图 1.
1, 8-萘酰亚胺的化学结构
Figure 1.
Chemical structure of 1, 8-naphthalimide

近年来, 生物体内重要的活性分子的可视化是分析领域的研究热点, 相比于其它检测技术, 小分子荧光探针因具有高灵敏度、高专一性、可实时进行复杂生物系统内生物分子监测及成像的优点而受到重点关注[1~4].随着双光子显微技术的发展以及双光子染料的不断开发, 双光子成像技术已成为研究生命科学的重要工具.相比于单光子荧光成像技术, 双光子成像技术由于可实现长波长激发从而具有高分辨率、强组织穿透性以及低的组织自发荧光干扰等显著的优越性, 可进一步进行细胞、组织切片、活体斑马鱼以及老鼠成像, 获得更好的实时3D观测与成像效果, 推动了生命科学、生物医学以及药学的发展[5~7].
1, 8-萘酰亚胺作为典型的D-π-A双光子荧光染料, 具有光稳定性、大斯托克斯/反斯托克斯位移等优点, 目前被广泛应用于生物体可视化、有机光电材料等研究领域[8~10].相比于其它双光子荧光染料, 1, 8-萘酰亚胺一般以经济、易制的4-溴-1, 8-萘酰酸酐为起始原料, 合成路线简单, 结构容易修饰, 易于设计并合成荧光探针[11, 12].从结构上来看, 1, 8-萘酰亚胺具有较好的共轭体系(图 1), 共轭体系两端分别是供电子基团(萘环上的取代基, 例如氨基、甲氧基等)以及吸电子基团(酰亚胺), 从而形成D-π-A电子结构[13], 因此该染料的荧光量子产率和双光子吸收截面(Two-photon absorption cross-section)比较高, 可被广泛用于深层次组织双光子成像.基于上述的优越性, 1, 8-萘酰亚胺染料作为热门的双光子染料被用于活细胞中的酶、活性碳簇(Reactive carbon species, RCS)、活性氧簇(Reactive oxygen species, ROS)、活性氮簇(Reactive nitrogen species, RNS)、生物硫醇、离子等生物活性分子的可视化.鉴于此, 本文将按照分子内电荷转移(Intramolecular charge transfer, ICT)、光致电子转移(photoinduced electron transfer, PET)和荧光共振能量转移(Fluorescence resonance energy transfer, FRET)等不同的探针荧光发光机制进行分类, 总结该染料在双光子成像领域上的应用, 并对未来研究方向进行展望.
ICT机制荧光探针是由识别基团(供电子基团或吸电子基团)和荧光基团通过共轭基团连接从而形成的D-π-A共轭体系, 在外界光的激发下, 荧光分子中的电荷从供电基团(供体)向吸电基团(受体)转移, 这一过程就是ICT(图 2)[14], 当该探针与被检测物相互作用后会引起探针中吸电基团(供电基团)的吸电能力发生改变时, 分子内电荷分布发生变化(即ICT增强或减弱), 同时通常伴随着紫外-可见吸收光谱和荧光发射光谱发生蓝移或红移. 1, 8-萘酰亚胺具有萘环平面结构, 形成较大的共轭体系, 属于D-π-A类荧光染料.如图 1所示, 取代基R2对酰亚胺的吸电子能力影响很小, 而萘环上不同的取代基R1对萘环的吸供电子能力的影响大, 因此研究人员往往将识别基团安装在萘环部分[10], 当识别基团与被分析物反应或结合时, 萘环上的取代基吸电或供电能力变化, 紫外-可见吸收光谱和荧光发射光谱发生蓝移或红移, 因此很容易利用这类染料的ICT机制进行荧光探针的设计.目前, 许多文献报道了基于ICT机制的1, 8-萘酰亚胺类荧光探针, 这类探针通过荧光关-开、开-关或者比率响应等信号变化来检测被分析物, 同时可对生物样品内的活性分子进行双光子成像.
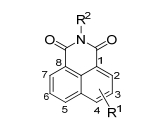
过氧化氢(H2O2)作为生物体内含量最高的ROS, 是一种重要的氧化应激标记和信号转导分子, 研发准确快速检测生物系统内的H2O2含量的荧光探针对于诊断人体疾病具有十分重要的意义[15, 16]. Chang课题组[17]设计并合成基于1, 8-萘酰亚胺的H2O2双光子比率荧光探针1(图 3).他们利用硼酸酯基团为反应位点, 将硼酸酯基团通过氨基甲酸酯与1, 8-萘酰亚胺相连.与H2O2反应前, 探针1在375 nm处有最佳吸收, 在475 nm处发射蓝色荧光; 与H2O2作用后, 氨基甲酸酯基团变成了具有更强供电子能力的氨基基团, ICT增强, 最佳吸收和发射波长发生红移现象, 其最佳吸收红移到435 nm, 其发射波长红移至540 nm处发出绿色荧光, 比率(F540/F475)增强12倍, H2O2去保护反应的分解速率常数反应kobs为8.8×10-4 s-1.之后, 作者利用探针1的比率响应和双光子特性实现了RAW264.7内外源性H2O2的可视化以及定量检测, 在激发波长为820 nm下, 作者发现在受十四烷酸乙酸大戟二萜醇酯(PMA)诱导后, 除了吞噬小泡处的荧光强度变化很大以外, 细胞质周围的荧光强度无明显变化, 说明探针1可以实现在自然免疫应答下的活细胞内H2O2的双光子成像.
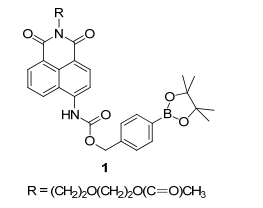
Zdolsek课题组[18]发现过量的内源或外源性H2O2可与溶酶体中的亚铁离子产生羟基自由基从而损害细胞, 为了检测细胞溶酶体内的H2O2含量, Lin课题组[19]研发了一种快速响应的溶酶体靶向的双光子H2O2荧光探针2(图 4), 该探针通过将硼酸酯基团直接连接在萘酰亚胺的4位上从而变成A-π-A的电子结构, 这类电子结构导致探针2拥有很弱的荧光.当探针2与H2O2反应后, 硼酸酯基团变成了羟基, 电子结构变成了D-π-A, ICT增强, 在550 nm处荧光强度有很大的增强(80倍).探针2与H2O2反应速度快(60 s内), 其它相关的ROS和RNS对信号干扰小.该探针具有吗啉基团, 可作为溶酶体靶向基团[20], 能够有效地监测细胞溶酶体内的内源或外源的H2O2浓度, 最后, 该课题组利用双光子显微镜在激发波长为780 nm下成功实现深层活体组织内H2O2的荧光成像, 并且得到探针在活体组织中的H2O2浓度的检测限为10 μmol·L-1.
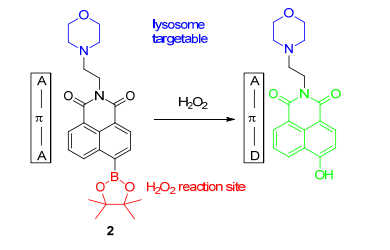
次氯酸(HClO)作为另一种重要的ROS, 主要来源于生物体内的H2O2和氯离子在血红素酶(MPO)的催化下的反应, 过高的HClO含量将会导致动脉粥样硬化、骨关节炎、风湿性关节炎甚至癌症[21, 22]. 2016年, Tang课题组[23]报道了首例能够检测皮摩尔级别HClO的双光子荧光探针3a和3b (Scheme 1), 利用双甲基硫代氨基甲酸酯实现ICT电子结构的有效抑制, HClO分解的Cl+使探针生成亚胺中间体, 随后发生水解等系列反应从而得到4-羟基-1, 8-萘酰亚胺, ICT抑制消失, 荧光恢复.在此处改成EtOH/PBS (PBS代表磷酸盐缓冲液, pH=7.4), 探针3a的检测限极低(7.6 pmol·L-1), 对HClO专一性好, 细胞毒性低.随后, 作者成功利用单、双光子成像技术将探针3a在巨噬细胞中进行内源HClO追踪, 并且, 该探针可通过检测HClO的浓度来区分癌细胞和正常细胞.探针3b以吗啉基团为溶酶体靶向基团, 成功检测了溶酶体细胞器的HCl.最后, 作者利用探针3a、3b双光子深层次的组织穿透能力, 在激发波长为800 nm下实现了小鼠癌组织和正常组织内HClO的检测, 证明探针可检测出癌组织内源性HClO, 可用于诊断癌症以及其它与HClO相关的疾病.
Zhao课题组[24]设计并合成了可逆的双光子HClO荧光探针4 (Eq. 1).该探针原先具有很强的荧光, 当它的4位取代基硫醚被HClO氧化成亚砜后, 吸电子能力变强, ICT被抑制, 缓冲液里的探针荧光被淬灭, 探针的颜色同时也消失了, 随后加入GSH可实现荧光恢复.鉴于此, 虽然已有可检测一些氧化还原循环的荧光探针[25~29], 但是探针4是首例可循环检测HClO/GSH氧化还原的双光子荧光探针.该探针在检测HClO时, 在发射波长为505 nm下的荧光强度与HClO (3~150 μmol·L-1)成线性关系, 检测下限为0.674 μmol·L-1, 其它的ROS以及RNS等生物分子干扰小, 最后, 作者利用探针4双光子的特性, 在800 nm下进行活细胞内外源性HClO的检测, 同时证明该探针可进行溶酶体内HClO的监测.
|
|
(1) |
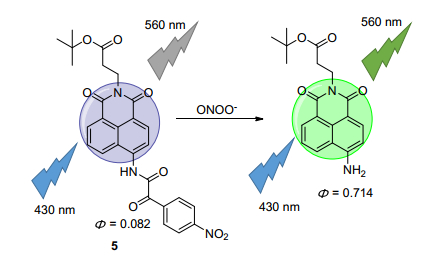
过氧硝酸盐(ONOO-)作为一种高活性的RNS, 可参与到硝化酪氨酸信号中, 同时, 由于它能硝化脂质、蛋白质以及DNA从而被认为是有毒物质[30, 31]. Tang课题组[32]报道了双光子荧光ONOO-探针5(图 5).作者利用吸电子基团酰胺基团抑制ICT过程导致探针荧光淬灭, 当探针与ONOO-反应后, 释放出4-氨基-1, 8-萘酰亚胺染料, ICT抑制消失, 荧光恢复.该探针的荧光强度与ONOO-的浓度呈线性关系, 检测限为25 nmol·L-1, 对ONOO-专一性好, 细胞毒性低(IC50=63 μmol·L-1).随后, 作者利用探针5进行活细胞中外源性ONOO-的检测, 并且利用双光子特性证明了ONOO-是药物导致肝组织损伤的生物标记物.
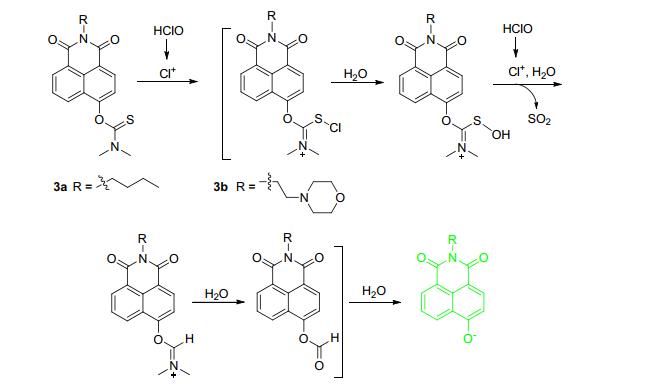
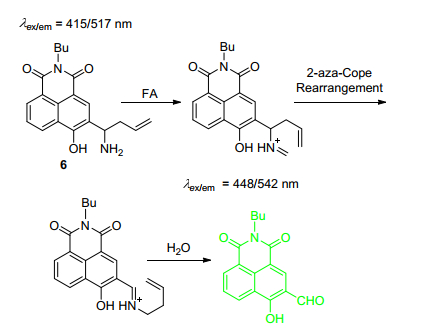
甲醛(FA)作为RCS中最小的一类活性分子, 是公认的致癌物质[33, 34]. Zhu课题组[35]报道了一种新型双光子荧光探针6 (Scheme 2), 他们通过达夫反应在4位羟基引入醛基, 随后引入homoallylamino基团(FA识别基团)[33, 34], 当FA与探针6反应后, homoallylamino上的氨基先与FA生成亚胺, 随后发生aza-cope重排, 水解后变成强吸电基团醛基, ICT效应降低, 最佳紫外-可见吸收峰与荧光发射波长均发生蓝移.在PBS缓冲溶液(pH=7.4, 体积分数为0.5%的DMSO)中, 探针6荧光强度与FA浓度(0~0.5 mmol·L-1)之间具有良好的线性关系, 检测限至5 μmol·L-1, 其它RCS以及相关生物活性分子干扰小, 适合在中性条件下进行FA检测.经实验证明, 探针6细胞毒性小, 在激发波长为820 nm下可用于HeLa细胞以及活体斑马鱼甲醛FA成像研究.
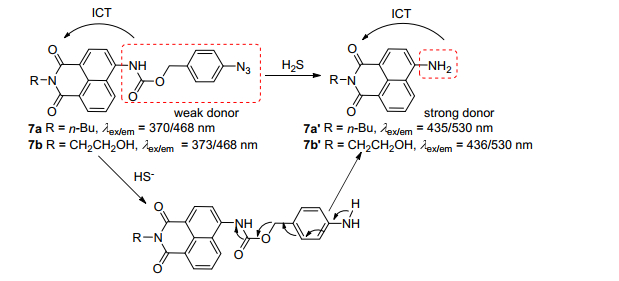
作为第三重要的气体递质, 硫化氢(H2S)在人类心血管、神经、免疫、内分泌和胃肠系统起着重要的调控作用[36, 37]. Song课题组[38]报道了两个H2S双光子比率探针7a、7b (Scheme 3), 他们利用吸电子基团4-叠氮苯氨基甲酸酯为硫化氢应答位点, 当H2S与探针反应后, N3基团被还原成氨基, 随后释放出带有强供电子氨基的1, 8-萘酰亚胺, ICT效应变强, 荧光最佳发射波长从468红移到530 nm, 实现比率探针的效果.该探针对H2S有很好的专一性, 检测下限为50~85 nmol·L-1, 细胞毒性低.最后作者利用探针的双光子特性进行MCF-7中外源性的H2S成像, 并验证了探针7b能够定位到细胞内线粒体中.
H2S是血浆膜可透的细胞内和细胞间气体递质, 作为细胞内的气体递质时主要与它在细胞内的产生、分布和浓度相关, 作为细胞间的气体递质时H2S的浓度是基于它在细胞间的传递的效果[39], 至今仍未有文章报道细胞膜上的H2S检测.因此, Wang课题组[40]研发了细胞膜靶向的双光子H2S探针8 (Eq. 2), 作者通过引入脂质C16长链和亲水性磺酸基团使探针嵌在细胞膜上, 利用4'位上的叠氮基团阻止ICT从而使探针荧光淬灭(Φ=0.01), 当H2S参与还原反应时, ICT恢复, 荧光产生(Φ=0.22), 荧光强度与H2S浓度(0~10 μmol·L-1)之间存在很好的线性关系, 检测限低至0.75 μmol·L-1.双光子成像实验证明, 该探针可被成功用于活细胞和深层组织的膜上的内源性H2S的成像研究.
|
|
(2) |
氟离子(F-)是人体内不可或缺的, 摄入过量的F-会导致胃肾紊乱, 牙齿和骨骼氟中毒, 尿石病, 甚至死亡[41, 42]. Kim课题组[43]报道新型比率双光子F-荧光探针9 (Eq. 3), 该探针以Si—O键为F-的反应位点, 通过氨基甲酸酯连接到1, 8-萘酰亚胺的4位上, 从而得到无色的溶液.当F-存在时, Si—O键断裂, 4-氨基-1, 8-萘酰亚胺释放, ICT效应增强, 溶液颜色从无色变成淡绿色(最佳紫外可见光吸收波长从365 nm红移421 nm), 荧光从蓝色红移到绿色(最佳发射波长从449 nm到508 nm), 实现紫外可见光吸收和荧光双比率增强的效果.随后, 作者对探针9与F-反应前后的双光子吸收截面进行了检测, 在双光子激发波长为740 nm的条件下, 荧光强度比率(F515/F465)增强15倍.
钯离子(Pd2+)作为一种广泛使用的重金属, 对人体健康以及环境影响甚大. Zhou课题组[44]以炔丙基醚为Pd2+的识别位点, 利用氧醚键使得ICT效应降低从而设计并合成拥有低波长的荧光探针10, 加入Pd2+后, 得到具有裸露的羟基基团产物, ICT效应增强, 紫外可见光吸收以及荧光波长都红移, 得到比率检测(F550/F445)的效果(Eq. 4).该探针检测限低至0.28 μmol·L-1, 专一性好, 反应时间短(<5 min), 基于以上数据, 作者在激发波长为820 nm时, 对活细胞内以及组织切片中的Pd2+进行双光子成像, 为环境中或生物体内的Pd2+检测提供了新的检测工具.
|
|
(3) |
|
|
(4) |
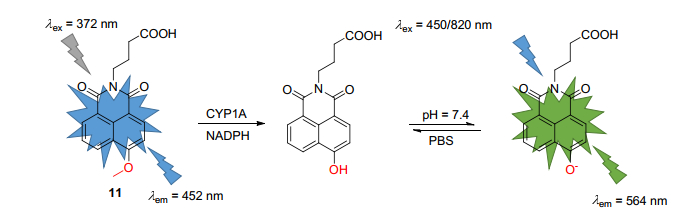
酶对生命至关重要, 尤其是生命研究、疾病与药物作用相关研究上, 我们无法观察酶的本身结构, 只能观察到酶的生化结果, 因此, 发展能够在体内直接监测酶活性的灵敏的双光子小分子探针是急需的[45].近三年来, Yang课题组利用双光子染料1, 8-萘酰亚胺为母核, 利用ICT机制开发了能够进行酶活性检测及双光子成像的荧光探针. 2015年, Yang课题组[46]报道了首例专一检测细胞色素P450 1A (CYP1A)活性的比率型双光子荧光探针11 (Scheme 5).该课题组合成一系列的O-烷基化的1, 8-萘二甲酰亚胺衍生物, 用于评价这些衍生物是否对CYP1A具有专一性, 经过筛选得到, 探针11只对CYP1A具有选择性.该探针与CYP1A反应后, 4位甲氧基变成供电基团羟基, ICT增强, 荧光发射波长红移.经后续实验证明, 探针11能够用于实时监测复杂生物系统的CYP1A的活性, 可利用活体组织制备的酶来源来用于快速筛选CYP1A的活性调节化合物.最终, 探针11被成功用于活细胞以及组织中CYP1A的双光子成像, 展示了高成像分辨率和深层细胞组织成像的优点.
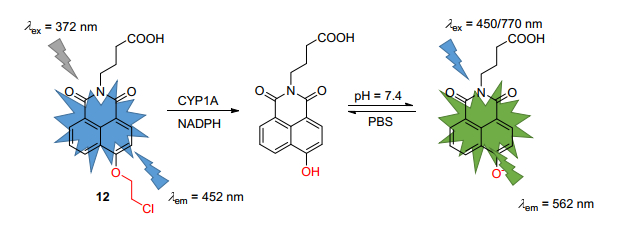
随后, Yang课题组[47]又研发了对CYP1A1具有专一性的双光子比率探针12 (Scheme 5). CYP1A1和CYP1A2在氨基酸序列上具有很高同源性(72.55%), 它们的底物以及抑制剂谱高度重合[48, 49], 因此开发能够专一监测CYP1A1活性的探针还是个挑战.根据探针11的工作原理, 作者通过分子docking筛选以及合成化合物进行表型筛选, 旨在寻找能够专一检测CYP1A1的荧光探针.经过合成以及筛选, 作者发现探针12对CYP1A1的选择性高于其余CYP类酶包括CYP1A2, 探针的荧光设计原理与探针11类似, 都是基于ICT, 即在CYP1A1的存在下, 荧光最佳发射波长452红移到562 nm.经专一性实验证明, 探针12对于CYP1A1的荧光强度高于CYP1A2 30倍, 高于其余CYP酶类15倍.随后, 该探针被用于CYP1A1抑制剂的筛选, 并被用于检测细胞内源性的CYP1A1的活性.最终, 在激发波长770 nm下进行大鼠肝组织的双光子成像, 证明肝组织中具有CYP1A1.
众多癌细胞中羧酸酯酶2 (CEs 2)往往会过量表达, 常常被用于抗癌药物前药的激活[50, 51], 但是至今没有报道过能够专一性识别CEs 2的荧光探针, 因此, Yang课题组[52]又报道了基于ICT的特异性识别组织以及活细胞内的CEs 2的双光子比率探针13 (Eq. 5).该探针与CEs 2发生水解作用后, 由酰胺基团(吸电子基团)水解成氨基基团(供电子基团), ICT效率增强, 从而使荧光最佳发射波长从452 nm红移到542 nm.通过实验证明, 该探针的荧光强度与羧酸酯酶2 (0.5~10 μg)的浓度成线性关系, 检测限为12 ng/mL, 同时该探针对CEs 2的特异性良好, 可用于人体肝组织微粒体中CEs 2的定量检测.最后, 在激发波长为800 nm下, 探针13被用于活细胞以及肝组织中内源性CEs 2的双光子成像, 证明CEs 2在老鼠肝组织中十分丰富.
|
|
(5) |
二肽基肽酶Ⅳ (DPP-Ⅳ)是一种多功能丝氨酸蛋白酶, 在分泌和免疫功能, 细胞代谢, 生长和附着起到调控作用[53]. Yang课题组[54]报道了能够选择性检测复杂生物系统的DPP-Ⅳ的双光子比率探针14 (Eq. 6).在DPP-Ⅳ的存在下, 探针水解释放出带有裸露氨基的1, 8-萘酰亚胺, ICT效应变强, 探针颜色由无色变黄色, 最佳荧光发射波段红移80 nm.该探针对DPP-Ⅳ的选择性高, 检测限为0.78 ng/mL, 灵敏度比商品化DPP-Ⅳ荧光探针高4倍.最后, 作者将探针14应用于血浆和组织匀浆中DPP-Ⅳ的检测, 并在激发波长805 nm下进行活细胞以及深层次小鼠肾组织的双光子成像, 成功检测到内源性DPP-Ⅳ的活性.
|
|
(6) |
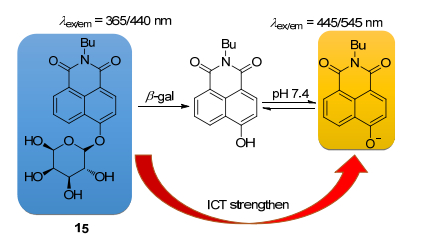
由lacZ基因表达的大肠杆菌β-半乳糖苷酶(β-gal)被广泛用于检查肿瘤细胞异种移植动物模型中肿瘤生长的报告标记[55], 之前的β-gal类荧光探针大多是单纯的关-开型[56, 57]. Han课题组[58]利用萘酰亚胺ICT机制以及双波长吸收特点实现了比率和反斯托克斯位移对β-gal的检测(Scheme 6).探针15通过几步温和的反应就能得到, 最佳吸收/发射波段为365/440 nm, 当探针与β-gal反应后, 产物具有比较强的ICT性质, 从而最佳吸收/发射波段红移到445/545 nm.与之前的β-gal探针进行比较, 探针15的反应速率更快.最后, 作者成功将探针应用于带有lacZ7基因的活细胞以及小鼠内的β-gal成像研究.
内质网氨基肽酶1 (ERAP1)是金属肽酶, 属于M1肽酶家族, 在体内抗原加工过程中起到重要的作用[59, 60], 此外, ERAP1的不当表达也会导致诸多疾病. Zhang课题组[61]利用L-亮氨酸为应答位点合成了内质网靶向的双光子探针16 (Eq. 7).作者利用甲基磺酰胺为内质网靶向基团, 将L-亮氨酸通过吸电子基团氨基甲酸酯连接在萘酰亚胺4位上, 当探针16与ERAP1发生水解、自发成环反应后释放出环脲和裸露羟基的1, 8-萘酰亚胺, ICT增强, 黄色荧光恢复.经实验发现, 探针荧光强度和LAP浓度(5~40 U/L)成很好的线性关系, 检测限为0.21 U/L, 专一性好, 在复杂生物系统内稳定性高, 细胞毒性小.基于以上结果, 作者先利用探针进行Hela细胞中内源性ERAP1的成像, 证明探针能够定位到内质网; 然后通过ERAP1活性检测来诊断细胞内质网的氧化还原态; 最后, 在激发波长为820 nm下, 进行肿瘤组织中内源性ERAP1成像, 在50~120 μm厚度下能观察到荧光信号, 远大于单光子(厚度50~70 μm)的穿透能力, 证明双光子探针16在用于ERAP1相关疾病的诊断中具有良好前景.
|
|
(7) |
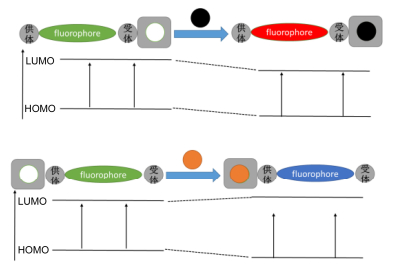
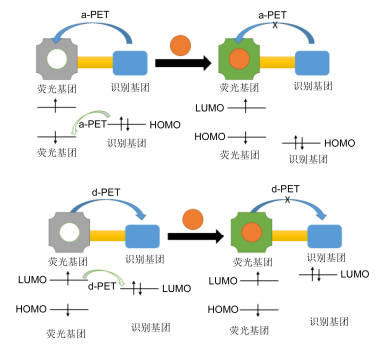
PET过程主要有a-PET和d-PET两类(图 6)[14], a-PET是指当荧光基团受到光激发从而使HOMO上的电子被激发到LUMO, 由于识别基团的HOMO能级处于荧光基团的HOMO和LUMO能级之间, 识别基团HOMO上的电子转移到荧光基团的HOMO, 从而使得荧光基团上LUMO上的电子不能回到基态, 从而荧光淬灭[62]. d-PET过程是指当荧光基团受到光激发从而使HOMO上的电子被激发到LUMO, 由于识别基团的LUMO能级处于荧光基团的HOMO和LUMO能级之间, 因此转到识别基团LUMO上, 从而荧光淬灭[63].这类分子与待测物结合或反应后识别基团的HOMO/LUMO会发生变化, PET效应消失, 从而实现由关-开效应, 许多离子和生物活性大分子探针就是基于这种机制设计的.近年来, 基于PET的1, 8-萘酰亚胺类荧光探针也有许多文献报道.
Chen课题组[64]报道了新型溶酶体靶向的双光子HClO类荧光探针17 (Eq. 8), 通过苯硫脲产生的PET效应从而导致探针荧光淬灭, 当HClO存在时, S原子被氧化成O原子, PET效应消失, 在发射波长538 nm处荧光增强.经实验证明, 该探针的荧光强度与HClO的浓度成线性关系, 检测下限为5 nmol·L-1, 远低于常规的检测下限(57.1 nmol·L-1), 同时该探针反应时间短(小于30 s), 对应于其它被检测物专一性好, 细胞毒性小.最后, 作者在激发波长为800 nm下利用探针17进行Hela细胞内溶酶体HClO的双光子成像.
诸多文献报道了内质网应激与很多新陈代谢疾病例如糖尿病、肥胖等相关, 这些疾病会诱导ROS的产生[65, 66], 基于此背景, Tang课题组[67]报道了内质网靶向的双光子超氧阴离子(O2·-)荧光探针18 (Eq. 9), 用于实时追踪内质网以及糖尿病小鼠中的O2·-的浓度.该探针以甲基磺胺为内质网靶向基团, 苯并噻唑啉为O2·-识别基团, 同时该基团也被用于实现PET效应, 导致探针荧光淬灭.当O2·-与苯并噻唑啉发生脱氢反应后, PET效应被抑制, 荧光恢复, 在O2·-为40 μmol·L-1浓度下, 荧光探针的量子产率从0.012到0.19.经实验证明, 该探针的检测下限为60 nmol·L-1, 对于其它生物活性分子例如ROS等对探针的荧光强度基本没有干扰, 并且细胞毒性低.最后, 作者选用激发波长为750 nm, 利用双光子的高分辨率以及深层穿透能力, 对细胞中内质网以及糖尿病小鼠肝组织进行内源性O2·-的双光子成像, 证明了糖尿病小鼠肝组织含有的内源性O2·-浓度要高于正常小鼠.
|
|
(8) |
|
|
(9) |
细胞内的单线态氧(1O2)在病理和生理过程中起到重要的作用, 一方面, 它被认为参与了细胞信号转导和基因表达的诱导, 另一方面, 它能氧化DNA、蛋白质、油脂, 可用于破坏恶性细胞[68, 69]. Zhang课题组[70]报道了能够检测线粒体内1O2的双光子探针19 (Eq. 10).该探针以三苯基膦盐为线粒体靶向基团, 1, 9-甲基蒽为PET过程的电子受体, 同时也是1O2捕捉器, 当探针19与1O2反应生成内过氧化物后, PET效应消失, 荧光具有很大的增强.经实验证明, 该探针对1O2的专一性好, 反应速度快, 细胞毒性小, 光稳定性好.由于线粒体是光动力治疗的主要场所, 作者将探针用于细胞内1O2的检测, 最后, 作者在激发波长为820 nm下, 成功实现在光动力治疗下活体组织内1O2的双光子成像.
|
|
(10) |
据文献报道, 一氧化氮(NO)具有调节溶酶体的功能[71], Jin课题组报道了首例能够检测细胞溶酶体内NO的双光子荧光探针20 (Eq. 11)[72].该探针以吗啉为溶酶体靶向基团, NO捕捉器-邻苯二胺为PET过程的供电基团, 当邻苯二胺和NO反应后, PET效应消失, 荧光增强.探针与NO反应后的产物pKa=5.61, 适合溶酶体酸性的条件下进行NO的检测.在PBS缓冲液[pH=5, 体积分数为20% CH3CN]中, 在5 equiv.的NO存在下, 探针在发射波长530 nm处荧光强度增强16倍, 灵敏度好, 检测下限低至5 nmol·L-1, 对于其它相关的生物活性分子的抗干扰能力好, NO专一性强.最后, 作者将探针20用于检测活细胞溶酶体外源性或内源性NO的双光子成像, 并利用流式细胞仪成功将探针用于细胞内NO的定量检测.
Lin课题组[73]报道了首例双光子FA探针21 (Eq. 12).该探针利用肼基团作为FA的反应位点, 同时也是肼基团作为电子供体从而发生PET淬灭荧光, 当肼基团与FA生成亚胺后, PET过程消失, 荧光恢复.在含有体积分数为1% DMSO的PBS (pH=7.4)缓冲液中, 在100 μmol·L-1甲醛存在下, 30 min后荧光增强900倍, 检测限为71 nmol·L-1.随后, 作者发现探针对FA的专一性好, 适合在pH酸性和中性下检测, 细胞毒性小, 光稳定性好.最后, 作者选用激发波长为880 nm, 成功进行了Hela细胞中外源或内源的FA双光子成像, 并首次实现了活体组织内FA的双光子成像, 证明了亚硫酸氢钠能够在复杂生物体系内进行FA的清除, 为甲醛相关疾病诊断以及治疗提供了新的手段.
|
|
(11) |
|
|
(12) |
随后, Lin和Kim课题组[74]合作开发并报道了带有生物素基团的双光子FA探针22 (Eq. 13), 该探针是在探针21的基础上, 引入了具有生物活性的生物素基团[75].在体积分数为1% DMSO的PBS (pH=7.4)缓冲液中, FA加入后在发射波长为541 nm处荧光强度增强约140倍, 检测下限为0.78 μmol·L-1, 反应速率快(小于8 min), 甲醛专一性好.基于之前的实验数据, 作者利用探针分别检测生物素受体阳性细胞系(4T-1)和生物素受体阴性细胞系(NIH 3T3 cells)中外源或者内源的FA浓度, 结果证明只有在4T-1中具有荧光, 而NIH 3T3 cells中几乎没有荧光, 实现了选择性定位于4T-1细胞的FA浓度的追踪.最后, 作者利用探针成功实现了活体组织切片中FA的双光子成像.
丙酮醛(MGO), 作为一个具有两个羰基基团的RCS, 它的毒性机理类似于甲醛, 也能与DNA、蛋白质以及其它生物活性分子反应从而致病[76, 77]. Zhou课题组[78]利用邻苯二胺为活性基团以及电子供体的MGO荧光探针23 (Eq. 14), 邻苯二胺可淬灭1, 8-萘酰亚胺的荧光, 当该基团和MGO反应后, PET效应消失, 荧光恢复.在PBS缓冲液[pH=7.4, 体积分数为10% DMSO]中, MGO的加入可使荧光强度(发射波长为528 nm)增强约33倍, 检测下限为77 nmol·L-1, 相对于其他醛类化合物、离子等生物活性物质, MGO使探针荧光强度增强最多.最后, 作者先测试了探针的细胞毒性(IC50=101.3 μmol·L-1), 然后利用双光子共聚焦显微镜, 成功实现Hela细胞里MGO含量的检测.
|
|
(13) |
|
|
(14) |
细胞以及血浆内生物硫醇的浓度与诸多疾病相关[79, 80], Zhang课题组[81]研发了能够应用于细胞以及组织的关-开的双光子硫醇类探针.利用2, 4-二硝基磺酰基作为PET电子受体以及硫醇的活性位点设计并合成荧光探针24 (Eq. 15), 当硫醇存在时, 磺酰基离去, 荧光由关至开.探针的荧光强度与硫醇浓度成线性关系, 其它氨基酸对探针信号无干扰, 探针对于细胞毒性小.基于以上数据, 作者在激发波长为820 nm下进行Hela细胞内源性硫醇的检测, 并通过对照单双光子进行组织切片的成像结果, 发现双光子穿透能力强, 可实现更深层次的组织成像(50~250 μm), 证明了双光子探针可用于复杂生物系统中硫醇的成像及监测.
生物硫醇主要包括谷胱甘肽(GSH)、半胱氨酸(Cys)和高半胱氨酸(Hcy), 由于这三种硫醇分子的结构类似, 能够专一性检测Cys是个挑战.细胞内的Cys浓度紊乱会导致生长缓慢、肝损伤和神经退行性疾病等健康问题[82, 83]. Liang课题组[84]报道了双光子Cys类荧光探针25 (Eq. 16), 作者利用马来酰亚胺作为Cys反应活性位点以及电子供体从而使荧光探针荧光淬灭.当Cys和马来酰亚胺发生加成反应后, PET效应消失, 荧光增强.在PBS [pH=7.4, 体积分数为0.1% EtOH]缓冲液中, 探针25对Cys的检测限低(4.8 nmol·L-1), 反应速度快(小于9 min)以及选择性良好, 最后, 作者在激发波长为760 nm下进行细胞中外源或内源Cys双光子成像.
|
|
(15) |
|
|
(16) |
FRET过程是指两个荧光基团(能量供体和能量受体, 也可以是一个荧光团, 一个荧光淬灭剂)之间发生的非辐射能量转移的过程.能量供体和能量受体发生FRET必须满足两个条件, 第一, 能量供体的发射波长与能量受体的吸收必须有很大的重叠; 第二, 能量供体和能量受体的距离必须合适(小于10 nm).当能量受体是荧光淬灭剂时, FRET类探针往往是关-开型, 当能量受体是荧光基团时, FRET类探针往往是比率型[85]. 1, 8-萘酰亚胺根据萘环上的取代基不同往往具有不同的吸收波长和发射波长, 灵活多变, 给研究人员诸多选择, 他们可根据探针设计的目标, 将萘酰亚胺作为能量供体或能量受体与其它荧光基团进行组合从而实现生物活性分子在复杂生物系统中的检测.
Qian课题组[86]选用罗丹明(能量受体)和1, 8-萘酰亚胺(能量供体)通过苯环连接组成FRET结构从而实现双光子比率型HClO荧光探针26的设计及合成(Eq. 17).作者选用的吡啶-1, 8-萘酰亚胺衍生物的发射波长与罗丹明-氨基硫脲的吸收波长有很大的重合, 由于罗丹明处于螺环结构, 因此探针只有吡啶-1, 8-萘酰亚胺的荧光, 罗丹明没有荧光, 当HClO存在时, 硫脲成环, 螺环结构转变成开环结构, 吡啶-1, 8-萘酰亚胺荧光能量转移到罗丹明上, 萘酰亚胺荧光消失, 罗丹明荧光恢复, 从而实现比率型检测HClO的效果.该探针检测限为96 nmol·L-1, 检测速度快, 对HClO专一性好, pH (4~11)适用性广, 并实现双光子激发下对HClO的检测.最后, 作者利用探针进行A549细胞内HClO的检测.
不同于探针26, 在FRET探针体系中1, 8-萘酰亚胺也可作为能量受体实现生物活性物质的监测. Tang课题组[87]以香豆素为能量供体、1, 8-萘酰亚胺为能量受体组合而成的FRET类FA探针27 (Eq. 18), 该探针以homoallylicamine基团为连接两个荧光基团的链, 由于FRET机制, 香豆素荧光被1, 8-萘酰亚胺淬灭, 当FA加入后, 两者分离随后香豆素的荧光恢复.经实验证明, 该探针具有很好的FA专一性, 检测限为3 μmol·L-1, 适合在pH 4~6之间检测, 细胞毒性低(IC50=234 μmol·L-1).最后, 作者先利用探针进行溶酶体定位, 随后进行Hela细胞内FA的定量检测, 并首次证明N-乙酰半胱氨酸可清除细胞内的FA, 是潜在的FA相关疾病的药物.在激发波长为800 nm下, 探针27可在老鼠腹部组织中进行FA的双光子成像, 为溶酶体FA浓度的诊断以及治疗提供新的有力工具.
传统的FRET探针往往只能实现比率型或者关-开型信号变化, 即只能实现单通道信号增强, 这类机制缺少自验证的效果从而不能准确地检测复杂体系中的生物分子[88, 89]. Zhu课题组[90]利用两个相连的萘酰亚胺为FRET组合, 设计并合成了双增强型双光子GSH荧光探针28 (Eq. 19), 由于萘酰亚胺属于聚集导致淬灭型的荧光染料, 当两个不同激发的萘酰亚胺通过二硫键相连后, 容易在缓冲液中发生聚集作用, 两个荧光染料的荧光强度均被降低, 当GSH存在时, 二硫键断裂, 两个染料的荧光均增强.该探针对GSH的检测下限为2.9 μmol·L-1, 对GSH专一性良好.最后, 作者利用单双光子激光对Hela细胞中的GSH进行共聚焦成像, 为双增强型探针的发展做了良好的铺垫.
|
|
(17) |
|
|
(18) |
|
|
(19) |
近年来, 由于双光子成像技术的高速发展以及1, 8-萘酰亚胺具有易合成、光稳定性好、大/反斯托克斯位移等优点, 以1, 8-萘酰亚胺为母核的双光子探针发展迅速, 本综述以不同的荧光发光机制来分类, 总结并讨论了1, 8-萘酰亚胺类荧光探针在生命科学、疾病诊断、药物筛选等领域中的设计理念以及双光子成像应用.
当然, 现有的1, 8-萘酰亚胺类荧光染料也有些许不足, 主要体现在: (1)发射波长往往在黄光波段, 组织穿透能力不够强, 限制了该染料在更深层次组织中成像的应用; (2)萘环部分多取代基的研究较少; 限制了染料的应用谱; (3)溶解度较差, 限制了部分探针在细胞成像中的应用.因此, 把握1, 8-萘酰亚胺双光子特性, 开发荧光性能更加优异的1, 8-萘酰亚胺类荧光探针, 将是双光子成像领域中的研究热点.
Li, X.; Gao, X.; Shi, W.; Ma, H. Chem. Rev. 2014, 114, 590. doi: 10.1021/cr300508p
Chen, X.; Tian, X.; Shin, I.; Yoon, J. Chem. Soc. Rev. 2011, 40, 4783. doi: 10.1039/c1cs15037e
Tang, Y.; Lee, D.; Wang, J.; Li, G.; Yu, J.; Lin, W.; Yoon, J. Chem. Soc. Rev. 2015, 44, 5003. doi: 10.1039/C5CS00103J
Kikuchi, K. Chem. Soc. Rev. 2010, 39, 2048. doi: 10.1039/b819316a
Kim, H. M.; Cho, B. R. Chem. Rev. 2015, 115, 5014. doi: 10.1021/cr5004425
Kim, D.; Ryu, H. G.; Ahn, K. H. Org. Biomol. Chem. 2014, 12, 4550. doi: 10.1039/C4OB00431K
黄池宝, 陈绍英, 化学进展, 2017, 29, 1215.Huang, C.; Chen, S. Prog. Chem. 2017, 29, 1215 (in Chinese).
Loving, G.; Imperiali, B. J. Am. Chem. Soc. 2008. 130, 13630. doi: 10.1021/ja804754y
Prezhdo, O. V.; Uspenskii, B. V.; Prezhdo, V. V.; Boszczyk, W.; Distanov, V. B. Dyes Pigm. 2009, 83, 324. doi: 10.1016/j.dyepig.2009.05.010
Banerjee, S.; Veale, E. B.; Phelan, C. M.; Murphy, S. A.; Tocci, G. M.; Gillespie, L. J.; Frimannsson, D. O.; Kelly, J. M.; Gunnlaugsson, T. Chem. Soc. Rev. 2013, 42, 1601. doi: 10.1039/c2cs35467e
Duke, R. M.; Veale, E. B.; Pfeffer, F. M.; Kruger, P. E.; Gunnlaugsson, T. Chem. Soc. Rev. 2010, 39, 3936. doi: 10.1039/b910560n
Qian, X.; Xiao, Y.; Xu, Y.; Guo, X.; Qian, J.; Zhu, W. Chem. Commun. 2010, 46, 6418. doi: 10.1039/c0cc00686f
Alexiou, M. S.; Tychopoulos, V.; Ghorbanian, S.; Tyman, J. H. P.; Brown, R. G.; Brittain, P. I. J. Chem. Soc., Perkin Trans. 2 1990, 837. doi: 10.1002/chin.199037042/full
de Silva, A. P.; Gunaratne, H. Q. N.; Gunnlaugsson, T.; Huxley, A. J. M.; McCoy, C. P.; Rademacher, J. T.; Rice, T. E. Chem. Rev. 1997, 97, 1515. doi: 10.1021/cr960386p
Finkel, T.; Holbrook, N. J. Nature 2000, 408, 2392. https://www.researchgate.net/publication/12237821_Oxidants_oxidative_stress_and_biology_of_ageing_Nature
Lippert, A. R.; Bittner, V.; Chang, C. J. Acc. Chem. Res. 2011, 44, 793. doi: 10.1021/ar200126t
Srikun, D.; Miller, E. W.; Domaille, D. W.; Chang, C. J. J. Am. Chem. Soc. 2008, 130, 4596. doi: 10.1021/ja711480f
Zdolsek, J.; Zhang, H.; Roberg, K.; Brunk, U. Free Radical Res. Commun. 1993, 18, 71. doi: 10.3109/10715769309147344
Ren, M.; Deng, B.; Wang, J. Y.; Kong, X.; Liu, Z. R.; Zhou, K.; He, L.; Lin, W. Biosens. Bioelectron. 2016, 79, 237. doi: 10.1016/j.bios.2015.12.046
Xu, Z.; Liu, T.; Spring, D. R.; Cui, J. Org. Lett. 2013, 15, 2310. doi: 10.1021/ol400973v
Winterbourn, C. C.; Hampton, M. B.; Livesey, J. H.; Kettle, A. J. J. Biol. Chem. 2006, 281, 39860. doi: 10.1074/jbc.M605898200
Pattison, D. I.; Davies, M. J. Biochemistry 2006, 45, 81522. http://europepmc.org/abstract/MED/16800640
Zhu, B.; Li, P.; Shu, W.; Wang, X.; Liu, C.; Wang, Y.; Wang, Z.; Wang, Y.; Tang, B. Anal. Chem. 2016, 88, 12532. doi: 10.1021/acs.analchem.6b04392
Zhang, B.; Yang, X.; Zhang, R.; Liu, Y.; Ren, X.; Xian, M.; Ye Y., Zhao Y. Anal. Chem. 2017, 89, 10384. doi: 10.1021/acs.analchem.7b02361
Wang, B.; Li, P.; Yu, F.; Song, P.; Sun, X.; Yang, S.; Lou, Z.; Han, K. Chem. Commun. 2013, 49, 1014. doi: 10.1039/C2CC37803E
Lou, Z.; Li, P.; Han, K. Acc. Chem. Res. 2015, 48, 1358. doi: 10.1021/acs.accounts.5b00009
Yu, F.; Li, P.; Wang, B.; Han, K. J. Am. Chem. Soc. 2013, 135, 7674. doi: 10.1021/ja401360a
Yu, F.; Li, P.; Li, G.; Zhao, G.; Chu, T.; Han, K. J. Am. Chem. Soc. 2011, 133, 11030. doi: 10.1021/ja202582x
Lou, Z.; Li, P.; Pan, Q.; Han, K. Chem. Commun. 2013, 49, 2445. doi: 10.1039/c3cc39269d
Weidinger, A.; Kozlov, A. V. Biomolecules 2015, 5, 472. doi: 10.3390/biom5020472
Radi, R. J. Biol. Chem. 2013, 288, 26464. doi: 10.1074/jbc.R113.472936
Li, Y.; Xie, X.; Yang, X.; Li, M.; Jiao, X.; Sun, Y.; Wang, X.; Tang, B. Chem. Sci. 2017, 8, 4006. doi: 10.1039/C7SC00303J
Brewer, T. F.; Chang, C. J. J. Am. Chem. Soc. 2015, 137, 10886. doi: 10.1021/jacs.5b05340
Roth, A.; Li, H.; Anorma, C.; Chan, J. J. Am. Chem. Soc. 2015, 137, 10890. doi: 10.1021/jacs.5b05339
Xie, Z.; Ge, J.; Zhang, H.; Bai, T.; He, S.; Ling, J.; Sun, H.; Zhu, Q. Sens. Actuators, B 2017, 241, 1050.
Lin, J.-Y.; Zhang, M.-W.; Wang, J.-G.; Li, H.; Wei, H.-Y.; Liu, R.; Dai, G.; Liao, X. -X. Exp. Ther. Med. 2016, 11, 577. doi: 10.3892/etm.2015.2950
Lin, V. S.; Chen, W.; Xian, M.; Chang, C. J. Chem. Soc. Rev. 2015, 44, 4596. doi: 10.1039/C4CS00298A
Liu, X. L.; Du, X. J.; Dai, C. G.; Song, Q. H. J. Org. Chem. 2014, 79, 9481. doi: 10.1021/jo5014838
Kimura, H.; Shibuya, N.; Kimura, Y. Antioxid. Redox Signaling 2012, 17, 45. http://www.ncbi.nlm.nih.gov/pubmed/22229673
Fu, Y. J.; Yao, H. W.; Zhu, X. Y.; Guo, X. F.; Wang, H. Anal. Chim. Acta 2017, 994, 1. doi: 10.1016/j.aca.2017.09.030
Zhou, Y.; Zhang, J.; Yoon, J. Chem. Rev. 2014, 114, 5511. doi: 10.1021/cr400352m
张惠敏, 吴彦城, 尤嘉宜, 曹梁, 丁沙, 蒋凯, 汪朝阳, 有机化学, 2016, 36, 2559. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201611004&dbname=CJFD&dbcode=CJFQZhang, H.; Wu, Y.; You, J.; Cao, L.; Ding, S.; Jiang, K.; Wang, Z. Chin. J. Org. Chem. 2016, 36, 2559 (in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201611004&dbname=CJFD&dbcode=CJFQ
Zhang, J. F.; Lim, C. S.; Bhuniya, S.; Cho, B. R.; Kim, J. S. Org. Lett. 2011, 13, 1190. doi: 10.1021/ol200072e
Zhou, L.; Hu, S.; Wang, H.; Sun, H.; Zhang, X. Spectrochim. Acta, Part A 2016, 166, 25. doi: 10.1016/j.saa.2016.05.013
Qian, L.; Li, L.; Yao, S. Q. Acc. Chem. Res. 2016, 49, 626. doi: 10.1021/acs.accounts.5b00512
Dai, Z. R.; Ge, G. B.; Feng, L.; Ning, J.; Hu, L. H.; Jin, Q.; Wang, D. D.; Lv, X.; Dou, T. Y.; Cui, J. N.; Yang, L. J. Am. Chem. Soc. 2015. 137, 14488.
Dai, Z. R.; Feng, L.; Jin, Q.; Cheng, H.; Li, Y.; Ning, J.; Yu, Y.; Ge, G. B.; Cui, J. N.; Yang, L. Chem. Sci. 2017, 8, 2795. doi: 10.1039/C6SC03970G
Kaminsky, L. S.; Spivack, S. D. Mol. Aspects Med. 1999, 20, 70.
Wei, C.; Caccavale, R. J.; Weyand, E. H.; Chen, S.; Iba, M. M. Cancer Lett. 2002, 178, 25. doi: 10.1016/S0304-3835(01)00809-6
Satoh, T.; Hosokawa, M. J. Pestic. Sci. 2010, 35, 218. doi: 10.1584/jpestics.R10-02
Pratt, S. E.; Durland-Busbice, S.; Shepard, R. L.; Donoho, G. P.; Starling, J. J.; Wickremsinhe, E. R.; Perkins, E. J.; Dantzig, A. H. Mol. Cancer Ther. 2013, 12, 481. doi: 10.1158/1535-7163.MCT-12-0654
Jin, Q.; Feng, L.; Wang, D. D.; Dai, Z. R.; Wang, P.; Zou, L. W.; Liu, Z. H.; Wang, J. Y.; Yu, Y.; Ge, G. B.; Cui, J. N.; Yang, L. ACS Appl. Mater. Interfaces 2015, 7, 28474. doi: 10.1021/acsami.5b09573
Hildebrandt, M.; Reutter, W.; Arck, P.; Rose, M.; Klapp, B. F. Clin. Sci. 2000, 99, 932.
Zou, L. W.; Wang, P.; Qian, X. K.; Feng, L.; Yu, Y.; Wang, D. D.; Jin, Q.; Hou, J.; Liu, Z. H.; Ge, G. B.; Yang, L. Biosens. Bioelectron. 2017, 90, 283. doi: 10.1016/j.bios.2016.11.068
Spergel, D. J.; Krȕth, U.; Shimshek, D. R.; Sprengel, R.; Seeburg, P. H. Prog. Neurobiol. 2001, 63, 673. doi: 10.1016/S0301-0082(00)00038-1
Kamiya, M.; Asanuma, D.; Kuranaga, E.; Takeishi, A.; Sakabe, M.; Miura, M.; Nagano, T.; Urano, Y. J. Am. Chem. Soc. 2011, 133, 12960. doi: 10.1021/ja204781t
Asanuma, D.; Sakabe, M.; Kamiya, M.; Yamamoto, K.; Hiratake, J.; Ogawa, M.; Kosaka, N.; Choyke, P. L.; Nagano, T.; Kobayashi, H.; Urano, Y. Nat. Commun. 2015, 6, 6463. doi: 10.1038/ncomms7463
Zhang, X.; Wu, H.; Li, P.; Qu, Z.; Tan. M.; Han. K. Chem. Commun. 2016, 52, 8283. doi: 10.1039/C6CC04373A
Hattori, A.; Matsumoto, H.; Mizutani, S.; Tsujimoto, M. J. Biochem. 1999, 125, 931. doi: 10.1093/oxfordjournals.jbchem.a022371
Shastri, N.; Schwab, S.; Serwold, T. Annu. Rev. Immunol. 2002, 20, 463. doi: 10.1146/annurev.immunol.20.100301.064819
Xu, S.; Liu, H. W.; Hu, X. X.; Huan, S. Y.; Zhang, J.; Liu, Y. C.; Yuan, L.; Qu, F. L.; Zhang, X. B.; Tan, W. Anal. Chem. 2017, 89, 7641. doi: 10.1021/acs.analchem.7b01561
Ueno, T.; Urano, Y.; Kojima, H.; Nagano, T. J. Am. Chem. Soc. 2006, 128, 10640. doi: 10.1021/ja061972v
Ueno, T.; Urano, Y.; Setsukinai, K.; Takakusa, H.; Kojima, H.; Kikuchi, K.; Ohkubo, K.; Fukuzumi, S.; Nagano, T. J. Am. Chem. Soc. 2004, 126, 14079. doi: 10.1021/ja048241k
Zhang, P.; Wang, H.; Zhang, D.; Zeng, X.; Zeng, R.; Xiao, L.; Tao, H.; Long, Y.; Yi, P.; Chen, J. Sens. Actuators, B 2018, 255, 2223.
Cnop, M.; Foufelle, F.; Velloso, L. A. Trends Mol. Med. 2012, 18, 59. doi: 10.1016/j.molmed.2011.07.010
Harding, H. P.; Ron, D. Diabetes 2002, 51, S455. doi: 10.2337/diabetes.51.2007.S455
Xiao, H.; Liu, X.; Wu, C.; Wu, Y.; Li, P.; Guo, X.; Tang, B. Biosens. Bioelectron. 2017, 91, 449. doi: 10.1016/j.bios.2016.12.068
Weishaupt, K. R.; Gomer, C. J.; Dougherty, T. J. Cancer Res. 1976, 36, 2326. doi: 10.1007/BF01946419.pdf
Castano, A. P.; Mroz, P.; Hamblin, M. R. Nat. Rev. Cancer 2006, 6, 535. doi: 10.1038/nrc1894
Liu, H. W.; Xu, S.; Wang, P.; Hu, X. X.; Zhang, J.; Yuan, L.; Zhang, X. B.; Tan, W. Chem. Commun. 2016, 52, 12330. doi: 10.1039/C6CC05880A
da Silva, T. R. M.; de Freitas, J. R.; Silva, Q. C.; Figueira, C. P.; Roxo, E.; Leao, S. C.; de Freitas, L. A. R.; Veras, P. S. T. Infect. Immun. 2002, 70, 5628. doi: 10.1128/IAI.70.10.5628-5634.2002
Yu, H.; Xiao, Y.; Jin, L. J. Am. Chem. Soc. 2012, 134, 17486. doi: 10.1021/ja308967u
Tang, Y.; Kong, X.; Xu, A.; Dong, B.; Lin, W. Angew. Chem., Int. Ed. 2016, 55, 3356. doi: 10.1002/anie.201510373
Lee, Y. H.; Tang, Y.; Verwilst, P.; Lin, W.; Kim, J. S. Chem. Commun. 2016, 52, 112472. http://www.researchgate.net/publication/306269873_A_biotin-guided_formaldehyde_sensor_selectively_detecting_endogenous_concentrations_in_cancerous_cells_and_tissues
Ren, W. X.; Han, J. Y.; Uhm, S.; Jang, Y. J.; Kang, C.; Kim, J. H.; Kim, J. S. Chem. Commun. 2015, 51, 10403. doi: 10.1039/C5CC03075G
Matafome, P.; Sena, C.; Seica, R. Endocrine 2013, 43, 472. doi: 10.1007/s12020-012-9795-8
Singh, R.; Barden, A.; Mori, T.; Beilin, L. Diabetologia 2001, 44, 129. doi: 10.1007/s001250051591
Tang, T.; Zhou, Y.; Chen, Y.; Li, M.; Feng, Y.; Wang, C.; Wang, S.; Zhou, X. Anal. Methods 2015, 7, 2386. 2 doi: 10.1021/nl302049k
Bulaj, G.; Kortemme, T.; Goldenberg, D. P. Biochemistry 1998, 37, 8965. doi: 10.1021/bi973101r
Weerapana, E.; Wang, C.; Simon, G. M.; Richter, F.; Khare, S.; Dillon, M. B. D.; Bachovchin, D. A.; Mowen, K.; Baker, D.; Cravatt, B. F. Nature 2010, 468, 790. doi: 10.1038/nature09472
Zhu, X.; Li, Y.; Zan, W.; Zhang, J.; Chen, Z.; Liu, X.; Qi, F.; Yao, X.; Zhang, X.; Zhang, H. Photochem. Photobiol. Sci. 2016, 15, 412. doi: 10.1039/C5PP00468C
Chen, X. Q.; Zhou, Y.; Peng, X. J.; Yoon, J. Y. Chem. Soc. Rev. 2010, 39, 2120. doi: 10.1039/b925092a
Wang, G. L.; Jiao, H. J.; Zhu, X. Y.; Dong, Y. M.; Li, Z. J. Analyst 2013, 138, 2000. doi: 10.1039/c3an36878e
Liu, Y.; Liu, Y.; Liu, W.; Liang, S. Spectrochim. Acta, Part A 2015, 137, 509. doi: 10.1016/j.saa.2014.08.072
Yuan, L.; Lin, W.; Zheng, K.; Zhu, S. Acc. Chem. Res. 2013, 46, 1462. doi: 10.1021/ar300273v
Yao, S.; Qian, Y. Sens. Actuators, B 2017, 252, 877. https://www.researchgate.net/publication/317620079_A_naphthalimide-rhodamine_two-photon_fluorescent_turn-on_probe_for_hypochlorous_acid_by_desulfurization-cyclization_and_fluorescence_resonance_energy_transfer
Xie, X.; Tang, F.; Shangguan, X.; Che, S.; Niu J.; Xiao, Y.; Wang, X.; Tang, B. Chem. Commun. 2017, 53, 6520. doi: 10.1039/C7CC03050A
Lee, D. E.; Koo, H.; Sun, I. C.; Ryu, J. H.; Kim, K.; Kwon, I. C. Chem. Soc. Rev. 2012, 41, 2656. doi: 10.1039/C2CS15261D
Yuan, Y.; Zhang, R.; Cheng, X.; Xu, S.; Liu, B. Chem. Sci. 2016, 7, 4245. doi: 10.1039/C6SC00055J
Shen, W.; Ge, J.; He, S.; Zhang, R.; Zhao, C.; Fan, Y.; Yu, S.; Liu, B.; Zhu, Q. Chem. -Asian J. 2017, 12, 1532. doi: 10.1002/asia.v12.13

图 4 溶酶体靶向探针2与H2O2的反应机理
Figure 4 Reaction mechanism of lysosome-targetable probe 2 with H2O2

图式 1 双光子探针3a和3b与HClO的反应机理
Scheme 1 Reaction mechanism of two-photon probes 3a and 3b with HClO

图式 5 双光子探针12与CYP1A1的反应机理
Scheme 5 Reaction mechanism of two-photon probe 12 with CYP1A1

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




















 下载:
下载:







