
Scheme 1.
Retrosynthesis of syn-difluorianted amino acid 1 based on benzylic fluorination

The introduction of fluorine often imparts organic molecules with desired properties owing to its unique properties. The formation of C—F bond has thus been an intensive research subject and gained impressive development in the past ten years.[1] Benzylic C—H fluorination has emerged as a valuable tool for the introduction of fluorine.[2] In this context, the research groups of Lectka, [3~6] Groves, [7] Inoue, [8] Tang[9] and Britton[10] have realized benzylic C—H bond fluorination with transition-metal catalysts, including copper, manganese, iron, silver and decatungstate catalysts. In a parallel, the research groups of Chen, [11] Inoue[8] Lec-tka, [12, 13] Kappe[14] group achieved benzylic C—H bonds fluorination with the aid of visible light in the presence of an organocatalyst such as diarylketones, N, N-dihydroxypy- romellitimide and 1, 2, 4, 5-tetracyanobenzene. Method with- out using transition-metal catalysts and organocatalysts has also been developed.[15] In addition, the research groups of Groves[16] and Carrol[17] have developed successful proto-cols for benzylic fluorination with 18F fluoride for positron emission tomography (PET). However, the powerfulness of benzylic C—H fluorination has not been tested in any multi-step synthesis. We report herein the successful application of benzylic C—H fluorination as a key step for the formal synthesis of syn-α, β-difluoro-γ-amino acid 1, a very challenging target.
α, β-Difluoro-γ-amino acids are useful entities for biological application owing to the so-called fluorine conformational effect.[18~20] These molecules have found applications as subtype-selective γ-aminobutyric acid (GABA) receptor ligands and as components of shape-controlled peptides.[21~23] In order to meet the demand for application of α, β-difluoro-γ-amino acids, we and others[21, 24] have devoted to develop scalable synthetic routes. However, all of the previous methods need to use nucleophilic fluorinating reagents such as triethylamine trifluoride and Deoxo-Fluor, which are both corrosive and toxic, thus potentially dangerous for handling. This has contributed largely to the difficulty for the synthesis of syn-α, β-difluoro-γ-amino acid 1 (the syn-isomer is much more difficult to synthesize than the anti-isomer), which could only be obtained in tens of milligram previously.[24]
We envisioned that a more practical route for 1 was possible if benzylic C—H fluorination could be utilized for the introduction of fluorine atom to 3, which could be readily prepared by an enantioselective electrophilic fluorination and reductive amination of 4 (Scheme 1).[25~28] This route could avoid nucleophilic fluorinating reagents by using safe and easy-to-handle electrophilic reagents, such as Selectfluor and NFSI.
As outlined in Scheme 1, the synthesis of 3 commenced with α-fluorination and in-situ reductive amination of phenylpropyl aldehyde 4. This reaction could be conducted on a multi-gram scale, with no decrease of enantiomeric excess. Removal of the benzyl group of 5 and protection of the resulting amine with phthalic anhydride afforded the desired product 3 in an overall yield of 60% from 4.
Initially, we carried out a model benzylic C—H fluori-nation using 7, the non-fluorinated analogue of 3. To have a good understanding of different methods (e.g., ease of setting up, workup, efficiency etc.), we evaluated the reported standard conditions of representative methods. As shown in Table 1, the desired fluorination underwent smoothly under most conditions to afford the desired 8. Even without optimization, the yields under conditions in Entries 1~5 are synthetically useful (given the difficulties encountered in the introduction of the second fluorine in the previous approachs.[21, 24] It is important to point out that the solvent needed to be strictly degassed before use, otherwise sig-nificant amount of ketone 9 was obtained as the side product, which would decrease the yield and complicate the isolation of desired product 8.
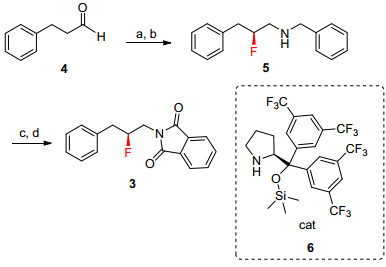
Conditions: (a) NFSI, cat. 6 (5 mol%), then benzylamine; (b) KBH3CN, pH < 3, MeOH, 93%, 99% ee; (c) Pd/C, H2, AcOH, r.t.; 24 h; (d) phthalic anhydride, toluene, reflux, 64%.
 下载:
导出CSV
下载:
导出CSV
 | ||
| Entry | Conditionsa | Yieldb/% |
| 1 | 9-Fluorenone (5 mol%)[11] Selectfluor (2 equiv), CFL, 25 ℃, 48 h | 77 |
| 2 | Fe(acac)2 (10 mol%), [5] Selectfluor (2.2 equiv.), 50 ℃, 24 h | 51 |
| 3 | NDHPI (2.5 mol%), [8]Selectfluor (1.2 equiv.), 50 ℃ 24 h | 33 |
| 4 | Xanthone (5 mol%) Selectfluor (1.2 equiv.), Black-light lamp, 25℃, 24 h[14] | 75 |
| 5 | Xanthone (5 mol%) Selectfluor (1.2 equiv.), Black-light lamp, 40℃, 24 h [14] | 43 |
| 6 | K2S2O8 (1.5 equiv.), Selectfluor (1.5 equiv.), 24 h[15] | 18 |
| 7 | Copper (I) bisimine complex (10 mol%), KB(C6F5)4 (10 mol%), NHPI (10 mol%), Selectfluor (2.2 equiv.), 24 h [3] | 0 |
| 8 | Manganese-salen complext, PhIO, TREAT•HF (0.5 equiv.), AgF (3.0 equiv.), 24 h[7] | 0 |
| aReactions were conducted on 0.07 mmol scale; b yields and ratio were calculated based on the integration of 19F NMR. | ||
Encouraged by the positive results obtained with 7, we next proceeded to conduct the benzylic fluorination of 3. To our surprise, we found that the mono-fluorinated 3 was actually quite difficult for fluorination. As shown in Table 2, the previously established viable methods for 7, such as conditions in Entries 1~4, gave either trace of no desired product. Optimization of the reaction conditions in Entries 1~3 proved to be fruitless. For example, increasing the reaction temperature had only marginal effect on the reac-tion efficiency (Entries 5~6, Table 2). These reactions along with the results obtained Entries 1~4 in Table 1 indicate that a vicinal fluorine atom may have a detrimental effect for the benzylic C—H fluorination.[29] To probe this fluorine effect further, we have synthesized compounds 10a~10c and 11a~11c and attempted the benzylic C—H fluorination under Chen's conditions.[11] As shown in Eq. 1, the non-fluorinated compounds 10a~10c underwent the desired reaction successfully to furnish 12a~12b in 55%~63% yields. In sharp contrast, no trace products could be observed for the fluorinated compounds 11a~11c.
下载:
导出CSV
 | |||
| Entry | Conditionsa | Yieldb/% | Molar ratiob of 2:14 |
| 1 | 9-Fluorenone (5 mol%), [11] Selectfluor (2 equiv), CFL, 2 ℃, 48 h | 0 | NA |
| 2 | Fe(acac)2 (10 mol%), [5] Selectfluor (2.2 equiv.), 50 ℃, 24 h | < 1 | NA |
| 3 | NDHPI (2.5 mol%), [8]Selectfluor (1.2 equiv.), 50 ℃, 24 h | < 1 | NA |
| 4 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 25℃, 24 h[11, 14] | 0 | NA |
| 5 | 9-Fluorenone (5 mol%), Selectfluor (2 equiv.), CFL, 40 ℃, 48 h | 4 | 1.1:1 |
| 6 | 9-Fluorenone (5 mol%), Selectfluor (2 equiv.), CFL, 60 ℃, 48 h | 3 | 1.1:1 |
| 7 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 40℃, 24 h[11, 14] | 38 | 1.76:1 |
| 8 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 60 ℃, 24 h | 38 | 1.19:1 |
| 9 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 60 ℃, 24 h | 68 | 1.58:1 |
| 10 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 70 ℃, 24 h | 56 | 1.42:1 |
| 11 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 80 ℃, 24 h | 43 | 1.32:1 |
| 12 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 60 ℃, 24 h | 79c, 71d | 1.52:1b, 1.53:1c |
| a Reactions were conducted on 0.07 mmol scale; b yields and ratio were calculated based on the integration of 19F NMR; c yield on 500 mini-gram scale; d yield on 1 gram scale. | |||
This "negative fluorine effect" thus posed significant challenge for the introduction of the second fluorine atom into compound 3.[30] To our delight, however, the modified conditions of Chen's method[11, 14] (Entry 4, Table 2) showed a promise for overcoming this fluorine effect, as the yield of the reaction was increased to 38% when the reaction tem-perature was increased (Entry 7, Table 2). After extensive investigation (not shown), the products were obtained in 68% yield with 1.58:1 of syn/anti ratio, when a large amount of photocatalyst and fluorinating reagent were used at 60 ℃ (Entries 8~9). Further optimization proved that a high temperature was not good for this reaction. With the optimized conditions in hand (Entry 9, Table 2), the remaining tasks were to scale the reaction up and separate the desired syn-isomer 2 from anti-isomer 14. To our delight, when the reaction was conducted on a 500 mini-gram and one gram scale, the yields were even better than that on small scale. However, the formation of ketone 15 was unavoidable, which was isolated in about 5% yield. As expected, the purification of 2 was not a trivial job, because compounds 2, 14 and 15 have almost the same polarity. At last, we managed to solve the problem by running two successive column purifications, first with V(n-hexane):V(CH2Cl2):V(ethyl acetate)=35:2:2 to isolate pure 2 and ketone 15, then with V(n-hexane):V(dioxane)=15:1 to get 14. Thus, 450 mini-gram of difluorinated 2 could be readily obtained from one gram of 3.
Conversion of compound 2 to the difluorinated amino acid 1 is a well-established chemistry (Eq. 2).[21b] Based on this fact, this formal synthesis of α, β-difluoro-γ-amino acid 1 requires 7 steps from commercial available reagents and has an overall yield of 18%. This result is significant as the previous synthesis of 1 requires 8 steps with overall yield around 5%. Moreover, no toxic and corrosive nucleophilic fluorinating reagents is used. Thus, the successful utilization of benzylic C—H fluorination has enabled a practical synthesis of a challenged target, α, β-difluoro-γ-amino acid 1.
|
|
(1) |
|
|
(2) |
α-Fluoro-γ-amino acid 17 is a useful component for prodrug development.[31] Previous synthesis also requires nucleophilic fluorinating reagents. In this work, α-fluoro-γ-amino acid 17 was obtained readily from 8 in an overall yield of 79%, thus offering additional benefit to our work (Scheme 3).
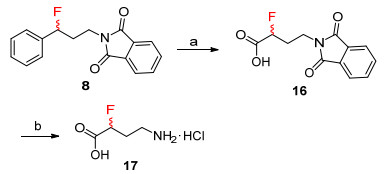
Conditions: (a) NalO4, RuCl3, CH2Cl2/MeCN/H2O, r.t., 80%; (b) NH2NH2•3H2O, EtOH, reflux, 99%
In conclusion, benzylic C—H fluorination has been successfully utilized as the key step for the formal synthesis of syn-α, β-difluoro-γ-amino acid 1, a very challenging but useful target. Because no corrosive and toxic nucleophilic fluorinating reagents are used and the overall yield is 18% (the previous approach is about 5%), the approach developed in this work could be viewed as more practical than the previous one. The key benzylic C—H reaction developed by Chen and co-workers[11, 14] was scaled up to one gram without decreasing its original efficiency. A striking fluorine effect was observed: a carbon with one fluorine atom on the adjacent carbon is much less reactive than an ordinary carbon for the benzylic C—H fluorination. We believe that the evaluation and optimization of different methods for benzylic C—H fluorination would provide valuable information for chemists who want to use them for target synthesis in future.
1H NMR, 13C NMR, and 19F NMR spectra were recorded on a JEOL FT-NMR spectrometer (1H NMR, 400 MHz; 2H NMR, 61 MHz; 13C NMR, 100 MHz; 19F NMR, 377 MHz). 1H NMR chemical shifts were determined relative to Me4Si (δ 0.0) as an internal standard. 13C NMR chemical shifts were determined relative to CDCl3 (δ 77.0). 19F NMR chemical shifts were determined relative to PhCF3 (δ —62.7) as an external standard. Infrared spectra were recorded on a SHIMADZU IRAffinity-1 FT-IR Spectrometer. Mass spectra were obtained on a JEOL JMS-DX303HF mass spectrometer. High-resolution mass spectra were obtained on a JEOL JMS-DX303HF mass spectrometer. Melting points were determined on a Stanford Research Systems MPA100 OptiMelt Automated Melting Point System. All reactions were carried out under nitrogen. Products were purified by chromatography on silica gel BW-300 (Fuji Silysia Chemical Ltd.) or aluminum oxide (Merck, 90 active stage I, 0.063~0.200 mm). Analytical thin-layer chromatography (TLC) was performed on pre-coated silica gel glass plates (Merck silica gel 60 F254, 0.25 mm thickness). Compounds were visualized with UV lamp or treatment with an ethanolic solution of phospho-molybdic acid followed by heating.
A 250 mL of round-bottom flask equipped with a magnetic stir bar was charged with Selectfluor (6.25 g, 17.65 mmol), xanthone (346.3 mg, 1.76 mmol) and (S)-2-(2- fluoro-3-phenylpropyl)isoindoline-1, 3-dione (3) (1.0 g, 3.53 mmol). Then anhydrous acetonitrile (120 mL) was added. The reaction mixture was degassed five times by freeze-pump-thaw cycles, and irradiated with a 26 W of black light bulb at 60 ℃ for 24 h. The reaction was quenched with H2O (20 mL) and extracted three times with EtOAc, dried over Na2SO4, filtered and concentrated in vacuo. Purification by flash column chromatography with silica gel [V(n-hexane):V(CH2Cl2):V(ethyl acetate)=35:2:2] to afford 2 as a white solid (457 mg, 43%), 15 as a colorless oil (50 mg, 5%) and a mixture of 14 (major) and 3 (minor). Then the mixture of 14 and 3 was purified by flash column chromatography with silica gel [V(n-hexane):V(dioxane) =15:1] to afford 14 as a white solid (297 mg, 28%) and recovered starting material 3 (100 mg).[11, 14]
2-((2R, 3R)-2, 3-Difluoro-3-phenylpropyl)isoindoline- 1, 3-dione (2):[21a] 1H NMR (400 MHz, CDCl3) δ: 7.85 (dd, J=3.1, 5.4 Hz, 2H), 7.73 (dd, J=3.0, 5.4 Hz, 2H), 7.45~7.35 (m, 5H), 5.56 (ddd, J=4.6, 19.3, 46.1 Hz, 1H), 5.11 (ddddd, J=3.6, 4.5, 9.1, 19.3, 48.3 Hz, 1H), 4.16 (ddd, J=9.1, 11.9, 21.2 Hz, 1H), 3.73 (ddd, J=3.6, 14.7, 29.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 167.8, 134.2, 131.8, 129.4 (d, J=1.9 Hz), 128.8, 126.6, 126.5, 123.5, 92.4 (dd, J=19.8, 180.5 Hz), 90.1 (dd, J=23.0, 185.4 Hz), 38.3 (dd, J=6.4, 23.8 Hz); 19F NMR (367 MHz, CDCl3) δ: —188.42 (ddd, J=12.7, 19.7, 46.4 Hz, 1F), —199.91 (m, 1F); 19F {1H} NMR (367 MHz, CDCl3) δ: —188.42 (d, J=12.3 Hz, 1F), —199.90 (d, J=12.9 Hz, 1F).
2-((2S, 3R)-2, 3-Difluoro-3-phenylpropyl)isoindoline-1, 3-dione (14):[21a] 1H NMR (400 MHz, CDCl3) δ: 7.81 (dd, J=3.1, 5.6 Hz, 2H), 7.70 (dd, J=3.1, 5.5 Hz, 2H), 7.44~7.31 (m, 5H), 5.68 (ddd, J=4.3, 12.9, 46.4 Hz, 1H), 5.11 (ddddd, J=3.9, 3.9, 8.5, 16.5, 47.8 Hz, 1H), 4.16 (ddd, J=8.8, 13.3, 14.4 Hz, 1H), 3.87 (ddd, J=3.4, 14.4, 29.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 167.8, 134.1, 131.8, 129.4 (d, J=1.9 Hz), 128.7, 126.1, 126.0, 123.4, 92.3 (dd, J=23.0, 177.2 Hz), 90.2 (dd, J=25.6, 181.7 Hz), 37.4 (dd, J=6.6, 23.6 Hz); 19F NMR (367 MHz, CDCl3) δ: —193.10 (ddd, J=16.5, 16.5, 46.5 Hz, 1F), —195.16 (m, 1F); 19F {1H} NMR (367 MHz, CDCl3) δ: —193.10 (d, J=16.5 Hz, 1F), —195.16 (d, J=16.9 Hz, 1F).
(R)-2-(2-Fluoro-3-oxo-3-phenylpropyl)isoindoline-1, 3-dione (15): [α]D25(c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=7.6 Hz, 2H), 7.88 (dd, J=3.1, 5.4 Hz, 2H), 7.75 (dd, J=3.0, 5.5 Hz, 2H), 7.66~7.60 (m, 1H), 7.55~7.40 (m, 2H), 5.99 (ddd, J=3.4, 9.0, 49.3 Hz, 1H), 4.35 (ddd, J=8.9, 14.6, 14.6, Hz, 1H), 4.18 (ddd, J=3.5, 14.8, 30.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 193.7 (d, J=17.3 Hz), 167.8, 134.3, 133.9, 131.8, 129.1, 129.0, 128.9, 123.5, 89.5 (d, J=188.0 Hz), 39.4 (d, J=23.1 Hz); 19F NMR (367 MHz, CDCl3) δ: —194.0 (ddd, J=14.6, 29.09, 49.4 Hz, 1F); 19F {1H} NMR (367 MHz, CDCl3) δ: —194.05 (s, 1F); IR (neat) νmax: 2928, 2855, 1772, 1717, 1486, 1467, 1457, 1419, 1395, 1363, 1279, 1223, 1188, 1080, 971, 888, 758, 720, 700, 647, 530 cm-1; HRMS (ESI) calcd for C17H13FNO3(M+H)+ 298.0874, found 298.0875.
Supporting Information The Supporting Information (PDF file of synthetic procedures, characterization data and NMR spectra) is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Champagne, P. A. ; Desroches, J. ; Hamel, J. D. ; Vandamme, M. ; Paquin, J. F. Chem. Rev. 2015, 115, 9073.
(b) Rong, J. ; Ni, C. ; Wang, Y. ; Kuang, C. ; Gu, Y. ; Hu, J. Acta Chim. Sinica 2017, 75, 105(in Chinese).
(荣健, 倪传法, 王云泽, 匡翠文, 顾玉诚, 胡金波, 化学学报, 2017, 75, 1, 105. )
(c) Zhang, P. ; Lu, L. ; Shen, Q. Acta Chim. Sinica 2017, 75, 744(in Chinese).
(张盼盼, 吕龙, 沈其龙, 化学学报, 2017, 75, 744. )
(d) He, J. ; Lou, S. ; Xu, D. Chin. J. Org. Chem. 2016, 36, 1218(in Chinese).
(何将旗, 娄绍杰, 许丹倩, 有机化学, 2016, 36, 1218. )
(e) Gu, Y. ; Lu, C. ; Gu, Y. ; Shen, Q. Chin. J. Chem. 2018, 36, 55.
Koperniku, A.; Liu, H.; Hurley, P. B. Eur.J.Org. Chem. 2016, 871. https://www.researchgate.net/publication/308993152_Denitrogenative_hydrofluorination_of_aromatic_aldehyde_hydrazones_using_difluoroiodotoluene
Bloom, S.; Pitts, C. R.; Miller, D. C.; Haselton, N.; Holl, M. G.; Urheim, E.; Lectka, T. Angew.Chem., Int.Ed. 2012, 51, 10580. doi: 10.1002/anie.201203642
Pitts, C. R.; Bloom, S.; Woltornist, R.; Auvenshine, D. J.; Ryzhkov, L. R.; Siegler, M. A.; Lectka, T. J.Am.Chem.Soc. 2014, 136, 9780. doi: 10.1021/ja505136j
Bloom, S.; Pitts, C. R.; Woltornist, R.; Griswold, A.; Holl, M. G.; Lectka, T. Org.Lett. 2013, 15, 1722. doi: 10.1021/ol400424s
Bloom, S.; Sharber, S. A.; Holl, M. G.; Knippel, J. L.; Lectka, T. J. Org.Chem. 2013, 78, 11082. doi: 10.1021/jo401796g
Liu, W.; Groves, J. T. Angew.Chem., Int.Ed. 2013, 52, 6024. doi: 10.1002/anie.201301097
Amaoka, Y.; Nagatomo, M.; Inoue, M. Org.Lett. 2013, 15, 2160. doi: 10.1021/ol4006757
Xu, P.; Guo, S.; Wang, L.; Tang, P. Angew.Chem., Int. Ed. 2014, 53, 5955. doi: 10.1002/anie.201400225
Nodwell, M. B.; Bagai, A.; Halperin, S. D.; Martin, R. E.; Knust, H.; Britton, R. Chem.Commun. 2015, 51, 11783. doi: 10.1039/C5CC04058B
Xia, J.-B.; Zhu, C.; Chen, C. J.Am. Chem.Soc. 2013, 135, 17494. doi: 10.1021/ja410815u
Bloom, S.; Knippel, J. L.; Lectka, T. Chem. Sci. 2014, 5, 1175. doi: 10.1039/C3SC53261E
Bloom, S.; McCann, M.; Lectka, T. Org.Lett. 2014, 16, 6338. doi: 10.1021/ol503094m
Cantillo, D.; de Frutos, O.; Rincon, J. A.; Mateos, C.; Kappe, C. O. J.Org.Chem. 2014, 79, 8486. doi: 10.1021/jo5016757
Ma, J.-J.; Yi, W.-B.; Lu, G.-P.; Cai, C. Org. Biomol.Chem. 2015, 13, 2890. doi: 10.1039/C4OB02418D
Huang, X.; Liu, W.; Ren, H.; Neelamegam, R.; Hooker, J. M.; Groves, J. T. J.Am.Chem.Soc. 2014, 136, 6842. doi: 10.1021/ja5039819
Carroll, L.; Evans, H. L.; Spivey, A. C.; Aboagye, E. O. Chem.Commun. 2015, 51, 8439. doi: 10.1039/C4CC05762G
Zimmer, L. E.; Sparr, C.; Gilmour, R. Angew. Chem., Int.Ed. 2011, 50, 11860. doi: 10.1002/anie.v50.50
Hunter, L. Beilstein J.Org.Chem. 2010, 6.
O'Hagan, D. Chem.Soc.Rev. 2008, 37, 308. doi: 10.1039/B711844A
(a) Hunter, L. ; Jolliffe, K. A. ; Jordan, M. J. T. ; Jensen, P. ; Macquart, R. B. Chem. -Eur. J. 2011, 17, 2340.
(b) Yamamoto, I. ; Jordan, M. J. T. ; Gavande, N. ; Doddareddy, M. R. ; Chebib, M. ; Hunter, L. Chem. Commun. 2012, 48, 829.
Hu, X. G.; Thomas, D. S.; Griffith, R.; Hunter, L. Angew.Chem.Int.Ed. 2014, 53, 6176. doi: 10.1002/anie.201403071
Hunter, L.; Butler, S.; Ludbrook, S. B. Org. Biomol.Chem. 2012, 10, 8911. doi: 10.1039/c2ob26596f
Patel, A. R.; Hu, X. G.; Lawer, A.; Ahmed, M. I.; Au, C.; Jwad, R.; Trinh, J.; Gonzalez, C.; Hannah, E.; Bhadbhade, M. M.; Hunter, L. Tetrahedron 2016, 72, 3305. doi: 10.1016/j.tet.2016.04.070
Beeson, T. D.; MacMillan, D. W. C. J.Am. Chem.Soc. 2005, 127, 8826. doi: 10.1021/ja051805f
Steiner, D. D.; Mase, N.; Barbas, C. F. Angew. Chem., Int.Ed. 2005, 44, 3706. doi: 10.1002/(ISSN)1521-3773
Marigo, M.; Fielenbach, D. I.; Braunton, A.; Kjoersgaard, A.; Jorgensen, K. A. Angew.Chem., Int.Ed. 2005, 44, 3703. doi: 10.1002/(ISSN)1521-3773
Fadeyi, O. O.; Lindsley, C. W. Org.Lett. 2009, 11, 943. doi: 10.1021/ol802930q
(a) Ni, C. F. ; Hu, J. B. Chem. Soc. Rev. 2016, 45, 5441.
(b) Ni, C. F. ; Hu, M. Y. ; Hu, J. B. Chem. Rev. 2015, 115, 765.
(a) Ni, C. ; Li, Y. ; Hu, J. J. Org. Chem. 2006, 71, 6829.
(b) Zhang, W. ; Ni, C. ; Hu, J. Top. Curr. Chem. 2012, 308, 25.
Ma, H.; Chen, G.; Wang, T.; Li, Q.; Liu, Y. Chem. Biol.Drug Des. 2016, 88, 363. doi: 10.1111/cbdd.2016.88.issue-3

Scheme 1 Retrosynthesis of syn-difluorianted amino acid 1 based on benzylic fluorination

Scheme 2 Synthesis of mono-fluorinated substrate 3
Conditions: (a) NFSI, cat. 6 (5 mol%), then benzylamine; (b) KBH3CN, pH < 3, MeOH, 93%, 99% ee; (c) Pd/C, H2, AcOH, r.t.; 24 h; (d) phthalic anhydride, toluene, reflux, 64%.

Scheme 3 Synthesis of α-fluoro-γ-amino acid from 8
Conditions: (a) NalO4, RuCl3, CH2Cl2/MeCN/H2O, r.t., 80%; (b) NH2NH2•3H2O, EtOH, reflux, 99%
Table 1. Model fluorination of non-fluorinated substrate 7
| ||
| Entry | Conditionsa | Yieldb/% |
| 1 | 9-Fluorenone (5 mol%)[11] Selectfluor (2 equiv), CFL, 25 ℃, 48 h | 77 |
| 2 | Fe(acac)2 (10 mol%), [5] Selectfluor (2.2 equiv.), 50 ℃, 24 h | 51 |
| 3 | NDHPI (2.5 mol%), [8]Selectfluor (1.2 equiv.), 50 ℃ 24 h | 33 |
| 4 | Xanthone (5 mol%) Selectfluor (1.2 equiv.), Black-light lamp, 25℃, 24 h[14] | 75 |
| 5 | Xanthone (5 mol%) Selectfluor (1.2 equiv.), Black-light lamp, 40℃, 24 h [14] | 43 |
| 6 | K2S2O8 (1.5 equiv.), Selectfluor (1.5 equiv.), 24 h[15] | 18 |
| 7 | Copper (I) bisimine complex (10 mol%), KB(C6F5)4 (10 mol%), NHPI (10 mol%), Selectfluor (2.2 equiv.), 24 h [3] | 0 |
| 8 | Manganese-salen complext, PhIO, TREAT•HF (0.5 equiv.), AgF (3.0 equiv.), 24 h[7] | 0 |
| aReactions were conducted on 0.07 mmol scale; b yields and ratio were calculated based on the integration of 19F NMR. | ||
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimization of the fluorination with mono-fluorinated 7
| |||
| Entry | Conditionsa | Yieldb/% | Molar ratiob of 2:14 |
| 1 | 9-Fluorenone (5 mol%), [11] Selectfluor (2 equiv), CFL, 2 ℃, 48 h | 0 | NA |
| 2 | Fe(acac)2 (10 mol%), [5] Selectfluor (2.2 equiv.), 50 ℃, 24 h | < 1 | NA |
| 3 | NDHPI (2.5 mol%), [8]Selectfluor (1.2 equiv.), 50 ℃, 24 h | < 1 | NA |
| 4 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 25℃, 24 h[11, 14] | 0 | NA |
| 5 | 9-Fluorenone (5 mol%), Selectfluor (2 equiv.), CFL, 40 ℃, 48 h | 4 | 1.1:1 |
| 6 | 9-Fluorenone (5 mol%), Selectfluor (2 equiv.), CFL, 60 ℃, 48 h | 3 | 1.1:1 |
| 7 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 40℃, 24 h[11, 14] | 38 | 1.76:1 |
| 8 | Xanthone (5 mol%), Selectfluor (1.2 equiv.), Black-light lamp, 60 ℃, 24 h | 38 | 1.19:1 |
| 9 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 60 ℃, 24 h | 68 | 1.58:1 |
| 10 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 70 ℃, 24 h | 56 | 1.42:1 |
| 11 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 80 ℃, 24 h | 43 | 1.32:1 |
| 12 | Xanthone (50 mol%), Selectfluor (5.0 equiv.), Black-light lamp, 60 ℃, 24 h | 79c, 71d | 1.52:1b, 1.53:1c |
| a Reactions were conducted on 0.07 mmol scale; b yields and ratio were calculated based on the integration of 19F NMR; c yield on 500 mini-gram scale; d yield on 1 gram scale. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

