13, 14]和金属有机化学[15~17]等诸多领域都具有潜在的应用价值.一方面, 邻碳硼烷具有不同寻常的反应性, 比如在一定的条件下可脱去两个C—H单元上的取代基而形成具有特殊反应性的邻碳硼炔(图 1, B)[18, 19].邻碳硼炔也称为1, 2-脱氢邻碳硼烷, 是一种活泼中间体, 与苯炔[20, 21]具有某些相似的化学反应性[22~26].另一方面, 由于完全填充的键合分子轨道系统的存在, 邻碳硼烷又具有一定的热稳定性.因此, 邻碳硼烷的衍生化和官能化成了该领域的一个核心和关键问题.
近十年来, 人们在邻碳硼烷的C—H和B—H活化和官能化方面取得了显著成就[27~40], 尤其在该反应的条件优化、产率提升、化学和区域选择性控制等方面取得了长足发展[41~46].据报道, 这些通过邻碳硼烷的C—H和B—H活化而得到的官能化产物在催化合成[47~50]、聚合物制备[16, 51]、药物设计[6, 10, 52]、癌症治疗[10, 11, 53]、金属有机骨架[54]、电子设备[55~57]等方面都有着广阔的应用前景[58~59].尤为引人注目的是, 邻碳硼炔作为一种有用的合成子, 可与多种不饱和有机分子发生环加成反应, 用以制备种类繁多的有机硼烷化合物[60~65].所得到的有机硼烷化合物在光电材料、化学传感器、催化合成等诸多领域都具有潜在的应用价值[2, 42, 66].
目前关于邻碳硼烷衍生化的实验研究成果已有多篇综述可查, 但其中绝大多数为英文文献[2, 16, 42, 66~70], 相应的中文综述[71, 72]屈指可数, 且鲜有涉及理论研究成果的综述类报道.为了深化读者对邻碳硼烷衍生化和功能化的认识, 本文以邻碳硼烷和邻碳硼炔与不饱和化合物的[2+2+2]、[2+2+1]、[2+2]、[3+2]、[4+2]、[5+2]环加成反应为重点, 对近十年来国内外课题组在该成领域所取得的实验和理论研究成果进行了综述.
[2+2+2]环加成反应是构筑六元环化合物的重要而有效的方法之一, 已被广泛应用于苯、萘、蒽、菲及其衍生物的合成[73~76].基于对过渡金属碳硼烷和过渡金属碳硼炔化合物中金属碳笼键(M—Ccage)的大量研究, 2006年至2012年间, 香港中文大学的Xie研究组[60, 77, 81]陆续报道了一系列邻碳硼烷与不饱和烃的[2+2+2]环加成反应, 如镍催化碳硼炔与炔烃的三组分[2+2+2]环加成反应、Pd/Ni共催化邻碳硼炔与2 equiv.炔烃的[2+2+2]环加成反应等, 并成功制备得到了相应的苯基碳硼烷和二氢苯基碳硼烷衍生物.这些反应大多具有反应条件温和、立体选择性和区域选择性好等优点.
鉴于镍苯炔络合物能与炔烃反应生成取代萘的实验事实, 基于对镍苯炔和镍碳硼炔络合物的对比分析, 2006年, Deng等[60]率先用1, 2-二氢邻碳硼烷与2 equiv.的炔烃、在Ni(PPh3)2Cl2和nBuLi存在的条件下, 于四氢呋喃(THF)溶剂中通过[2+2+2]环加成反应制备得到了相应的1, 2-苯并邻碳硼烷衍生物(Scheme 1).当以不对称炔烃为反应底物时, 该反应主要得到炔烃“头-尾-头-尾”相接的产物P1, 具有很好的区域选择性.作者推测邻碳硼烷A可能在nBuLi的作用下转化为1, 2-二锂邻碳硼烷C1, C1与Ni(PPh3)2Cl2进一步作用生成镍碳硼炔络合物B1.之后一分子的炔烃插入B1的镍碳笼(Ni—Ccage)键形成碳硼烷基镍环戊烯中间体D1.紧接着, 第二分子的炔烃继续插入镍环戊烯中间体D1的Ni—C键形成七元镍环化合物E1.最后, E1发生还原消除形成1, 2-苯并邻碳硼烷衍生物P1.由于该反应主要形成炔烃“头-尾-头-尾”相接的产物, 作者认为体积庞大的碳硼烷基对控制该反应的区域选择性有重要作用.这些推测被后来Mu等[82]的理论计算所证实.
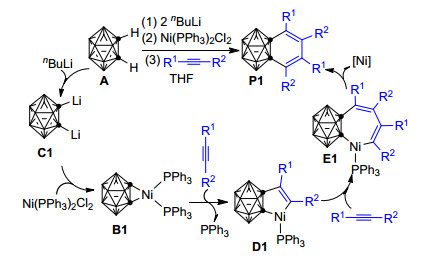
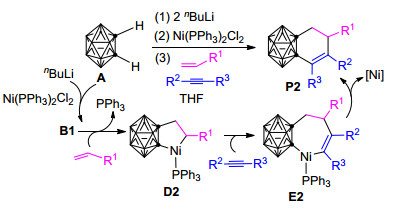
2009年, Qiu等[77]又报道了碳硼炔与烯烃和炔烃在Ni(PPh3)2Cl2和nBuLi共同作用下的[2+2+2]环加成反应.该反应在THF溶剂中、110 ℃的条件下可以有效实现二氢苯基碳硼烷衍生物的合成(Scheme 2).作者认为该反应中活泼烯烃的取代基R1上的杂原子(如O、N等)所带有的孤对电子可能与Ni(PPh3)2Cl2中Ni原子的空d轨道配位, 从而使烯烃插入镍碳硼炔络合物B1, 所形成的五元环中间体D2 (Scheme 2)更加稳定.换言之, 与炔烃相比, 活泼烯烃更易于优先插入镍碳硼炔络合物B1的Ni—Ccage键形成五元镍环中间体D2.之后, 炔烃进一步插入镍环戊烷中间体D2的Ni—C键形成七元镍环化合物E2.最后, E2发生还原消除形成二氢苯基碳硼烷衍生物P2.该反应中, 由邻碳硼烷A形成镍碳硼炔中间体B1的过程与上述Scheme 1中邻碳硼烷与炔烃的反应相同, 自D2之后的反应机理也与上述反应类似.可能由于碳硼烷基镍环戊烯D1与碳硼烷基镍环戊烷D2的性质差异, 炔烃插入D2的Ni—C键比插入D1的Ni—C键更加困难, 因此该反应的最佳实验温度比邻碳硼烷与炔烃的反应温度高出了20 ℃[77].
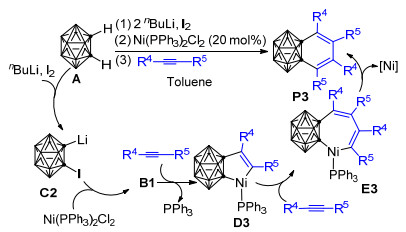
2010年, Qiu等[78]又以Ni(PPh3)2Cl2为催化剂、在nBuLi和I2存在的条件下, 实现了用1, 2-二氢邻碳硼烷与2 equiv的炔烃制备1, 2-苯并邻碳硼烷衍生物的反应(Scheme 3)[78].与上述Ni(PPh3)2Cl2和nBuLi共同促进的反应不同, 作者推测该反应中的1, 2-二氢邻碳硼烷可能首先与nBuLi和I2反应形成1-碘-2-锂-邻碳硼烷前体C2, 之后C2在催化剂Ni(PPh3)2Cl2的帮助下脱去LiI形成镍碳硼炔络合物B1.自B1之后的反应历程与Scheme 1中的反应历程类似:两分子炔烃依次插入镍碳硼炔络合物B1中的Ni—Ccage键和五元环镍环戊烯中间体D3中的Ni—C键, 形成七元镍环化合物E3, 最终E3通过还原消除形成具有良好区域选择性的1, 2-苯并邻碳硼烷衍生物P3.
随后, 该研究组[79]又发现:在nBuLi存在条件下, 由Pd/Ni共催化可以实现1, 3-脱氢邻碳硼烷与2 equiv炔烃的[2+2+2]环加成反应, 并成功制备得到相应的C, B-取代邻碳硼烷衍生物P4 (Scheme 4).基于前人的研究成果[60, 83, 84]和本实验中的系列控制实验结果, 作者推测该反应中经历了一个由Pd到Ni的转金属化过程.整个反应的可能机理为:反应底物1-氢-2-R1-3-碘-邻碳硼烷A1首先在nBuLi的作用下形成1-锂-2-R1-3-碘-邻碳硼烷C3, 之后Pd(0)催化剂氧化插入B—I键, 并消除一分子的LiI转化为钯邻碳硼炔络合物B2. B2与Ni(0)催化剂经转金属化形成活性更高的镍邻碳硼炔络合物B3. B3的性质与Scheme 1中的B1类似, 其与炔烃的后续反应历程、反应选择性也与B1和炔烃的反应类似, 最终通过还原消除反应, 在实现催化剂再生的同时生成相应的区域选择性1, 3-苯并碳硼烷衍生物P4.本实验中由Pd到Ni的转金属化一方面克服了Ni(0)催化剂无法活化碳硼烷中B—I键的困难, 另一方面也解决了钯邻碳硼炔络合物B2对炔烃插入惰性的的难题, 为邻碳硼烷中碳原子和硼原子的同时官能化提供了一种新思路, 同时也从侧面证实了金属-1, 3-脱氢邻碳硼烷可以作为一种新型的硼亲核试剂.作者认为该反应中炔烃对镍邻碳硼炔络合物B3中Ni—Bcage键插入的绝对优势主要得益于Ni—Bcage键的亲核性比Ni—Ccage键强, 而整个反应的区域选择性则是电子效应和空间位阻共同作用的结果, 但目前尚未见到有关该反应的区域选择性的理论研究报道.
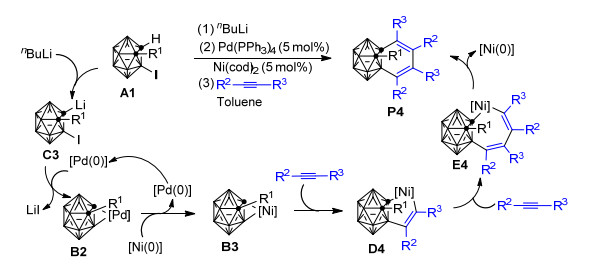
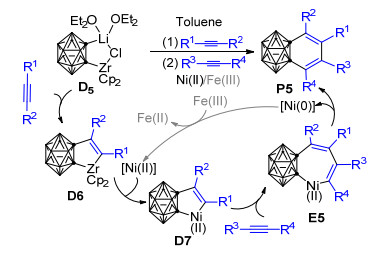
受Pd/Ni共催化1, 3-脱氢邻碳硼烷与炔烃[2+2+2]环加成反应的启发, 基于之前所做的研究, Ren等[80]接着报道了一种以“一锅法”或“两步”的方式将含有碳硼烷基单元的锆环戊烯转化为镍环戊烯或铁环戊烯化合物、并进一步与炔烃发生三组分[2+2+2]环加成反应的新方法(Scheme 5).该方法可用于在110 ℃的甲苯溶液中制备一系列苯并碳硼烷衍生物. Qiu[77, 78]、Deng[60]和Ren等[80, 81]的实验结果显示, 当以Ni(Ⅱ)为促进剂时, 该反应可能经过了以下历程:首先碳硼烷基二茂锆前体D5和炔烃作用生成锆环戊烯中间体D6, 随后D6与Ni(Ⅱ)试剂作用、通过Zr到Ni的转金属化生成五元镍环中间体D7.紧接着, 另一分子炔烃插入D7的Ni—C键形成七元镍环中间体E5, 最后E5发生还原消除得到1, 2-苯并邻碳硼烷衍生物P5.当以FeCl3为促进剂时, 由Zr到Fe的转金属化与由Zr到Ni的转金属化类似, 之后炔烃的插入反应也跟使用Ni(Ⅱ)为促进剂的反应历程类似.值得注意的是, 实验发现当有过量FeCl3存在时, 催化量的镍与1 equiv.镍对该反应具有同样的促进效果, 这可能是由于FeCl3有助于将发生还原消除后的Ni(0)重新氧化为Ni(Ⅱ)所致. Ren等还进一步尝试了NiCl2促进碳硼炔与炔烃和非活化烯烃的[2+2+2]环加成反应, 并取得了成功.他们发现在THF溶剂中, NiCl2能够有效促成五元锆环戊烷与炔烃反应生成二氢苯并碳硼烷衍生物, 而且当使用非对称炔烃时能够得到较好的区域选择性产物[81].上述实验的成功意味着基于锆环化物的反应取得了新进展, 这对于深入认识基于碳硼烷基二茂锆的反应具有十分重要的意义.但关于转金属化的微观反应机理还有待进一步的理论研究来加以确证和支撑.
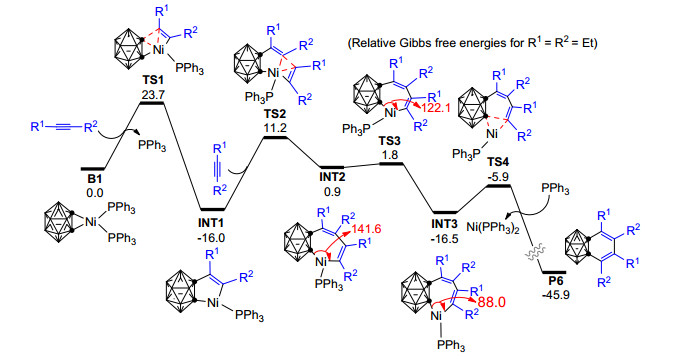
为了深入探讨邻碳硼烷/邻碳硼炔与炔烃的三组分[2+2+2]环加成反应的微观反应机理和反应选择性问题, 2015年, Mu等[82]应用密度泛函理论对镍催化碳硼炔与两分子炔烃的[2+2+2]环加成反应开展了理论研究.计算结果显示, 该反应的最有可能机理为:第一分子炔烃亲核进攻镍碳硼炔络合物B1形成镍环戊烯中间体INT1, 随后第二分子炔烃插入INT1中的Ni—C键, 形成镍环庚二烯中间体INT2, 后经INT2的异构化(INT2→INT3)和催化剂再生(INT3→P6)过程生成苯并碳硼烷衍生物P6(图 2).其中第二分子炔烃的插入过程(TS2)为整个反应的决速步, 该步在IDSCRF-B3LYP/DZVP计算水平上需要克服27.3 kcal·mol-1的Gibbs自由能垒, 与实验上90 ℃时反应4 d产率为67%很好吻合. Mu等的工作从理论计算的角度对该反应的微观机理、最优反应路径和决速步等给出了详细的解释, 在验证了Xie等[60]基于实验提出的反应机理的同时, 还从反应动力学层面对实验中所观测到的区域选择性结果给出了合理的理论解释.
五元杂环是众多天然产物和药用分子的基本骨架.作为构筑五元环化合物的常用手段之一, [2+2+1]环加成反应同样为合成结构多样的碳硼烷衍生物提供了行之有效的方法.基于之前对Zr/Ni共促邻碳硼炔与炔烃的三组分[2+2+2]环加成反应的研究成果, 2013年, Quan等[61]开发了一种全新的Zr/Ni共促邻碳硼烷与炔烃和非活化烯烃的[2+2+1]环加成反应, 进而开启了邻碳硼烷与不饱和烃的[2+2+1]三组分环加成反应的新征程.
该研究小组首先从Zr/Ni共促邻碳硼炔与炔烃和非活化烯烃的[2+2+2]环加成反应[80, 81]中分离得到了相应的[2+2+1]五元环产物, 如Scheme 6中所示的P7[61].之后他们通过条件优化, 发现在Ni(PMe3)2Cl2存在条件下, 碳硼烷基锆环戊烷配合物D8与炔烃和烯烃可通过[2+2+1]环加成反应选择性地形成相应的碳硼烷衍生物P7 (Scheme 6).该研究小组还发现:炔烃上的取代基对五元环碳硼烷衍生物P7的形成起着关键作用——炔烃上的三甲基硅烷基(TMS)对于二氢亚甲基环戊二烯碳硼烷衍生物P7的形成是必不可少的.另外, 该[2+2+1]环加成反应产物的顺反(Z/E)选择性跟炔烃上的取代基R2和TMS的相对大小有直接关系:当R2的体积比TMS的体积大时, 主要得到顺式(Z)构型产物; 而当R2的体积比TMS的体积小时, 主要得到反式(E)构型产物.同时, 如果R2上带有能与Ni配位的给电子原子基团时, 也主要得到Z-构型产物.这些实验结果说明炔烃所带有的取代基大小和Ni(PMe3)2Cl2上的膦配体对该反应的选择性控制有重要作用.基于已有的实验结果, 作者推测该反应的可能机理为:碳硼烷基锆环戊烷D8与镍配合物作用, 经转金属化形成五元镍环戊烷中间体D9.之后炔烃与镍络合, 经三甲基硅烷基的1, 2-迁移形成镍亚乙烯基中间体F1.最后, 中间体F1中亚乙烯基卡宾发生迁移形成中间体G1, 并经过还原消除转化为二氢亚甲基环戊二烯碳硼烷衍生物P7[61].
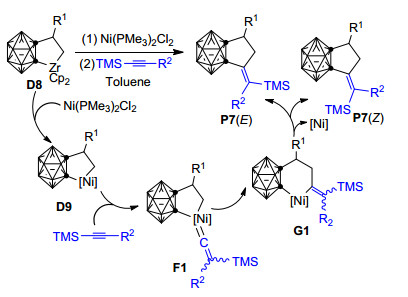
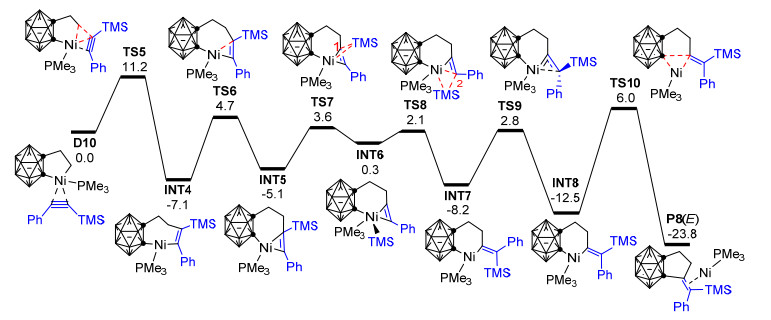
为了更好地理解上述反应的取代基效应和Z/E选择性问题, 同时验证Scheme 6中的镍亚乙烯基中间体F1是否确实位于最优反应路径上, 2014年, Zhang等[85]应用密度泛函理论的B3PW91方法、在B3PW91/ccpVTZ&6-31G**计算水平上对该反应进行了理论研究.他们的计算结果表明: Quan等基于实验结果所推测的通过镍亚乙烯基中间体F1的反应机理从动力学和热力学角度看均不是最有利的, 该反应最有可能的反应机理如图 3所示.四甲基硅烷基芳基炔烃首先通过过渡态TS5插入碳硼烷基镍环戊烷D10中的Ni—C键, 形成η1-型烯基镍配合物INT4, 之后烯基配位模式由η1重排为η2并异构化为中间体INT5.紧接着, TMS基团依次发生从C(1)到Ni (TS7)、Ni到C(2) (TS8)的1, 2-σ迁移, 经过中间体INT6转化为乙烯基碳硼烷基镍环己烷中间体INT7.中间体INT7通过双键旋转过渡态TS9转化为中间体INT8, 最后INT8发生还原消除形成碳硼烷基环戊烷衍生物P8 (E).作者认为不饱和的镍金属中心对双键旋转过渡态TS9起到了关键的稳定作用, 从而使反式碳硼烷基环戊烷衍生物P8 (E)的形成得以实现.当四甲基硅烷基炔烃上带有较大的芳基取代基时, 由于双键旋转受阻, 将主要形成Z-构型的[2+2+1]环加成产物.这些结果说明该反应的Z/E选择性主要受炔烃上TMS基团和芳基大小的影响, 与Quan等的实验结果[61]很好吻合.另外, 作者还对该反应发生[2+2+1]和[2+2+2]环化反应的化学选择性进行了探讨, 发现以烷基炔烃为底物时, 由于受电子效应的影响, 碳硼烷基锆环戊烷(Scheme 6, D8)将与烷基炔烃主要发生[2+2+2]环加成反应形成苯并碳硼烷衍生物. Zhang等的理论研究不仅阐明了该反应的微观机理, 还对该反应的化学和Z/E选择性给出了很好的理论解释, 有效深化了人们对邻碳硼烷与不饱和烃环加成反应的认识, 但目前关于碳硼烷基锆环戊烷(Scheme 6, D8)向碳硼烷基镍环戊烷(D9)的转金属化机理尚未见有文献报道.
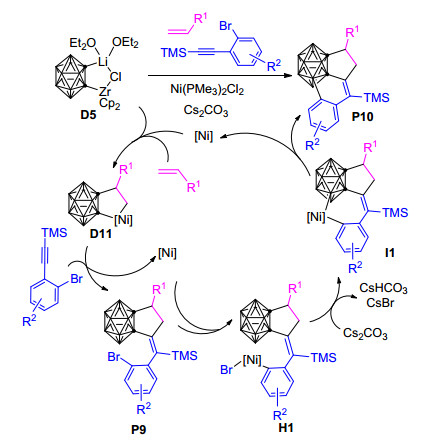
基于上述邻碳硼烷与烯烃和三甲基硅烷基芳基炔烃的[2+2+1]环加成反应的实验和理论研究成果, Quan等[86]进一步尝试了Zr/Ni共促碳硼炔与烯烃和2-溴苯基三甲基硅烷基乙炔的“一锅法”[2+2+1]环加成反应, 发现该反应在形成碳硼烷基环戊烷衍生物P9之后, 苯基上的Br取代基能够与邻碳硼烷基α位上的B—H结合, 脱掉一分子的HBr形成一个新的六元环, 最终制备得到一系列的C, C, B-取代的碳硼烷基稠合三环衍生物P10 (Scheme 7).该工作代表了通过B—H活化实现碳硼笼上的硼与苯基上的碳直接偶联的第一个实例, 对制备新的碳硼烷基稠合多环化合物提供了很好的思路, 但相应的微观反应历程、动力学控制因素以及所添加碱Cs2CO3的作用机制等, 都还有待于更深入的实验和理论研究来加以揭示.
由于四元环化合物广泛存在于具有药用价值或生物活性的分子骨架中, 高效构建四元环骨架一直都是合成化学领域的热点问题之一. [2+2]环化反应作为构筑四元环化合物的重要方式, 也被成功应用到邻碳硼烷与不饱和化合物的[2+2]环加成反应中, 并制备得到一系列碳硼烷基稠合的环丁烯和环丁烷衍生物[62, 87].
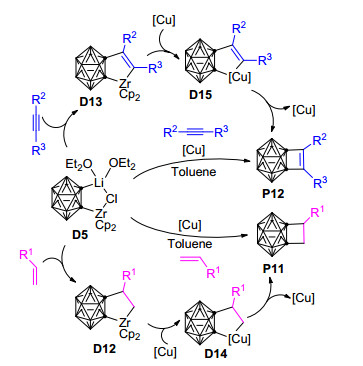
与碳硼烷基镍环戊烷和碳硼烷基镍环戊烯[60, 77, 78, 80]不同, 碳硼烷基锆环戊烷和碳硼烷基锆环戊烯对于不饱和有机分子如炔烃的进一步插入是惰性的[88], 因此, 只有通过与金属卤化物如Ni(PMe3)2Cl2、FeCl3等相互作用并进行转金属化, 才有可能产生活性反应中间体, 进而合成结构多样的功能碳硼烷衍生物.换言之, 由Zr到Ni的转金属化在Zr/Ni共促邻碳硼烷与炔烃/烯烃和炔烃的三组分[2+2+2]或[2+2+1]环加成反应中起着关键作用, 对于这些反应的实现和完成都有着十分重要的影响.基于上述Zr/Ni共促邻碳硼烷与炔烃/烯烃和炔烃的三组分[2+2+2]和[2+2+1]环加成反应的研究结果, 2014年, Yuan等[62]开发了一种通过Zr/Cu共促邻碳硼烷与炔烃/烯烃的[2+2]环加成反应制备碳硼烷基环丁烯和环丁烷的新方法(Scheme 8).该反应在甲苯溶剂中, 室温下反应24~48 h可得到不错的产率, 且对烷基和芳基取代的烯烃和炔烃具有较好的兼容性, 是制备碳硼烷基环丁烯和环丁烷衍生物的行之有效的方法.参考之前Pd/Ni和Zr/Ni共促反应的可能机理(Schemes 4~7)[61, 79, 80, 86], 此Zr/Cu共促反应的可能机理为:碳硼烷基二茂锆配合物D5首先与烯烃或炔烃反应转化为碳硼烷基锆环戊烷中间体D12或碳硼烷基锆环戊烯中间体D13.之后中间体D12或D13与Cu(OTf)2作用, 通过转金属化形成相应的碳硼烷基铜环戊烷中间体D14或碳硼烷基铜环戊烯中间体D15.最后, D14和D15发生还原消除反应得到相应的碳硼烷基环丁烷(P11)或碳硼烷基环丁烯(P12)产物.
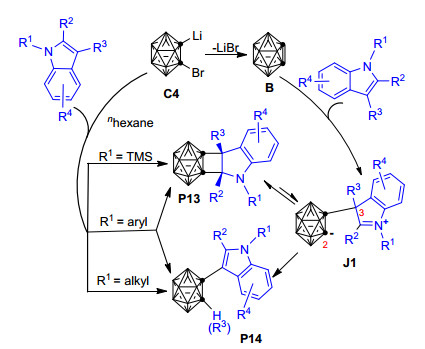
继Yuan等[62]实现Zr/Cu共促邻碳硼烷与炔烃/烯烃的[2+2]环加成反应制备碳硼烷基环丁烯和环丁烷衍生物之后, Zhao等[87]又发现邻碳硼炔能与N-TMS取代吲哚通过[2+2]环加成反应生成相应的四元环产物(Scheme 9, P13), 而N-烷基取代吲哚则倾向于发生常规的C—H插入反应生成P14.当以N-芳基吲哚为底物时, 实验上能同时检测到[2+2]环加成产物P13和C—H插入产物P14, 两个产物的比例取决于芳环上的取代基的性质.一般而言, 芳环上带吸电子基有利于[2+2]环加成产物的形成, 而带给电子基则有利于C—H插入产物的生成.为了解释上述实验结果, 作者进一步开展了相应的控制实验和同位素标记实验, 并推测该反应的可能机理为: 1-溴-2-锂-邻碳硼烷前体C4首先消去LiBr形成活性邻碳硼炔中间体B, 随后活泼的邻碳硼炔中间体B亲电进攻N-取代吲哚的C(3)位置形成两性离子中间体J1. J1通过分子内亲核加成形成[2+2]环加成产物P13, 或通过质子转移形成常规的C—H插入产物P14 (Scheme 9).作者认为N-芳基保护吲哚芳环上的吸电子基可能有利于增加两性离子中间体J1中C(2)位置的亲电性, 从而使[2+2]环加成反应更加容易进行.从目前的研究结果看, 该反应的化学选择性主要取决于N原子上的保护基团R1的性质:当R1为TMS基团时, 唯一形成[2+2]环加成产物P13; 当R1为烷基如Me时, 则主要形成C—H插入产物P14; 当R1为芳基时, 则可同时生成P13和P14[87].本实验是吲哚在无过渡金属和无光照条件下发生[2+2]环加成反应的首个应用实例, 为制备在医学和材料科学领域[12, 53, 54, 89]有重要应用价值的碳硼烷基官能化吲哚和其它杂环化合物提供了一种有效手段.
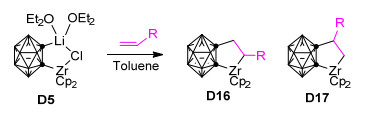
与[2+2+1]环加成反应相比, [3+2]环加成反应是一种更为常见和通用的构筑五元环化合物的重要方法, 但目前关于碳硼烷参与的[3+2]环加成反应的报道相对较少.邻碳硼烷可与不饱和分子如烯烃、酮、腈、亚胺等发生[3+2]环加成反应, 生成相应的碳硼烷基五元环化合物[63, 90, 91]. 2012年, Ren等[90]首次报道了碳硼烷基二茂锆D5与烯烃的[3+2]环加成反应(Scheme 10).该反应在甲苯中回流48 h能得到较高产率的碳硼烷基锆环戊烷衍生物D16或D17, 反应的区域选择性受烯烃中C=C双键极性的影响.通常, 当R为芳基取代基时主要得到α位产物D16, 而R为烷基取代基时优先生成β位产物D17.该反应是合成碳硼烷基锆环戊烷衍生物的有效途径, 对于Zr/Ni[61, 80, 81, 86]、Zr/Cu[62]共促邻碳硼烷与不饱和烃的[2+2+2]、[2+2+1]和[2+2]等环加成反应具有重要的奠基意义.
紧接着, Ren等[63]通过1, 2-二锂邻碳硼烷C5和金属化合物Cl2ML2 (M=Ti, Zr, Hf, L=[η2-R2C(NR1)2])的复分解反应制备得到了较高产率的金属碳硼炔络合物B4 (Scheme 11).该金属碳硼炔络合物B4可与酮、腈等极性不饱和分子经不饱和键插入M—Ccage键形成五元杂环产物D18, 也能与累积二烯如碳二亚胺、异氰酸酯、硫氰酸酯、二硫化碳、叠氮苯等反应形成种类繁多的碳硼烷基五元杂环产物, 但不与吡啶、炔烃和烯烃反应.上述实验结果表明:金属碳硼炔络合物B4与上述碳硼烷基二茂锆配合物D5 (Scheme 10)对极性不饱和分子具有相似的反应性, 但对吡啶、炔烃和烯烃的反应性却有着显著差异.作者推测该反应性差异可能是由配体的空间位阻引起的, 但具体的影响机制还有待进一步的实验与理论研究来加以探究和证实.
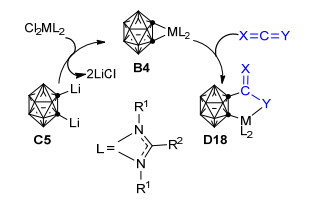
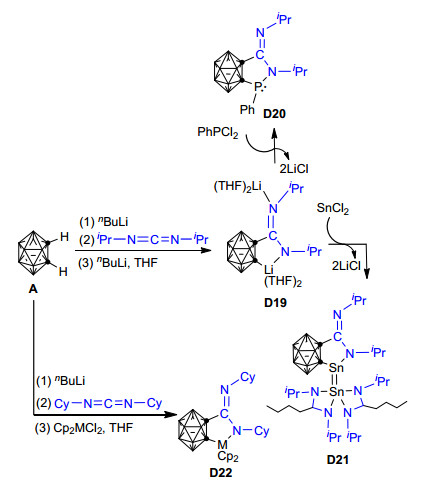
一年后, Edelmann等[91]对邻碳硼烷与亚胺衍生物经[3+2]环加成反应合成五元杂环化合物的反应进行了深入的研究, 发现邻碳硼烷A能与二异丙基碳二亚胺在正丁基锂存在的条件下转化为五元环的碳硼烷基脒基锂盐D19, 并且产率可高达83% (Scheme 12).该碳硼烷基脒基锂盐可进一步与PhPCl2或SnCl2反应, 生成结构新颖的碳硼烷基脒基磷氮杂环戊烷衍生物D20或双金属化的碳硼烷基脒基锡氮杂环戊烷衍生物D21, 这是关于以邻碳硼烷“一锅法”直接高效合成双金属化的碳硼烷基脒基锡氮杂环戊烷衍生物的首次报道.此外, 当正丁基锂和二茂锆(或二茂钛)化合物同时存在时, 邻碳硼烷A还可与二环己基碳二亚胺在THF溶剂中反应生成相应的五元金属氮杂环戊烷衍生物D22. Edelmann等的研究成果为从邻碳硼烷和脒基阴离子高效合成无机杂环化合物提供了一种新策略, 有效拓展了碳硼烷衍生物的合成途径和应用范围.
多年的研究表明, 苯炔及其衍生物可作为亲双烯体参与[2+1]、[2+2]或[4+2]环加成反应[20, 92~95].跟苯炔与多环芳烃的[4+2]环加成反应类似, 由邻碳硼烷前体1-溴-2-锂-邻碳硼烷、1-碘-2-锂-邻碳硼烷、1-三甲基硅烷基-2-醋酸碘苯基-邻碳硼烷等[18, 60, 77, 78, 96, 97]制备得到的邻碳硼炔活泼中间体B亦可与苯及其衍生物发生[4+2]环加成反应生成相应的六元环产物. 2010年, Wang等[98]用1-碘-2-锂-邻碳硼烷与苯溶液加热得到了相应的[4+2]环加成产物P15 (Scheme 13), 同时还伴随生成了环辛四烯基碳硼烷衍生物P17.
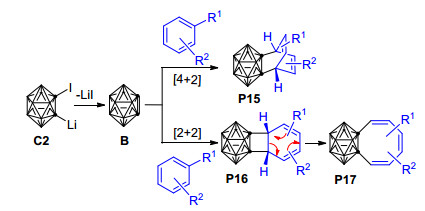
实验发现, 以1-碘-2-锂-邻碳硼烷为邻碳硼炔前体与苯的反应比以1-溴-2-锂-邻碳硼烷为前体的反应产率高, 说明1-碘-2-锂-邻碳硼烷是比1-溴-2-锂-邻碳硼烷更好的邻碳硼炔前体.另外, 根据在甲苯中(R1=Me, R2=H, Scheme 13)仅形成[4+2]环加成产物而在苯甲醚中(R1=OMe, R2=H)可同时形成[4+2]环加成产物和环辛四烯基碳硼烷衍生物的实验事实, 可推知电子效应对控制该反应的化学选择性具有一定的作用; 其他多组对比实验则表明空间因素对该反应的化学和区域选择性都有重要影响.基于一系列的控制实验结果, 作者推测该反应的可能机理如下: 1-碘-2-锂-邻碳硼烷前体C2首先消除一分子的LiI形成邻碳硼炔活泼中间体B, 之后B与苯甲醚发生[4+2]环加成反应生成产物P15, 或者发生[2+2]环加成反应生成产物P16. P16经6-π电开环过程最终形成环辛四烯基碳硼烷衍生物P17 (Scheme 13).芳烃与邻碳硼炔的脱芳构化反应为合成环辛四烯基碳硼烷衍生物提供了一种新策略.基于之前的研究成果, Wang等于2012年实现了由1-碘-2-锂邻碳硼烷前体C2所产生的邻碳硼炔B与烷基苯[99]、苯乙烯及其衍生物[100]的[4+2]环加成反应, 并成功制备得到了一系列相应的[4+2]环化产物(Scheme 14).在邻碳硼炔与烷基苯的反应中, 当苯环上带有位阻不同的烷基取代基时, 得到两种不同的[4+2]环化主产物, 说明空间位阻对于该反应的区域选择性具有明显的控制作用.比如以甲苯(R1=Me, R2=H)为反应底物时, 主要得到产物P19; 而当以叔丁基苯(R1=tBu, R2=H)为底物时, 则主要得到产物P18 (Scheme 14).此外, 由于该[4+2]环加成反应具有可逆性, 相应的[4+2]环加成产物可在热解条件下作为邻碳硼炔的来源.而在邻碳硼炔与苯乙烯及其衍生物的反应中, 苯乙烯衍生物的环外双键和一个相邻的芳环双键共同扮演了双烯角色, 与邻碳硼炔B通过[4+2]环加成反应形成了性质极为活泼的中间产物K1, 该中间产物可通过1, 3-氢迁移形成更稳定的二氢萘基邻碳硼烷P20, 或通过脱氢反应重新芳构化形成萘基邻碳硼烷衍生物P21 (Scheme 14).对比邻碳硼炔和苯炔与苯乙烯及其衍生物的反应可以发现:邻碳硼炔和苯炔与苯乙烯及其衍生物的反应具有相似的一面.但由于碳硼烷基的空间位阻作用, 邻碳硼炔和苯乙烯的反应又具有许多独特的性质.比如, 邻碳硼炔能与苯乙烯发生[2+2]环加成反应而苯炔不能, 苯炔能与相应的[4+2]环加成产物进一步反应而邻碳硼炔则不能等[100].
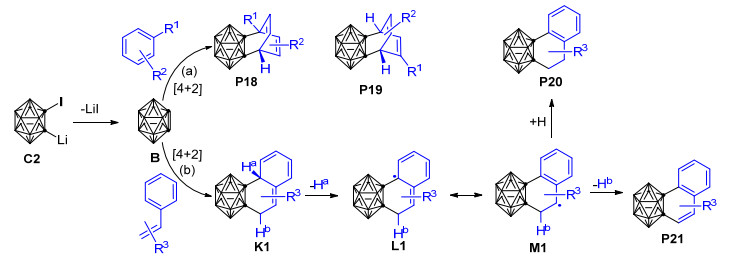
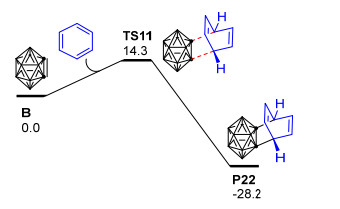
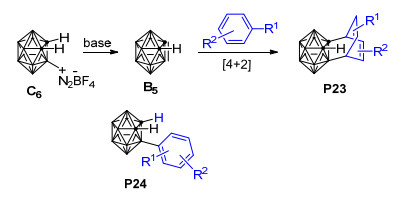
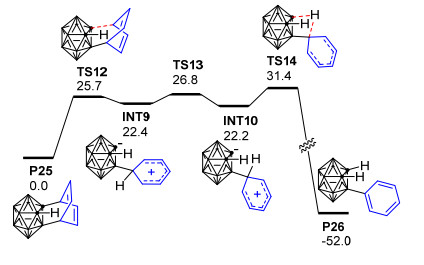
值得一提的是, 最近, Huo等[101]用密度泛函理论的M11方法和6-311+G(d)基组对邻碳硼炔B和苯的[4+2]环化反应开展了理论研究, 发现邻碳硼炔和苯可以通过一个协同过渡态TS11形成[4+2]环加成产物P22, 相应的自由能垒仅为14.3 kcal·mol-1(图 4).此外, 作者还对邻碳硼炔与苯之间可能发生的[2+2]环加成、C—C插入反应和C—H插入反应都进行了对比计算, 发现这些反应的能垒都比上述[4+2]环化反应的能垒高很多, 说明邻碳硼炔和苯主要发生[4+2]环化反应生成六元环碳硼烷衍生物.该理论计算结果与Wang等[98]关于邻碳硼炔和甲苯的实验结果吻合较好.目前, 虽然关于邻碳硼炔与苯甲醚发生[2+2]环加成反应的产物P16可经6-π电开环过程形成环辛四烯基碳硼烷衍生物P17的猜想(Scheme 13)[98]尚未得到理论计算的证实, 但1, 3-脱氢邻碳硼烷与苯的[2+2]环化产物异构化为环辛四烯基碳硼烷衍生物的过程已经被Zhao等[102]的计算结果所证实.这些实验和理论研究结果对于深入了解邻碳硼炔与苯及其苯衍生物的[4+2]环化反应具有重要的参考价值. 2014年, Zhao等[102]以非亲核碱二异丙基氨基锂处理3-重氮基邻碳硼烷四氟硼酸盐C6得到1, 3-脱氢邻碳硼烷B5, 并通过B5与苯的[4+2]环加成反应成功制备得到了产率高达72%的1, 3-脱氢邻碳硼烷衍生物(Scheme 15).与邻碳硼炔和苯的[4+2]环加成反应[98]相比, 上述1, 3-脱氢邻碳硼烷与苯的[4+2]环加成反应更加有效, 且电子效应对该反应的区域选择性控制起主导作用.另外, 对于带有苄型C—H键的芳烃, 除[4+2]环化产物P23之外, 实验中还检测到了具有高度区域选择性的芳香烯反应产物P24, 两种产物的比例取决于苄基质子的数量.为了解释该反应的化学选择性以及3-芳基-邻碳硼烷产物P24的形成机制, 作者使用密度泛函理论在B3LYP/6-31+G**水平上对1, 3-脱氢邻碳硼烷B5与苯的[4+2]环化产物P25异构化为3-芳基-邻碳硼烷P26的过程进行了计算(图 5).结果显示: [4+2]环化产物P25可以通过TS12断开cageC—Csp3键异构化为中间体INT9, INT9发生绕cageB—Csp3键的二面角旋转、越过过渡态TS13转化为中间体INT10.最后, INT10通过过渡态TS14实现质子转移, 从而转化为热稳定性更好的3-芳基-邻碳硼烷产物P26.整个过程在气相中需要越过的位垒为31.4 kcal·mol-1, 这在加热到250 ℃的实验条件下是完全可以实现的.另外, 作者还通过理论计算阐明了1, 3-脱氢邻碳硼烷与苯的[2+2]环化产物异构化为环辛四烯基碳硼烷衍生物的可能性.该工作代表了制备带有多重键性质的C—Bcage键的1, 3-脱氢邻碳硼烷的首个实例, 同时也为碳硼烷的笼碳和硼顶点同时功能化提供了新思路, 对邻碳硼烷的B—H和C—H同时官能化具有重要意义.
几乎与此同时, Zhang等[64]以1-碘-2-锂-邻碳硼烷为邻碳硼炔前体、环己烷为溶剂、在80 ℃条件下与6, 6'-二芳基亚甲基环戊二烯衍生物R1经[4+2]环加成反应制备得到了一系列结构新颖的碳硼烷基降冰片烯衍生物(Scheme 16).实验发现, 6, 6'-二芳基亚甲基环戊二烯衍生物中芳基上的取代基R1和R2对反应产率具有重要影响, 当R1=R2=H时反应产率可高达56%;而将R1和R2换成其他取代基之后, 产率都有不同程度的下降.
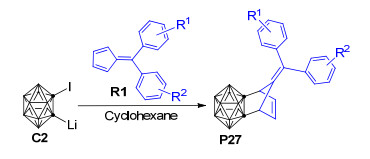
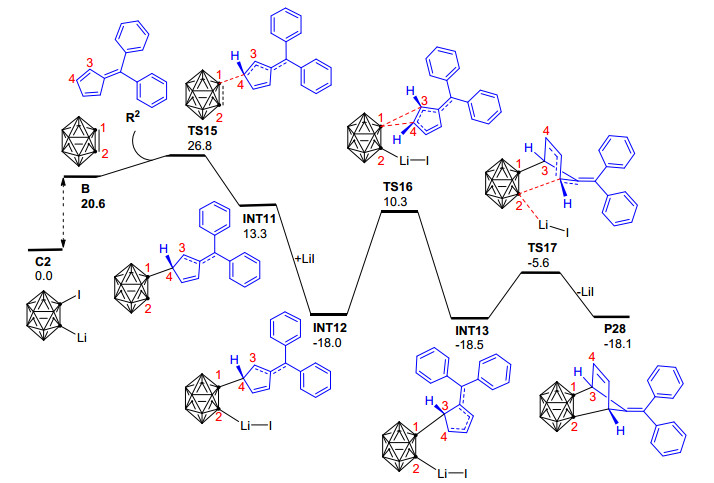
为了进一步探讨1-碘-2-锂-邻碳硼烷与6, 6'-二芳基亚甲基环戊二烯衍生物[4+2]环化反应的取代基效应和微观反应机理, 最近, Mu等[103]使用密度泛函理论的B3LYP方法对上述反应进行了理论研究.计算结果显示该反应最有可能的机理为: 1-碘-2-锂-邻碳硼烷前体C2脱去LiI所形成的邻碳硼炔中间体B与6, 6'-二芳基亚甲基环戊二烯反应, 经与分步Diels-Alder (D-A)反应类似的过程最终形成碳硼烷基降冰片烯衍生物P28(图 6).该反应与经典分步D-A反应的不同点在于:邻碳硼炔中间体B与6, 6'-二苯基亚甲基环戊二烯R2的反应首先发生在R2的C-4位置并形成中间体INT11, 之后INT11被LiI稳定化形成中间体INT12, INT12中的碳硼烷基发生1, 2-σ迁移才形成与D-A分步反应中间体类似的INT13.上述碳硼烷基的1, 2-σ迁移过程为整个反应的决速步, 在353 K的实验条件下需越过28.3 kcal·mol-1的自由能垒才能形成相应的碳硼烷基降冰片烯衍生物.作者还利用自然价键轨道理论(NBO)对相关过程的电子成键特性进行了分析, 明确了6, 6'-二芳基亚甲基环戊二烯衍生物在该反应中扮演的电子给体角色和邻碳硼炔的电子受体角色, 即该反应与正常电子需求的D-A反应具有相似的电子流动特征.该工作对于进一步改进相关的实验条件和提高反应产率具有一定的理论借鉴意义, 对深入探讨其他邻碳硼炔与不饱和化合物的[4+2]环化反应机理也有一定的参考价值.
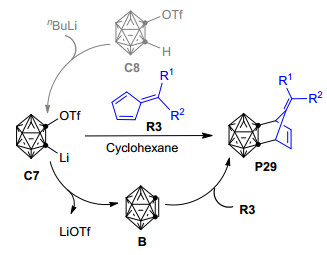
虽然邻碳硼炔与6, 6'-二芳基亚甲基环戊二烯衍生物的[4+2]环加成反应能够以良好的产率和区域选择性制备得到相应的碳硼烷基降冰片烯衍生物, 但该反应的底物仅限于6, 6'-二芳基亚甲基环戊二烯衍生物, 对于反应底物的兼容性还有很大提升空间.为了扩大反应底物的适用范围, 最近, Zhang等[104]报道了一种以1-三氟甲磺酰基-2-锂-邻碳硼烷C7为邻碳硼炔前体、通过与6-取代或6, 6'-二取代亚甲基环戊二烯衍生物R3发生[4+2]环化反应制备碳硼烷基降冰片烯衍生物P29的新方法(Scheme 17).与上述邻碳硼炔和6, 6'-二芳基亚甲基环戊二烯衍生物的[4+2]环加成反应[64]相比, 该反应的条件更为温和——在室温下就能进行; 同时产率大幅提高, 有的甚至高达80%.另外, 该反应具有非常好的底物兼容性, 不论以6-取代亚甲基环戊二烯衍生物还是以6, 6'-二取代亚甲基环戊二烯衍生物为底物, 都能得到较高产率的碳硼烷基降冰片烯衍生物.所得的碳硼烷基降冰片烯还可通过[3+2]/[4+2]环加成、硫醇化、环氧化反应等进一步官能化, 进而制得多官能化的碳硼烷衍生物.结合本文前述实验和理论研究报道以及其他相关实验结果[35, 105], 该反应的可能机理为: 1-三氟甲磺酰基-2-锂-邻碳硼烷C7首先脱去LiOTf形成邻碳硼炔中间体B, 之后B与6-取代或6, 6'-二取代亚甲基环戊二烯衍生物R3通过类似于图 6中的分步过程形成[4+2]环化产物P29.如果以1-三氟甲磺酰基-2-氢-邻碳硼烷C8为邻碳硼炔前体, 则C8首先与正丁基锂作用形成邻碳硼炔前体C7, 之后的反应过程与直接用C7为前体参与反应的过程相同.
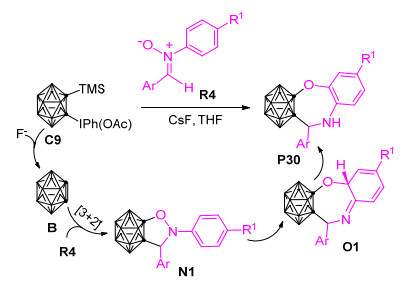
如前所述, 由邻碳硼烷前体制备得到的邻碳硼炔中间体是一种重要的合成子, 能够与多种不饱和化合物发生[2+2]、[4+2]、[2+2+2]等环加成反应转化为功能碳硼烷衍生物.但是, 目前基于邻碳硼炔的偶极环加成反应来合成邻碳硼烷基稠合杂环化合物的反应仍然极为少见. 2015年, Zhao等[65]首次报道了硝酮R4与邻碳硼炔通过[5+2]环加成反应制备邻碳硼烷基稠合七元杂环化合物P30的新方法(Scheme 18).在该反应中, 硝酮扮演了五原子偶合体角色, 且其中的两个杂原子(N和O)同时包含在了新形成的七元杂环中.为了探究该[5+2]环加成反应的微观机理, 作者开展了一系列的控制实验.基于相应的控制实验结果, 他们推测该反应的可能机理为:邻碳硼炔前体C9在F-存在条件下转化为活泼的邻碳硼炔中间体B, 之后B与二芳基硝酮R4发生[3+2]环加成反应形成五元环中间体N1. N1中的N—O键发生解离转化为七元环中间体O1.最后, O1发生芳构化形成邻碳硼烷基稠合七元杂环化合物P30.
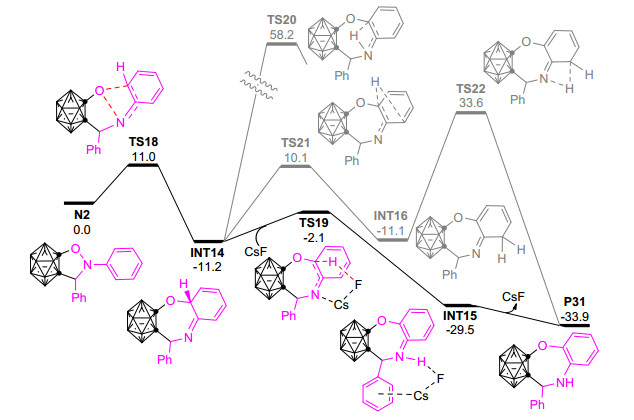
为了验证上述推测的合理性, Zhao等进一步利用密度泛函理论的DFT方法对邻碳硼炔与二苯基硝酮的反应进行了理论计算, 结果如图 7所示.邻碳硼炔与二苯基硝酮通过[3+2]环化反应生成的五元环中间体N2断开N—O键只需要越过11.0 kcal·mol-1的能垒(TS18), 与该反应在室温下进行的实验事实很好吻合; N—O键断裂后所形成的七元环中间体INT14比N2更加稳定, 也与实验中能分离得到与INT14结构类似的中间产物相符.形成中间体INT14后, 通过[1, 3]-氢迁移(TS20)或[1, 5]-氢迁移/[1, 3]-氢迁移(TS21, TS22)进行芳构化的能垒都非常高, 在相应的实验条件下很难发生; 相反, CsF辅助去质子化/质子化的过程(TS19)则非常容易, 为芳构化的最有可能路径[65].该结果很好地解释了碱性试剂在该反应中的必要性, 对于人们深入认识碱性添加物CsF在该反应中的作用具有重要意义.综合实验与理论研究结果, 该反应需依次经历[3+2]环加成反应、N—O键断裂、氧迁移和芳构化等四个过程才能最终形成[5+2]环化的邻碳硼烷基稠合七元杂环产物P31.该工作不但为合成可能具有重要生物活性的邻碳硼烷基稠合七元杂环化合物开拓了一种新方法, 同时也为人们深入认识硝酮的性质和反应性等提供了新见解.
最近十年, 人们在邻碳硼烷和邻碳硼炔的官能化领域取得了显著进步, 尤其是在基于邻碳硼炔的环加成反应方面呈现出了前所未有的繁荣景象, 主要体现在以下几个方面.
邻碳硼炔可由多种前体如1, 2-二氢邻碳硼烷、1, 2-二锂邻碳硼烷、1-溴-2-锂-邻碳硼烷、1-碘-2-锂-邻碳硼烷、1-三氟甲磺酰基-2-锂-邻碳硼烷、1-三氟甲磺酰基-2-氢-邻碳硼烷、1-三甲基硅烷基-2-醋酸碘苯基-邻碳硼烷等制备得到, 其中以1-碘-2-锂-邻碳硼烷为前体比以1-溴-2-锂-邻碳硼烷为前体产率有所提高[98], 而以1-三氟甲磺酰基-2-锂-邻碳硼烷为前体时, 与1-碘-2-锂-邻碳硼烷前体相比, 在产率、实验温度和底物兼容性方面都有显著改进[104].
就反应类型而言, 以邻碳硼炔为底物的多组分环化反应已经从最初的[2+2+2]、[2+2+1]、[2+2]环加成反应扩展到后来的[3+2]、[4+2]、[5+2]等多种类型, 且对反应底物的兼容性越来越好, 实验产率也在逐步提高, 由此制备得到的功能碳硼烷衍生物的结构和性质也愈加多样化.
邻碳硼烷和邻碳硼炔的C—H和B—H键选择性活化控制技术得到不断发展, 已经从单纯的C—H键活化逐渐发展为B—H键的选择性活化, 甚至是C—H和B—H键的同时活化, 从而制备得到结构新颖的C, B-或C, C, B-同时取代的碳硼烷基稠合多环产物.
基于量子化学的理论计算已经在该领域的研究中崭露头角, 目前已经通过理论计算确认了一部分邻碳硼炔与炔烃/烯烃的[2+2+2]、[2+2+1]、[4+2]环化以及邻碳硼炔与硝酮的[5+2]环化反应机理, 对于人们深入认识这些反应的取代基效应和反应选择性等起到了不可或缺的作用.但与苯炔相比, 关于邻碳硼烷和邻碳硼炔的研究仍然处于初级阶段.目前, 关于邻碳硼烷和邻碳硼炔B—H键的选择性官能化、Zr/Ni、Zr/Cu、Pd/Ni共促反应的转金属化机理研究等依然极具挑战性.因此, 努力优化反应前体和改进实验条件, 通过具有操作简单、原子经济性等优势的环加成反应来选择性制备高产率的多官能化邻碳硼烷衍生物将会是该领域的长期发展方向之一.同时, 随着理论研究与实验研究的不断交融和相互渗透, 理论计算有望在该领域的发展中发挥越来越重要的启迪和导向作用.
Zhang, J.; Xie, Z. Acc. Chem. Res. 2014, 47, 1623. doi: 10.1021/ar500091h
Qiu, Z.; Xie, Z. Dalton Trans. 2014, 43, 4925. doi: 10.1039/C3DT52711E
Eleazer, B. J.; Smith, M. D.; Popov, A. A.; Peryshkov, D. V. J. Am. Chem. Soc. 2016, 138, 10531. doi: 10.1021/jacs.6b05172
Dziedzic, R. M.; Martin, J. L.; Axtell, J. C.; Saleh, L. M. A.; Ong, T.-C.; Yang, Y. F.; Messina, M. S.; Rheingold, A. L.; Houk, K. N.; Spokoyny, A. M. J. Am. Chem. Soc. 2017, 139, 7729. doi: 10.1021/jacs.7b04080
Bregadze, V. I. Chem. Rev. 1992, 92, 209. doi: 10.1021/cr00010a002
Scholz, M.; Hey-Hawkins, E. Chem. Rev. 2011, 111, 7035. doi: 10.1021/cr200038x
Zhang, J.; Xie, Z. Chem.-Asian J. 2010, 5, 1742. doi: 10.1002/asia.201000175
Soloway, A. H.; Tjarks, W.; Barnum, B. A.; Rong, F.-G.; Barth, R. F.; Codogni, I. M.; Wilson, J. G. Chem. Rev. 1998, 98, 1515. doi: 10.1021/cr941195u
Armstrong, A. F.; Valliant, J. F. Dalton Trans. 2007, 4240. http://www.ncbi.nlm.nih.gov/pubmed/17893811
Sivaev, I. B.; Bregadze, V. V. Eur. J. Inorg. Chem. 2009, 1433. doi: 10.1002/ejic.200900003/full
Zhu, Y.; Peng, A. T.; Carpenter, K.; Maguire, J. A.; Hosmane, N. S.; Takagaki, M. J. Am. Chem. Soc. 2005, 127, 9875. doi: 10.1021/ja0517116
Chen, G.; Yang, J.; Lu, G.; Liu, P. C.; Chen, Q.; Xie, Z.; Wu, C. Mol. Pharmaceutics 2014, 11, 3291. doi: 10.1021/mp400641u
Hawthorne, M. F.; Zheng, Z. Acc. Chem. Res. 1997, 30, 267. doi: 10.1021/ar9501479
Jude, H.; Disteldorf, H.; Fischer, S.; Wedge, T.; Hawkridge, A. M.; Arif, A. M.; Hawthorne, M. F.; Muddiman, D. C.; Stang, P. J. J. Am. Chem. Soc. 2005, 127, 12131. doi: 10.1021/ja053050i
Xie, Z. Acc. Chem. Res. 2003, 36, 1. doi: 10.1021/ar010146i
Yao, Z.-J.; Jin, G.-X. Coord. Chem. Rev. 2013, 257, 2522. doi: 10.1016/j.ccr.2013.02.004
Popescu, A. R.; Teixidor, F.; Viñas, C. Coord. Chem. Rev. 2014, 269, 54. doi: 10.1016/j.ccr.2014.02.016
Gingrich, H. L.; Ghosh, T.; Huang, Q.; Jones, M. Jr. J. Am. Chem. Soc. 1990, 112, 4082. doi: 10.1021/ja00166a080
Ho, D. M.; Cunningham, R. J.; Brewer, J. A.; Bian, N.; Jones, M. Jr. Inorg. Chem. 1995, 34, 5274. doi: 10.1021/ic00125a028
Jones, M. Jr.; Levin, R. H. J. Am. Chem. Soc. 1969, 91, 6411. doi: 10.1021/ja01051a039
Kiran, B.; Anoop, A.; Jemmis, E. D. J. Am. Chem. Soc. 2002, 124, 4402. doi: 10.1021/ja016843n
Cunningham, R. J.; Bian, N.; Jones, M. Jr. Inorg. Chem. 1994, 33, 4811. doi: 10.1021/ic00100a001
Ghosh, T.; Gingrich, H. L.; Kam, C. K.; Mobraaten, E. C.; Jones, M., Jr. J. Am. Chem. Soc. 1991, 113, 1313. doi: 10.1021/ja00004a036
Deng, L.; Chan, H.-S.; Xie, Z. J. Am. Chem. Soc. 2005, 127, 13774. doi: 10.1021/ja054207+
Huang, Q.; Gingrich, H. L.; Jones, M. Jr. Inorg. Chem. 1991, 30, 3254. doi: 10.1021/ic00017a007
Ren, S.; Chan, H.-S.; Xie, Z. Organometallics 2009, 28, 4106. doi: 10.1021/om9002973
Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem. 2015, 127, 10769. doi: 10.1002/ange.201504481
Lu, J.-Y.; Du, Y.; Zhao, B.; Lu, J. Tetrahedron 2016, 72, 161. doi: 10.1016/j.tet.2015.11.019
Ni, H.; Qiu, Z.; Xie, Z. Angew. Chem., Int. Ed. 2017, 56, 712. doi: 10.1002/anie.201610810
Zheng, F.; Leung, T.-F.; Chan, K.-W.; Sung, H. H. Y.; Williams, I. D.; Xie, Z.; Jia, G. Chem. Commun. 2016, 52, 10767. doi: 10.1039/C6CC05283E
Lyu, H.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2016, 138, 12727. doi: 10.1021/jacs.6b07086
Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2015, 137, 3502. doi: 10.1021/jacs.5b01169
Liu, G.; Yan, H. Organometallics 2015, 34, 591. doi: 10.1021/om501016w
Zhao, D.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 3166. doi: 10.1002/anie.201511251
Cheng, R.; Zhang, J.; Zhang, J.; Qiu, Z.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 1751. doi: 10.1002/anie.201507952
Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 11840. doi: 10.1002/anie.201605880
Chan, T. L.; Xie, Z. Chem. Commun. 2016, 52, 7280. doi: 10.1039/C6CC03368G
Wang, Z.; Ye, H.; Li, Y.; Li, Y.; Yan, H. J. Am. Chem. Soc. 2013, 135, 11289. doi: 10.1021/ja4047075
Wang, S. R.; Xie, Z. Organometallics 2012, 31, 4544. doi: 10.1021/om300324n
Tang, C.; Xie, Z. Angew. Chem., Int. Ed. 2015, 54, 7662. doi: 10.1002/anie.201502502
Quan, Y.; Xie, Z. Angew. Chem., Int. Ed. 2016, 55, 1295. doi: 10.1002/anie.201507697
Qiu, Z.; Ren, S.; Xie, Z. Acc. Chem. Res. 2011, 44, 299. doi: 10.1021/ar100156f
Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 15513. doi: 10.1021/ja509557j
Qiu, Z.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2013, 135, 12192. doi: 10.1021/ja405808t
Wang, S. R.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2011, 133, 5760. doi: 10.1021/ja201126h
Zhao, D.; Zhang, J.; Xie, Z. Angew. Chem., Int. Ed. 2014, 53, 12902. doi: 10.1002/anie.201409141
Visbal, R.; Ospino, I.; Lopez-De-Luzuriaga, J. M.; Laguna, A.; Gimeno, M. C. J. Am. Chem. Soc. 2013, 135, 4712. doi: 10.1021/ja401523x
Guo, J.; Liu, D.; Zhang, J.; Zhang, J.; Miao, Q.; Xie, Z. Chem. Commun. 2015, 51, 12004. doi: 10.1039/C5CC03608A
Jin, G. F.; Hwang, J.-H.; Lee, J.-D.; Wee, K.-R.; Suh, I.-H.; Kang, S. O. Chem. Commun. 2013, 49, 9398. doi: 10.1039/c3cc45313h
Lee, J.-D.; Lee, Y.-J.; Son, K.-C.; Cheong, M.; Ko, J.; Kang, S. O. Organometallics 2007, 26, 3374. doi: 10.1021/om0701569
Brown, D. A.; Colquhoun, H. M.; Daniels, J. A.; MacBride, J. A. H.; Stephenson, I. R.; Wade, K. J. Mater. Chem. 1992, 2, 793. doi: 10.1039/jm9920200793
Zhang, X.; Zou, X.; Yan, H. Organometallics 2014, 33, 2661. doi: 10.1021/om500411b
Cioran, A. M.; Musteti, A. D.; Teixidor, F.; Krpetic, Ž.; Prior, I. A.; He, Q.; Kiely, C. J.; Brust, M.; Viñas, C. J. Am. Chem. Soc. 2012, 134, 212. doi: 10.1021/ja203367h
Grimes, R. N. Dalton Trans. 2015, 44, 5939. doi: 10.1039/C5DT00231A
Wee, K.-R.; Cho, Y.-J.; Jeong, S.; Kwon, S.; Lee, J.-D.; Suh, I.-H.; Kang, S. O. J. Am. Chem. Soc. 2012, 134, 17982. doi: 10.1021/ja3066623
Wee, K.-R.; Han, W.-S.; Cho, D. W.; Kwon, S.; Pac, C.; Kang, S. O. Angew. Chem. Int. Ed. 2012, 51, 2677. doi: 10.1002/anie.201109069
Shi, C.; Sun, H.; Tang, X.; Lv, W.; Yan, H.; Zhao, Q.; Wang, J.; Huang, W. Angew. Chem., Int. Ed. 2013, 52, 13434. doi: 10.1002/anie.201307333
卞德乾, 聂永, 苗金玲, 王亚峰, 张振伟, 有机化学, 2013, 33, 1774. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201308021&dbname=CJFD&dbcode=CJFQBian, D.; Nie, Y.; Miao, J.; Wang, Y.; Zhang, Z. Chin. J. Org. Chem. 2013, 33, 1774(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201308021&dbname=CJFD&dbcode=CJFQ
朱琳, 蒋其柏, 燕红, 无机化学学报, 2014, 30, 2246. http://industry.wanfangdata.com.cn/jt/Magazine?magazineId=wjhxxb&yearIssue=2014_10Zhu, L.; Jiang, Q.-B.; Yan, H. Chin. J. Inorg. Chem. 2014, 30, 2246(in Chinese). http://industry.wanfangdata.com.cn/jt/Magazine?magazineId=wjhxxb&yearIssue=2014_10
Deng, L.; Chan, H.-S.; Xie, Z. J. Am. Chem. Soc. 2006, 128, 7728. doi: 10.1021/ja061605j
Quan, Y.; Zhang, J.; Xie, Z. J. Am. Chem. Soc. 2013, 135, 18742. doi: 10.1021/ja410233e
Yuan, Y. G.; Ren, S. K.; Qiu, Z. Z.; Wang, S. W.; Xie, Z. W. Sci. China:Chem. 2014, 57, 1157. doi: 10.1007/s11426-014-5112-0
Ren, S.; Qiu, Z.; Xie, Z. Organometallics 2013, 32, 4292. doi: 10.1021/om400458r
Zhang, J.; Qiu, Z.; Xu, P.-F.; Xie, Z. ChemPlusChem 2014, 79, 1044. doi: 10.1002/cplu.201402129
Zhao, D.; Zhang, J.; Xie, Z. J. Am. Chem. Soc. 2015, 137, 13938. doi: 10.1021/jacs.5b09074
Zhao, D.; Xie, Z. Coord. Chem. Rev. 2016, 314, 14. doi: 10.1016/j.ccr.2015.07.011
Qiu, Z. Tetrahedron Lett. 2015, 56, 963. doi: 10.1016/j.tetlet.2015.01.038
Xie, Z. W. Sci. China:Chem. 2014, 57, 1061.
Qiu, Z. Z.; Xie, Z. W. Sci. China, Ser. B:Chem. 2009, 39, 1544. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jbxg200910003&dbname=CJFD&dbcode=CJFQ
Zhang, J.; Xie, Z. Pure Appl. Chem. 2013, 85, 661.
邱早早, 谢作伟, 中国科学B辑:化学, 2009, 39, 1053. http://www.cqvip.com/qk/88064X/200910Qiu, Z. Z.; Xie, Z. W. Sci. China, Ser. B:Chem. 2009, 39, 1053(in Chinese). http://www.cqvip.com/qk/88064X/200910
古林莎·果依其巴依, 张锐, 燕红, 无机化学学报, 2010, 26, 733. http://www.oalib.com/paper/5127311Gulinsha, G.; Zhang, R.; Yan, H. Chin. J. Inorg. Chem. 2010, 26, 733(in Chinese). http://www.oalib.com/paper/5127311
Patel, R. M.; Argade, N. P. Org. Lett. 2013, 15, 14. doi: 10.1021/ol3028658
Dateer, R. B.; Shaibu, B. S.; Liu, R.-S. Angew. Chem. Int. Ed. 2012, 51, 113. doi: 10.1002/anie.201105921
Shi, D.; Xie, Y.; Zhou, H.; Xia, C.; Huang, H. Angew. Chem. 2012, 124, 1274. doi: 10.1002/ange.201107495
Qiu, Z. Ph. D. Dissertation, Chinese University of Hong Kong, Hong Kong, 2012.
Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2009, 131, 2084. doi: 10.1021/ja809389k
Qiu, Z.; Wang, S. R.; Xie, Z. Angew. Chem., Int. Ed. 2010, 49, 4649. doi: 10.1002/anie.201001249
Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2010, 132, 16085. doi: 10.1021/ja1058789
Ren, S.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2012, 134, 3242. doi: 10.1021/ja211485t
Ren, S.; Qiu, Z.; Xie, Z. Angew. Chem., Int. Ed. 2012, 51, 1010. doi: 10.1002/anie.v51.4
Mu, W.-H.; Xia, S.-Y.; Li, J.-X.; Fang, D.-C.; Wei, G.; Chass, G. A. J. Org. Chem. 2015, 80, 9108. doi: 10.1021/acs.joc.5b01464
Sayler, A. A.; Beall, H.; Sieckhaus, J. F. J. Am. Chem. Soc. 1973, 95, 5790. doi: 10.1021/ja00798a074
Qiu, Z.; Deng, L.; Chan, H.-S.; Xie, Z. Organometallics 2010, 29, 4541. doi: 10.1021/om100669x
Zhang, J.; Quan, Y.; Lin, Z.; Xie, Z. Organometallics 2014, 33, 3556. doi: 10.1021/om5004545
Quan, Y.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 7599. doi: 10.1021/ja503489b
Zhao, D.; Zhang, J.; Xie, Z. J. Am. Chem. Soc. 2015, 137, 9423. doi: 10.1021/jacs.5b05426
Ren, S.; Chan, H.-S.; Xie, Z. J. Am. Chem. Soc. 2009, 131, 3862. doi: 10.1021/ja900563u
Naito, H.; Morisaki, Y.; Chujo, Y. Angew. Chem., Int. Ed. 2015, 54, 5084. doi: 10.1002/anie.v54.17
Ren, S.; Qiu, Z.; Xie, Z. Organometallics 2012, 31, 4435. doi: 10.1021/om300202p
Harmgarth, N.; Gräsing, D.; Dröse, P.; Hrib, C. G.; Jones, P. G.; Lorenz, V.; Hilfert, L.; Busse, S.; Edelmann, F. T. Dalton Trans. 2014, 43, 5001. doi: 10.1039/C3DT52751D
Brinkley, J. M.; Friedman, L. Tetrahedron Lett. 1972, 28, 4141. http://www.sciencedirect.com/science/article/pii/S0040403901942583
Reinecke, M. G.; Mazza, D. D. J. Org. Chem. 1989, 54, 2142. doi: 10.1021/jo00270a024
Barnett-Thamattoor, L.; Zheng, G.-X.; Ho, D. M.; Jones, M. Jr. Inorg. Chem. 1996, 35, 7311. doi: 10.1021/ic960284h
Pellissier, H.; Santelli, M. Tetrahedron 2003, 59, 701. doi: 10.1016/S0040-4020(02)01563-6
Lee, T.; Jeon, J.; Song, K. H.; Jung, I.; Baik, C.; Park, K.-M.; Lee, S. S.; Kang, S. O.; Ko, J. Dalton Trans. 2004, 933.
Jeon, J.; Kitamura, T.; Yoo, B.-W.; Kang, S. O.; Ko, J. Chem. Commun. 2001, 2110. http://www.ncbi.nlm.nih.gov/pubmed/12240187
Wang, S. R.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2010, 132, 9988. doi: 10.1021/ja1044488
Wang, S. R.; Xie, Z. Organometallics 2012, 31, 3316. doi: 10.1021/om300129t
Wang, S. R.; Xie, Z. Tetrahedron 2012, 68, 5269. doi: 10.1016/j.tet.2012.02.049
Huo, R.-P.; Guo, L.-H.; Zhang, F.-Q.; Zhang, X. Int. J. Quantum Chem. 2017, 117, e25372. doi: 10.1002/qua.v117.12
Zhao, D.; Zhang, J.; Xie, Z. Angew. Chem., Int. Ed. 2014, 53, 8488. doi: 10.1002/anie.v53.32
母伟花, 马瑶, 方德彩, 王蓉, 张海娜, 化学学报, 2018, 76, 55. doi: 10.11862/CJIC.2018.009Mu, W.; Ma, Y.; Fang, D.; Wang, R.; Zhang, H. Acta Chim. Sinica 2018, 76, 55(in Chinese). doi: 10.11862/CJIC.2018.009
Zhang, J.; Qiu, Z.; Xie, Z. Organometallics 2017, 36, 3806. doi: 10.1021/acs.organomet.7b00574
李欢欢, 博士论文, 南京大学, 南京, 2017.Li, H. Ph. D. Dissertation, Nanjing University, Nanjing, 2017 (in Chinese).

图式 1 邻碳硼烷与炔烃[2+2+2]环加成反应的可能机理
Scheme 1 Probable mechanism for the [2+2+2] cycloaddition of o-carborane and alkynes

图式 2 邻碳硼烷与烯烃和炔烃的[2+2+2]环加成反应及其可能的反应机理
Scheme 2 Probable mechanism for the [2+2+2] cycloaddition of o-carborane, alkenes and alkynes

图式 3 邻碳硼烷与炔烃的[2+2+2]环加成反应及其可能的反应机理
Scheme 3 Probable mechanism for the [2+2+2] cycloaddition of o-carborane and alkynes

图式 4 1, 3-脱氢邻碳硼烷与炔烃[2+2+2]环加成反应的可能机理
Scheme 4 Proposed mechanism for the [2+2+2] cycloaddition of 1, 3-dehydro-o-carborane and alkynes

图式 5 碳硼烷基二茂锆与炔烃的[2+2+2]环化反应的可能机理
Scheme 5 Proposed mechanism for the [2+2+2] cycloaddition of carboranylzirconacycle with alkynes

图 2 镍催化碳硼炔与二分子炔烃[2+2+2]环加成反应的势能剖面图(能量:kcal/mol)
Figure 2 Potential energy profiles for the nickel-catalyzed [2+2+2] cycloaddition of o-carboryne with alkynes (energy in kcal/mol)

图式 6 碳硼烷基锆环戊烷与烯烃和三甲基硅烷基炔烃的[2+2+1]环加成反应
Scheme 6 [2+2+1] cycloaddition of carboranylzirconacyclopentane, alkene and trimethylsilylalkyne

图 3 碳硼烷基镍环戊烷与烯烃和三甲基硅烷基芳基炔烃的[2+2+1]环加成反应的势能剖面图(能量:kcal/mol)
Figure 3 Potential energy profiles for the [2+2+1] cycloaddition of carboranylnickelacyclopentane, alkene and trimethylsilylalkyne (energy in kcal/mol)

图式 7 碳硼烷基二茂锆与烯烃和2-溴苯基三甲基硅烷基乙炔的[2+2+1]环加成反应及可能机理
Scheme 7 Proposed mechanism for the [2+2+1] cycloaddition of carboranylzirconacycle, alkenes and 2-bromophenyltri- methylsilylacetylenes

图式 8 碳硼烷基二茂锆与烯烃/炔烃的[2+2]环加成反应
Scheme 8 [2+2] cycloaddition of carboranylzirconacycle with alkenes or alkynes

图式 9 1-溴-2-锂-邻碳硼烷与吲哚的[2+2]环加成反应和C—H插入反应
Scheme 9 [2+2] cycloaddition and C—H insertion reaction of 1-bromo-2-lithio-o-carborane with indoles

图式 10 碳硼烷基二茂锆与烯烃的[3+2]环加成反应
Scheme 10 [3+2] cycloaddition reaction between carboranylzirconacycle and alkenes

图式 11 金属碳硼炔络合物与不饱和化合物的[3+2]环加成反应
Scheme 11 [3+2] cycloaddition reaction of meta-carboryne complexes with unsaturated molecules

图式 12 邻碳硼烷与碳二亚胺的[3+2]环加成反应
Scheme 12 [3+2] cycloaddition of o-carborane with carbodiimides

图式 13 1-碘-2-锂-邻碳硼烷与芳烃的[4+2]/[2+2]环加成反应
Scheme 13 [4+2]/[2+2] cycloadditions of 1-I-2-Li-o-carborane with aromatics

图式 14 邻碳硼炔与苯和苯乙烯衍生物的[4+2]环化反应
Scheme 14 [4+2] Cycloaddition of o-carboryne with benzenes and styrenes

图 4 邻碳硼炔与苯的[4+2]环化反应的势能剖面图(能量: kcal/mol)
Figure 4 Potential energy profiles for the [4+2] cycloaddition of carboryne and benzene (energy in kcal/mol)

图式 15 1, 3-脱氢邻碳硼烷与芳烃的[4+2]环加成反应
Scheme 15 [4+2] cycloaddition of 1, 3-dehydro-o-car-borane with arenes

图 5 1, 3-脱氢邻碳硼烷与苯的[4+2]环化产物转化为3-苯基-邻碳硼烷的势能剖面图(能量: kcal/mol)
Figure 5 Potential energy profiles for the transformation of [4+2] cycloaddition product into 3-phenyl-o-carborane (energy in kcal/mol)

图式 16 1-碘-2-锂-邻碳硼烷与亚甲基环戊二烯衍生物的[4+2]环化反应
Scheme 16 [4+2] Cycloaddition between 1-iodo-2-lithio-o-carborane and 6, 6'-diphenylfulvenes

图 6 邻碳硼炔与6, 6'-二苯基亚甲基环戊二烯[4+2]环加成反应的势能剖面图(能量: kcal/mol)
Figure 6 Potential energy profiles for the [4+2] cycloaddition between o-carboryne and 6, 6'-diphenylfulvene (energy in kcal/mol)

图式 17 1-三氟甲磺酰基-2-锂-邻碳硼烷与亚甲基环戊二烯衍生物的[4+2]环化反应
Scheme 17 [4+2] cycloaddition between 1-OTf-2-Li-o-car-borane and pentafulvenes

图式 18 硝酮与邻碳硼炔之间的[5+2]偶联环化反应
Scheme 18 [5+2] cycloaddition between nitrones and o-carboryne

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
