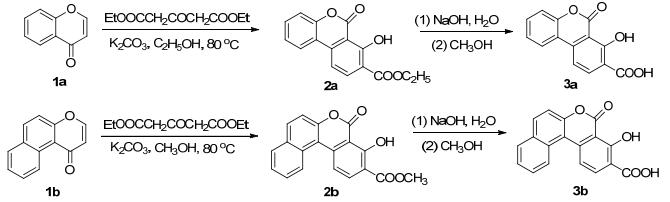
Scheme 1.
Synthetic routes of 3a and 3b

Due to the desirable optical and physical use, the design and synthesis of new solid fluorescence material have drawn a great attention in recent years.[1, 2] Simple and convenient synthesis strategy, photochemical stability and high quantum efficiency are crucial to the development of new fluorescence compound. Besides the structure of the fluorophore, [3, 4] fluorescence of organic compound in the solid state largely depends on the architecture of the crystal packing and intermolecular interaction, such as π…π stacking, hydrogen bonds and weak noncovalent forces.[5, 6]
6H-Benzo[c]chromen-6-ones are also known as coumarin derivatives, which contain biaryl lactones, and represent many biological effects, including antibacterial, [7] antioxidant, [8] anticancer and immune-modulating effects.[9, 10] They are usually isolated from plant sources and appeared as glycosides or aglycons.[11] So far, a few methods have been applied to their synthesis, such as palladium catalyzed coupling reactions, [12] Michael reactions, [13] Suzuki reactions[14] and retro-Michael aldol lactonization reactions reported recently.[15] So far, the photophysical properties of benzocoumarin derivatives are mainly focused on the type and position of electron-donating and/or electron-accepting substituents.[16] The strategies such as modulating molecular packing modes or intermolecular interactions are rarely reported, which contribute to the improvement of the fluorescent quantum efficiency.[17]
In this work, 7-hydroxy-6-oxo-6H-benzo[c]chromene- 8-carboxylic acid (3a) and 2, 3-benzo[f]-7-hydroxy-6- oxo-6H-benzo[c]chromene-8-carboxylic acid (3b) were synthesized from chromones (1) and 3-acetonedicarboxy- late by Michael reaction (Scheme 1). The structures of 2 and 3 were determined by 1H NMR, 13C NMR, ESI-MS and the X-ray single-crystal diffraction. Meanwhile, the photoluminescent experiment showed that the cyclic dimers of coumarin carboxylic acids have a significant effect on the fluorescence properties.
Crystallographic data of 3a and 3b are summarized in Table 1 (CCDC number: 1448872 and 1448877).
 下载:
导出CSV
下载:
导出CSV
| D—H…A | D—H | H…A | D—A | ∠D—H…A |
| 3a | ||||
| O3—H3…O4 | 0.820 | 1.841 | 2.576 | 148.50 |
| O1—H1…O2(ⅰ) | 0.820 | 1.870 | 2.674 | 166.80 |
| C9—H9…O4(ⅱ) | 0.930 | 2.517 | 3.364 | 140.83 |
| 3b | ||||
| O3—H3…O4 | 0.820 | 1.815 | 2.552 | 148.76 |
| O1—H1…O6 | 0.820 | 1.815 | 2.626 | 170.08 |
| C18—H18B…O4(ⅱ) | 0.930 | 2.488 | 3.303 | 146.35 |
| C19(ⅰ)—H19A(ⅰ)…O6 | 0.960 | 2.560 | 3.489 | 162.8 |
| C20—H20B…O3(ⅰ) | 0.960 | 2.754 | 3.524 | 136.41 |
| C20—H20C…O6(ⅰ) | 0.820 | 2.434 | 3.345 | 158.4 |
| a Symmetry codes: 3a (ⅰ) -x, -y, 1-z; (ⅱ) x, 2+y, z-0.5. 3b (ⅰ) 1-x, 1-y, 1-z; (ⅱ) 1-x, y, 0.5-z. | ||||
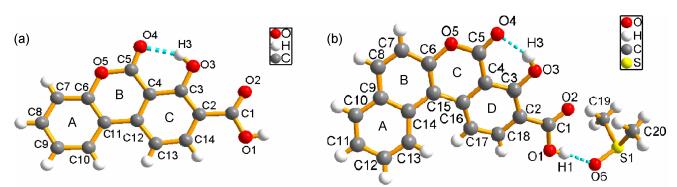
The molecular structures of 3a~3b are illustrated in Figure 1. Crystal structural investigation reveals that 3a possesses a 6-oxo-6H-benzo[c]chromene skeleton, a hydroxyl and a carboxylic group. The atoms of chromene moiety compose of ring A/B (C4~C5/O5/C6~C12) and benzyl ring composed of ring C (C2~C4/C12~C14), are planar with a small dihedral angle (1.6°), indicating that the 6-oxo-6H-benzo[c]chromene core has an approximate plane structure. In the crystal of 3b (Figure 1b), the coplanarity between the naphthalene ring (rings A and B) and the chrome moiety (rings C and D) is poor because of the space repulsion from the hydrogens in C13 and C17. The dihedral angle between two rings is 134°, this twist results in the reduction of the π conjugation. In addition, 3a~3b skeletons easily form strong innate intramolecular hydrogen bonds between the carbonyl unit and hydroxyl group, with the bond lengths of 0.255 and 0.275 nm. In the crystal structures of 3a and 3b, the carboxylic groups at C2 are nearly coplanar with its attached rings C/D, indicating the small torsion angles (172.7° and 178.7°).
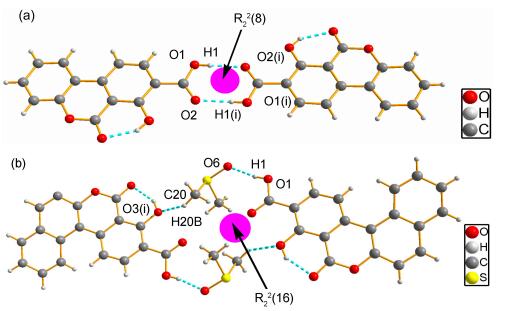
Crystal 3a forms in the triclinic centrosymmetric, space group P2(1)/c. 3a molecules adopt dimers via a pair of O1—H1…O2(ⅰ) intermolecular hydrogen bonds (symmetry code (ⅰ): -x, -y, 1-z). The carboxyl groups, acting as donor and acceptor in this contacts, create dimers in the R22(8) moieties, [18] which are typical for the carboxylic acids (Figure 2a), and bond lengths are 1.870 Å, bond angles are 166.8°. In the solid state, 3b molecules are self-assembled into dimers (Figure 2) through O1—H1…O6 and C20—H20B…O3(ⅰ) (symmetry code (ⅰ): 1-x, 1-y, 1-z). This motif can be described using graph-set notation as R22(16).
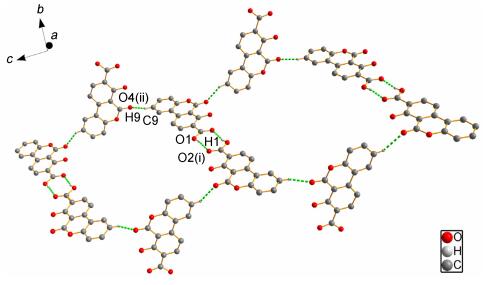
Furthermore, the dimers of 3a are linked by C9—H9…O4(ⅱ) (symmetry code (ⅱ): x, 2+y, z-0.5) intermolecular hydrogen bonds and form a 2D network along the c axis. As shown in Figure 3, the dimers are nearly perpendicular to connected molecules and the angle between them is 83°.
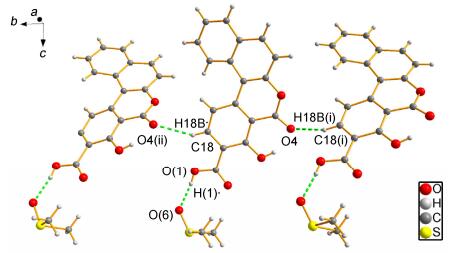
Compound 3b crystallizes in the orthorhombic and Pbca group with one lattice dimethyl sulfoxide (DMSO) in asymmetric unit. O6—H1…O6 and O3—H3…O4 are found in the asymmetric unit (Figure 2b). The dimeric unit of 3b is different from 3a, which is interlinked by O(1)—H(1)…O(6) and C18—H18A…O4B(ⅱ) (symmetry code (ⅱ): 1-x, y, 0.5-z) and form a chain structure along b axis (Figure 4).
The crystal packing is stabilized by π-π stacking interactions in 3a and 3b. In 3a crystal state, adjacent benzo[c]chromene rings are arranged in parallel fashion and aromatic stacking exist in the centers of rings C and A/B, the distance of interplanar centers of rings is 0.351 nm, which is in the range of 0.33~0.38 nm (Figure 5a).
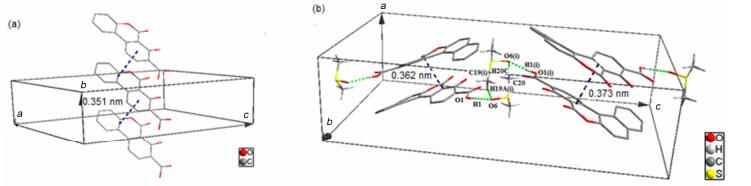
As shown in Figure 5b, the molecular packing of 3b exhibits intermolecular face-to-face interactions. The π-π stacking of 3b exists between C/D rings, which are arranged in anti-parallel stretches and forms a stacking fragment. Furthermore, the fragments are closely linked by two intermolecular hydrogen bonds, namely C19(ⅰ)—H19A(ⅰ)…O6 and C20—H20C…O6(ⅰ).
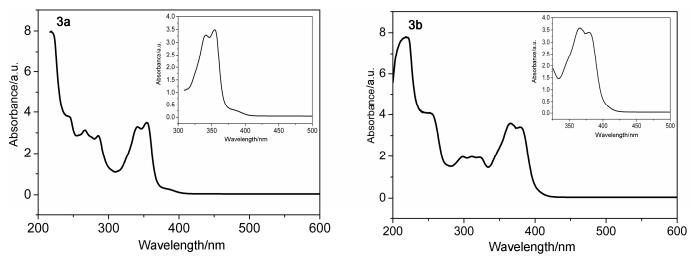
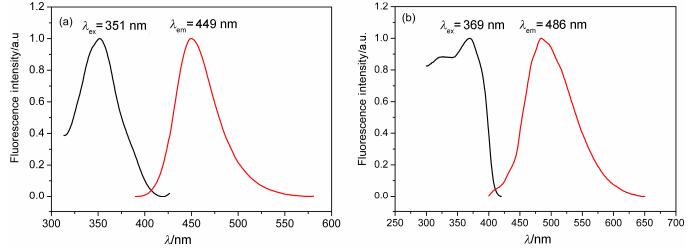
The photophysical properties of 3a and 3b have been measured using UV-vis absorption and fluorescence spectra, and the data are summarized in Table 2 and Figures 6, 7. In the UV-visible spectra (Figure 6), the UV-vis absorption maxima of 3a is at 354 nm and of 3b at 366 nm, which can be attributed to π-π* transitions of the conjugated aromatic segments.
下载:
导出CSV
| Compd. | Liquid state | Solid state | |||||
| λex/nm | λem/nm | ФF | λex/nm | λem/nm | ФF | ||
| 3a | 351 | 449 | 0.81 | 369 | 486 | 0.32 | |
| 3b | 370 | 453 | 0.84 | 388 | 487 | 0.20 | |
| 2a | 342 | 451 | 0.70 | 368 | 497 | 0.22 | |
The fluorescence spectra of 3a and 3b in the solid state and in 10-6 mol/L ethanol solution at room temperature are shown in Figures 7~8. Furthermore, the solid fluorescence quantum yields of 3a and 3b crystals are recorded on a Fluoromax-4 spectrofluorometer with centimeter-scale size and noodle shape (Table 2). It is well known that π conjugated framework, intermolecular electronic interactions and stacking modes of molecules in the solid-state exert great influence on the luminescent materials.[19] Compared with common coumarin (4H-chromen-4-one), the enlarged aromatic conjugated system and strong H-bonds lead to 3a and 3b have an excellent fluorescence not only in the ethanol solution but also in the crystal state.
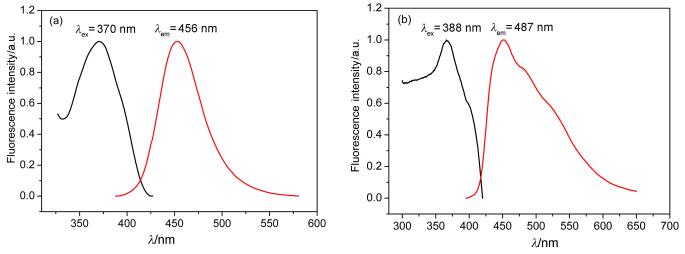
The fluorescence quantum yields of 3a and 3b in the crystals are 0.32 and 0.20, respectively. For the luminescence materials, the stronger the molecular rigidities, the higher fluorescence quantum yields they have. Although the conjugated system of 3b became bigger with naphtholcoumarin skeleton, its ФF is 0.2. To avoid steric conflicts, the naphthalene ring and coumarin moiety are rotated with 134° and this twist lead to the reduction of π-conjugation in 3b. In this case, strong intramolecular H-bonds between the carbonyl and hydroxyl group greatly increase the molecular rigidity and assist in activating the molecular planarization. The synthon was observed in 3a and 3b, which helped to improve the molecular stability. The interaction of O—H…O hydrogen bonding constituted 3a synthon is higher than that of C—H…O constituted 3b synthon, resulted in the better stability of 3a. Conventionally, carboxylic acid derivatives are known to form cyclic dimers.[20] The dimers of 3a crystals are formed via O1—H1…O2(ⅰ), which have the strongest intermolecular interactions (bond lengths are 0.1870 nm). And the angle between adjacent 3a molecules is about 7.8°, the atoms belonged to dimer located in the same plane and produced strong plane rigidity. In contrast with 3a, the fluorescence quantum yield of 2a is 0.22 because of a lack of carboxylic acid dimer. Another intermolecular hydrogen bond, C9—H9…O4(ⅱ), linked neighboring dimer into 2D network. Either O1—H1…O2 (ⅰ) or C9—H9…O4(ⅱ) are all composed between benzo[c]coumarin carboxylic skeleton without the participation of lattice solvent molecules. The strong intramolecular H-bonds greatly increase the molecular rigidity and assist in activating the molecular planarization. The crystal structures of 3b have formed different synthons by intermolecular action between benzo[c]chromene ring and lattice dimethyl sulfoxide (DMSO), and their rigid are weak.
Seen from the π…π interactions, which easily lead to fluorescence quenching, [21] the face-to-face aromatic stacking exist in the crystals of 3a and 3b. However, the edge-to-face neighboring mode of 3a weaken the π…π stacking to some extent. Follow these considerations, it could be concluded that the fluorescence of 3a is stronger than that of 3b in the solid, and this is in agreement with the facts of the fluorescent experiments. Furthermore, the fluorescence quantum yield of 3a is close to 3b in 10-6 mol/L ethanol solution because intermolecular interactions are weak in dilute solution.
Two fluorescence compounds, 3a and 3b were synthesized by Michael reaction with chromones and 3-acetonedicarboxylate. Especially, 3a owns a high fluorescence quantum efficiency in solid state. These results exhibit that the strategy of enlarged aromatic conjugated, regulated packing modes and strengthened intermolecular interactions is an effective way for exploring highly blue- emissive coumarin materials.
Melting points were measured with an X-5 micro-melting point apparatus and are uncorrected. NMR spectra were recorded with a Bruker AM 400 instrument using solvent peaks as reference in DMSO-d6 solutions. High-resolution mass spectrometry (HRMS) was recorded using the electron-spray ionization (ESI) technique. IR spectra were recorded with a Nicollet 170SX FT-IR spectrophotometer with KBr pellets. The crystal diffraction data were collected on a Bruker Smart-1000 CCD diffractometer. The liquid fluorescence spectra were recorded with a Hitachif-4600 luminescence spectrometer. The solid fluorescence spectra were recorded on a FluoroMax-4 fluorescence spectrometer equipped with an integrating sphere. Thin-layer chromatography (TLC) was performed on silica gel 60 GF254 plate. The silica gel (size 200~300 mesh) used for the column chromatography was purchased from Qingdao Haiyang Chemistry Plant (China).
According to a published procedure, [22] chromones (1a and 1b) were synthesized. Diethyl 1, 3-acetonedicarboxy- late (2 mmol) was dropped into the solution of chromone 1 (1 mmol) with potassium carbonate (1 mmol) as a base in ethanol (20 mL). Stirring was continued until plenty of yellow precipitates appeared (followed by TLC, approximately 4 h). The reaction mixture was adjusted to pH 6~7 with a solution of 3 mol/L HCl. After standing, the precipitate was filtered off and purified on silica gel column (petroleum ether-EtOAc) to give 2.
Thereafter, a solution of 2 (1 mmol) in methanol (30 mL) and 3 wt% NaOH solution (5 mL) was heated at reflux for 1 h. Then, the mixture was poured into H2O and the pH was adjusted to pH 6~7 with 3 mol/L HCl. The resulting precipitate was filtered to give the crude product 3. And recrystallized in the DMSO solution, pure product of 3 was obtained.
Ethyl 7-hydroxy-6-oxo-6H-benzo[c]chromene-8-carboxylate (2a): 0.222 g, 78% yield, white solid. m.p. 178.3~178.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 12.54 (s, 1H), 8.28 (d, J=8.4 Hz, 1H), 8.03 (d, J=7.9 Hz, 1H), 7.55 (dd, J=8.6, 7.7 Hz, 2H), 7.37 (dd, J=8.1, 8.4 Hz, 2H), 4.44 (q, J=7.1 Hz, 2H), 1.43(t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 165.8, 163.5, 151.4, 139.9, 138.8, 132.0, 125.4, 124.0, 117.9, 117.5, 116.8, 111.7, 107.5, 61.7, 14.4; IR (KBr) ν: 3432, 2912, 1694, 1615, 1519, 1351, 1051, 751, 601 cm-1; HRMS calcd for C16H13O5 [M+H]+ 285.0757, found 285.0766.
Methyl 4-hydroxy-5-oxo-5H-dibenzo[c, f]chromene-3- carboxylate (2b): 0.260 g, 81% yield, white solid. m.p. 180.0~181.70 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 12.35 (s, 1H), 8.77 (d, J=14.5 Hz, 1H), 8.40~8.09 (m, 4H), 7.77 (s, 1H), 7.64 (dd, J=18.4, 7.9 Hz, 2H), 3.90 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 167.6, 162.2, 150.1, 139.6, 136.9, 132.8, 130.9, 128.6, 127.1, 125.2, 124.4, 116.4, 116.0, 115.4, 111.3, 108.0; IR (KBr) ν: 2960, 1715, 1607, 1417, 1106, 1015, 824, 613 cm-1; HRMS calcd for C19H13O5 [M+H]+321.0757, found 321.0760.
7-Hydroxy-6-oxo-6H-benzo[c]chromene-8-carboxylic acid (3a): 0.202 g, 79% yield, white solid. m.p. 241.0~242.0 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.36~8.31 (m, 1H), 8.22 (d, J=8.4 Hz, 1H), 7.90 (d, J=8.6 Hz, 1H), 7.66~7.60 (m, 1H), 7.43 (dd, J=7.5, 6.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ: 168.6, 163.0, 160.7, 151.1, 139.9, 137.6, 132.0, 125.1, 124.6, 117.1, 115.9, 112.4, 107.7; IR (KBr) ν: 3749, 1722, 1622, 1422, 1171, 1121, 1004, 756, 587 cm-1; HRMS calcd for C14H9O5 [M+H]+257.0450, found 279.0446.
3-Benzo[f]-7-hydroxy-6-oxo-6H-benzo[c]chromene-8-carboxylic acid (3b): 0.119 g, 39% yield, white solid, m.p. 228.2~219.3 ℃; 1H NMR (600 MHz, DMSO-d6) δ: 8.64 (d, J=8.5 Hz, 1H), 8.23 (d, J=8.6 Hz, 1H), 8.11 (d, J=8.9 Hz, 1H), 8.04 (t, J=7.6 Hz, 2H), 7.69 (t, J=7.7 Hz, 1H), 7.58 (t, J=7.4 Hz, 1H), 7.48 (d, J=8.9 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ: 164.9, 162.7, 161.2, 149.9, 139.1, 137.5, 132.9, 131.1, 128.9, 128.6, 128.0, 125.4, 124.4, 116.4, 116.3, 115.8, 111.364, 107.6; IR (KBr) ν: 3573, 1730, 1605, 1463, 1363, 1255, 1080, 945, 813, 687 cm-1; HRMS calcd for C18H11O5 [M+H]+ 307.0606, found 307.0587.
Data for compounds 3a and 3b were measured from single crystals using a Bruker Smart-1000 CCD diffractometer meter with graphite-monochromated Mo-Kα radiation (λ=0.07107 nm) and intensity data for all compounds were collected by the narrow frame method at room temperature. All four data sets were corrected for absorption by the ψ-ω method. All structures were solved by direct methods with SHELXS-97[23] and further refined on F2 using SHELXL-97.[24] All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms on carbon atoms were calculated theoretically.
Supporting Information 1H NMR, 13C NMR, IR and HRMS data of 2a, 2b, 3a and 3b. Crystal data collection, spherical and multipole refinement of 3a and 3b. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
Hu, B. B.; Lu, P.; Wang, Y. G. New J. Chem. 2013, 37, 1645. doi: 10.1039/c2nj41063j
Yu, C.; Zhang, J.; Chen, L.; Li, J.; Liu, P.; Wang, W.; Yan, B. Talanta 2011, 85, 1627. doi: 10.1016/j.talanta.2011.06.057
Lee, E. Y.; Jang, S. Y.; Suh, M. P. J. Am. Chem. Soc. 2005, 127, 6374. doi: 10.1021/ja043756x
Hisaki, I.; Kometani, E.; Shigemitsu, H.; Saeki, A.; Seki, S.; Tohnai, N.; Miyata, M. Cryst. Growth Des. 2011, 11, 5488. doi: 10.1021/cg201075z
Seth, D. K.; Sakar, D.; Kar, T. CrystEngComm 2011, 13, 4528. doi: 10.1039/c1ce05037k
Zhou, T. L.; Jia, T.; Zhao, S. S.; Guo, J. H.; Zhang, H. Y.; Wang, Y. Cryst. Growth Des. 2012, 12, 179. doi: 10.1021/cg200920d
Ito, H.; Iguchi, A.; Hatano, T. J. Agric. Food Chem. 2008, 56, 393. doi: 10.1021/jf0726942
Bialonska, D.; Kasimsetty, S. G.; Khan, S. I.; Ferreira, D. J. Agric. Food Chem. 2009, 57, 10181. doi: 10.1021/jf9025794
Schmidt, J. M.; Tremblay, G. B.; Page, M.; Mercure, J.; Feher, M.; Dunn-Dufault, R.; Peter, M. G.; Redden, P. R. J. Med. Chem. 2003, 46, 1289. doi: 10.1021/jm034007d
Pandey, J.; Jha, A. K.; Hajela, K. Bioorg. Med. Chem. 2004, 12, 2239. doi: 10.1016/j.bmc.2004.02.018
Appel, B.; Saleh, N. N. R.; Langer, P. Chem.-Eur. J. 2006, 12, 1221. doi: 10.1002/(ISSN)1521-3765
Nguyen, V. T. H.; Langer, P. Tetrahedron Lett. 2005, 46, 1013. doi: 10.1016/j.tetlet.2004.12.030
Ullah, E.; Appel, B.; Fischer, C.; Langer, P. Tetrahedron 2006, 62, 9694. doi: 10.1016/j.tet.2006.07.084
Nikolov, P.; Petkov, I.; Marko, P. Z. Naturforsch. 2000, 55a, 741.
陈超越, 胡劲松, 柴飞飞, 谢凯云, 张晓梅, 石建军, 结构化学, 2014, 33, 395. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jghx201403009&dbname=CJFD&dbcode=CJFQChen, C. Y.; Hu, J. S.; Chai, F. F.; Yun, X. K.; Zhang, X. M.; Shi, J. J. Chin. J. Struct. Chem. 2014, 33, 395(in Chinese). http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jghx201403009&dbname=CJFD&dbcode=CJFQ
Tasior, M.; Kim, D.; Singha, S.; Krzeszewski, M.; Ahn, K. H.; Gryko, D. T. J. Mater. Chem. C 2015, 3, 1421.
Jiao, L.; Wu, Y.; Ding, Y.; Wang, S.; Zhang, P.; Yu, C.; Wei, Y.; Mu, X.; Hao, E. Chem.-Asian J. 2014, 9, 805. doi: 10.1002/asia.v9.3
Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, N. L. Angew. Chem., Int. Ed. 1995, 34, 1555. doi: 10.1002/(ISSN)1521-3773
孙静波, 张恭贺, 贾小宇, 薛鹏冲, 贾俊辉, 卢然, 化学学报, 2016, 74, 165. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb201602005Sun, J. B.; Zhang, G. H.; Jia, X. Y.; Xue, P. C.; Jia, J. P.; Lu, R. Acta Chim. Sinica 2016, 74, 165(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxxb201602005
Feng, Q.; Wang, M.; Dong, B.; Xu, C.; Zhao, J.; Zhang, H. CrystEngComm 2013, 15, 3623. doi: 10.1039/c3ce27102a
Cheng, X.; Wang, K.; Huang, S.; Zhang, H. Y.; Zhang, H. Y.; Wang, Y. Angew. Chem., Int. Ed. 2015, 54, 1. doi: 10.1002/anie.201410930
Yu, T. Z.; Zhao, Y. L.; Fan, D. W. J. Mol. Struct. 2006, 1~3, 18.
Sheldrick, G. M. SHELXS-97, Program for X-Ray Crystal Structure Solution, University of Götingen, Germany, 2007.
Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112.

Figure 4 Fragment of molecular structure of 3b with chain formed by C18—H18A…O4B(ⅱ) interaction propagating along the b direction

Figure 5 (a) Crystal packing of 3a showing π—π stacking interactions and (b) a side view of the mode of 3b molecules packing in a unit cell

Figure 7 Fluorescence spectra of 3a in 10-6 mol/L ethanol (a) and in crystal state (b) at room temperature

Figure 8 Fluorescence spectra of 3b in 10-6 mol/L ethanol (a) and in crystal state (b) at room temperature
Table 1. Hydrogen bond and other interactions geometry (Å, °)a
| D—H…A | D—H | H…A | D—A | ∠D—H…A |
| 3a | ||||
| O3—H3…O4 | 0.820 | 1.841 | 2.576 | 148.50 |
| O1—H1…O2(ⅰ) | 0.820 | 1.870 | 2.674 | 166.80 |
| C9—H9…O4(ⅱ) | 0.930 | 2.517 | 3.364 | 140.83 |
| 3b | ||||
| O3—H3…O4 | 0.820 | 1.815 | 2.552 | 148.76 |
| O1—H1…O6 | 0.820 | 1.815 | 2.626 | 170.08 |
| C18—H18B…O4(ⅱ) | 0.930 | 2.488 | 3.303 | 146.35 |
| C19(ⅰ)—H19A(ⅰ)…O6 | 0.960 | 2.560 | 3.489 | 162.8 |
| C20—H20B…O3(ⅰ) | 0.960 | 2.754 | 3.524 | 136.41 |
| C20—H20C…O6(ⅰ) | 0.820 | 2.434 | 3.345 | 158.4 |
| a Symmetry codes: 3a (ⅰ) -x, -y, 1-z; (ⅱ) x, 2+y, z-0.5. 3b (ⅰ) 1-x, 1-y, 1-z; (ⅱ) 1-x, y, 0.5-z. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Wavelengths of excitation and emission, fluorescence quantum yields of 3a~3b
| Compd. | Liquid state | Solid state | |||||
| λex/nm | λem/nm | ФF | λex/nm | λem/nm | ФF | ||
| 3a | 351 | 449 | 0.81 | 369 | 486 | 0.32 | |
| 3b | 370 | 453 | 0.84 | 388 | 487 | 0.20 | |
| 2a | 342 | 451 | 0.70 | 368 | 497 | 0.22 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们